

Enzymologists continuously assess their current understanding of how enzymes work against what they were taught as students, and what they hope to accomplish in their future endeavors. It has been a hundred years since the derivation of the Michaelis-Menten equation and the definition of the Michaelis constant, and the event of the original publication of this work has essentially passed from living memory.1 Enzymologists working today have been fortunate to experience an era of tremendous progress in our understanding of the origin of the catalytic rate enhancement for enzymes. This progress has created, in some, the sense that there will soon be full solutions to the problem of the origin of the catalytic rate enhancement for many enzymes.
Structural, kinetic and other mechanistic data are generally sufficient to define the chemical mechanism for an enzymatic reaction, where the mechanism is defined by the number of steps in a reaction and their sequence. The networks of protein-ligand interactions observed in X-ray crystal structures may be used to justify explanations for enzymatic rate enhancements. At the same time, it is difficult to formulate unique and unambiguous explanations for these rate enhancements. Therefore, while enzymologists may speak with confidence when explaining the origin of the rate enhancement for their favorite enzyme, such confidence can project a consensus about important issues of the origin of these rate enhancements that does not in fact exist. This collection of Current Topics is a sampling of ideas regarding the origin of, and explanations for, the catalytic rate enhancement of enzymes. These concepts must be thoroughly examined before arriving at the type of definitive conclusions needed to render future research on the subject pointless. The diversity of ideas expressed in ongoing discussions of enzyme mechanisms reflects a healthy level of disorder that is characteristic of a vibrant area of research, rich with opportunities for discovery and advancement. It is diversity that enzymologists should embrace as we work towards common goals.
This Collection of five articles consists of three contributions from laboratories that focus on experimental studies on enzyme mechanisms, and two from theoretical/computational laboratories where there is an abiding interest in enzyme mechanisms. They partly bridge the gap between experimental chemists who work with real enzymes, and computational chemists who work with improving models for enzymes created in silico. Sharon Hammes-Schiffer has prepared an excellent primer on the underlying physical principles of enzyme catalysis, which provide a basis for the development of computational methods to model enzyme action.2 There is a brief discussion of the ab initio computational methods used to calculate rate constants for turnover of enzyme-bound substrates, followed by a summary of work on three enzymes - dihydrofolate reductase, ketosteroid isomerase and soybean lipoxygenase. The use of general concepts such as hydrogen tunneling, proton donor-acceptor motion, hydrogen bonding, electrostatics and conformational motion in rationalizing the catalytic rate enhancement is described. The review emphasizes the role of equilibrium motions and conformational sampling in enzyme catalysis. It encourages the reader to think deeply about emerging global models for enzyme catalysis that emphasize the importance of electrostatic, hydrogen bonding, hydrophobic and other interactions responsible for stabilization of the enzymatic transition state, alongside the contribution of dynamic protein motions that occur on proceeding from the Michaelis complex to this transition state.
Perhaps the most exciting discovery to arise from comparisons of X-ray crystal structures of free and liganded forms of enzyme catalysts is that ligand binding can result in large changes in protein structure. It would require a profound lack of imagination to fail to appreciate that such conformational changes play a critical role in enzyme catalysis. Fortunately, enzymologists possess imagination in abundance, and this has prompted the development of models that connect the dynamics of enzyme conformational changes with catalytic function. The role of dynamics in catalysis by dihydrofolate reductase (DHFR) is of great current interest. Mutations distant from the active site of this enzyme were found to affect enzyme catalytic activity, but not the structure of enzyme-inhibitor complexes.3 Jiali Gao and coworkers have modeled seminal results from these studies using high-level QM-MM calculations.4 His contribution summarizes the different ways to interpret experimental results that provide evidence for a link between dynamics and enzyme catalytic activity. He settles on an interpretation adopted by most enzymologists, which emphasizes the role of allosteric conformational changes. Gao’s calculations reproduce the effects of the M42W/G121V double mutation at DHFR on the kinetic parameters and kinetic isotope effects for the enzyme-catalyzed reactions. The reduced activity is rationalized by a consideration of the effect of the mutations on the barrier to formation of the transition state for DHFR-catalyzed hydride transfer. Loop conformational motion is found to be severely restricted at this transition state, compared to the transition state for the wildtype enzyme. This restriction of loop motion is reflected by an increase in the entropic barrier to DHFR-catalyzed hydride transfer.
Enzymes that catalyze polar reactions, such as carbonyl group addition, glycosyl transfer and deprotonation of α–carbonyl carbon are faced with the problem of facilitating the formation of highly unstable tetrahedral adducts, oxocarbenium ions, and enolates, respectively. They were among the most intensively studied protein catalysts between 1960 and 1980, in work that was guided by the notion that comparisons of the reaction coordinate profiles for the nonenzymatic and the enzyme-catalyzed processes can provide important insight into the origin of the enzymatic rate enhancement. The activation barrier for a nonenzymatic reaction that proceeds through an unstable intermediate is composed mainly of the thermodynamic barrier to formation of the intermediate, ΔGo (Figure 1). The direct route toward lowering this barrier is to stabilize the enzyme-bound intermediate relative to the enzyme-bound substrate, ΔΔGo (Figure 1). This is in accord with Pauling’s proposal that the large rate enhancements for enzymes are due to the high specificity of the protein catalyst for binding the reaction transition state,5 which according to Hammond closely resembles the reactive intermediate.6 The ligand binding energy required to account for the rate enhancement of such enzymes is generally so large that it cannot be expressed entirely at the ground state Michaelis complex, since this would result in effectively irreversible ligand binding. An important question is the mechanisms by which enzymes bind their transition states with a much higher affinity than substrate, in cases where the structures of the substrate and transition state are similar. These mechanisms are discussed in the review by Amyes and Richard,7 which focuses on enzyme specificity in transition state binding of nonreacting portions of the substrate,8 such as the coenzyme A fragment of CoA esters,9 and of the phosphodianion moiety of phosphate esters.10
Figure 1.

Reaction coordinate profiles for nonenzymatic (upper profile) and enzymatic (lower profile) heterolytic bond cleavage at a reactant R to give a high energy reactive intermediate I. The activation barrier for formation of the intermediate, ΔG‡f, is equal to the sum of the large thermodynamic barrier to formation of the reactive intermediate, ΔGo, and the small barrier for the reaction in the reverse direction, ΔG‡r. If ΔG‡r is not changed on proceeding to the enzymatic reaction, then the stabilization of the intermediate at the active site of an enzyme catalyst (E•I) by an amount ΔΔGo will result in a corresponding reduction in the activation barrier for formation of the intermediate, such that ΔΔG‡f = ΔΔGo.
Dan Herschlag and Aditya Natarajan take a broader prospective in their contribution, which defines several fundamental challenges in mechanistic enzymology.11 This review illustrates how these challenges have been addressed in studies from the Herschlag laboratory. The de novo design of proteins with enzymatic activity poses a unique challenge for scientists in the 21-first century. Herschlag and Natarajan also present an evaluation of recent progress towards the design of proteins with retroaldolase activity, which illustrates the gap between exciting recent achievements in catalyst design and the ultimate goal of obtaining enzyme-like catalytic rate enhancements.
It is important to progress to general conclusions about how enzymes work, but yet to avoid over-generalizations that fail to recognize the different catalytic strategies utilized by different enzymes. For example, there are many enzymatic reactions that proceed by concerted mechanisms that avoid the formation of unstable reaction intermediates. In cases where the concerted reaction is nearly thermoneutral, the catalyst is faced with the problem of reducing a large intrinsic reaction barrier.12 In many cases it is unlikely that the enzymatic rate enhancement can be accounted for by a consideration of the specificity in transition state binding. For example, soybean lipoxygenase catalyzes a proton coupled electron transfer reaction, where formal electron transfer from a C-H bond to form a resonance stabilized radical is coupled to proton transfer to a hydroxyl ligand of the iron cofactor (Scheme). The reaction is treated nonadiabatically, and is described by a rate constant with a prefactor that includes a term describing the overlap between the reactant and product vibrational wavefunctions. In short, this reaction profile is radically different from those used to describe heterolytic bond cleavage to form a thermodynamically unstable polar reaction intermediate. The observation of primary kinetic deuterium isotope effects on soybean lipoxygenase-catalyzed reactions of kH/kD = 80 at room temperature provides strong evidence that the reaction proceeds by tunneling through the free energy reaction barrier, as shown in Figure 2.13 This raises questions about whether such tunneling is strongly favored by protein dynamic motions. Judith Klinman reviews the compelling evidence for a role of protein dynamics in promoting C-H bond cleavage catalyzed by soybean lipoxygenase.14
Scheme.
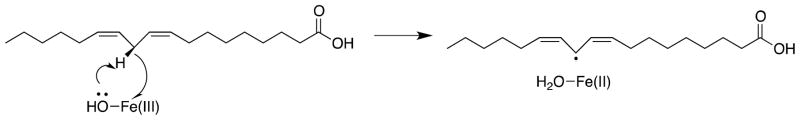
Figure 2.

Free energy profile for a thermoneutral reaction where ΔG‡f = ΔG‡r and for which there is tunneling through the barrier that avoids formation of the classical high-energy transition state.13
The issues that must be resolved in order to achieve a full understanding of enzymatic rate enhancements are too numerous to deal with in these several short reviews. Nevertheless, these articles aim to inform the reader of recent progress on selected problems in enzyme catalysis. They seek to provide a sense of the incredible diversity of enzyme-catalyzed reactions and the difficulties of generalizing results obtained for a particular class of enzymatic reaction across the full spectrum of enzyme catalysts.
Footnotes
The work from the author’s laboratory described in the discussed review was supported by Grant GM39754 from the National Institutes of Health.
References
- 1.Johnson KA, Goody RS. The Original Michaelis Constant: Translation of the 1913 Michaelis–Menten Paper. Biochemistry. 2011;50:8264–8269. doi: 10.1021/bi201284u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammes-Schiffer S. Catalytic Efficiency of Enzymes: A Theoretical Analysis. Biochemistry. 2013;52 doi: 10.1021/bi301515j. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee J, Goodey NM. Catalytic Contributions from Remote Regions of Enzyme Structure. Chem Rev. 2011;111:7595–7624. doi: 10.1021/cr100042n. [DOI] [PubMed] [Google Scholar]
- 4.Fan Y, Cembran A, Ma S, Gao J. Connecting Protein Conformational Dynamics with Catalytic Function As Illustrated in Dihydrofolate Reductase. Biochemistry. 2013;52 doi: 10.1021/bi301559q. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauling L. Nature of forces between large molecules of biological interest. Nature. 1948;161:707–709. doi: 10.1038/161707a0. [DOI] [PubMed] [Google Scholar]
- 6.Hammond GS. A Correlation of Reaction Rates. J Am Chem Soc. 1955;77:334–338. [Google Scholar]
- 7.Amyes TL, Richard JP. Specificity in Transition State Binding: The Pauling Model Revisited. Biochemistry. 2013;52 doi: 10.1021/bi301491r. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jencks WP. Binding energy, specificity, and enzymic catalysis: the Circe effect. Adv Enzymology Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 9.Whitty A, Fierke CA, Jencks WP. Role of binding energy with coenzyme A in catalysis by 3-oxoacid coenzyme A transferase. Biochemistry. 1995;34:11678–11689. doi: 10.1021/bi00037a005. [DOI] [PubMed] [Google Scholar]
- 10.Morrow JR, Amyes TL, Richard JP. Phosphate Binding Energy and Catalysis by Small and Large Molecules. Acc Chem Res. 2008;41:539–548. doi: 10.1021/ar7002013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herschlag D, Natarajan A. Fundamental Challenges in Mechanistic Enzymology: Where Are We in Understanding Enzymatic Rate Enhancements? Biochemistry. 2013;52 doi: 10.1021/bi4000113. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Richard JP, Amyes TL, Toteva MM. Formation and Stability of Carbocations and Carbanions in Water and Intrinsic Barriers to Their Reactions. Acc Chem Res. 2001;34:981–988. doi: 10.1021/ar0000556. [DOI] [PubMed] [Google Scholar]
- 13.Bell RP. The tunnel effect correction for parabolic potential barriers. Trans Faraday Soc. 1959;55:1–4. [Google Scholar]
- 14.Klinman J. Importance of protein dynamics during enzymatic C–H bond cleavage catalysis. Biochemistry. 2013;52 doi: 10.1021/bi301504m. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]