

Abstract
The phosphatidylinositol 3 kinase (PI3K) pathway regulates fundamental cellular processes such as metabolism, proliferation, and survival. A central component in this pathway is the p85α regulatory subunit, encoded by PIK3R1. Using whole-exome sequencing, we identified a heterozygous PIK3R1 mutation (c.1945C>T [p.Arg649Trp]) in two unrelated families affected by partial lipodystrophy, low body mass index, short stature, progeroid face, and Rieger anomaly (SHORT syndrome). This mutation led to impaired interaction between p85α and IRS-1 and reduced AKT-mediated insulin signaling in fibroblasts from affected subjects and in reconstituted Pik3r1-knockout preadipocytes. Normal PI3K activity is critical for adipose differentiation and insulin signaling; the mutated PIK3R1 therefore provides a unique link among lipodystrophy, growth, and insulin signaling.
Main Text
The phosphatidylinositol 3 kinase (PI3K) signaling pathway is central to the action of many hormones and growth factors, including insulin,1–3 and is essential for differentiation of preadipocytes to adipocytes.4–6 Binding of insulin to its receptor leads to intrinsic activation of tyrosine kinase and phosphorylation of insulin-receptor substrates (IRSs) 1 and 2. Phosphorylated IRSs then bind to the regulatory subunit of PI3K, p85α, which is encoded by PIK3R1 (MIM 171833). This results in activation of the p110 catalytic subunit (encoded by PIK3CA [MIM 171834]) and production of phosphatidylinositol 3,4,5-triphosphate (PIP3), which in turn activates its major downstream target, AKT.5 Germline mutations in genes encoding components of the PI3K signaling pathway have been linked to congenital diseases characterized by disturbances in metabolism and cellular growth. Thus, mutations in PTEN (MIM 601728) are associated with cancer predisposition, insulin sensitivity, and obesity,7 whereas activating AKT2 (MIM 164731) mutations cause a syndrome of hypoglycemia and asymmetrical growth.8 Furthermore, germline mutations resulting in gain of function of AKT3 (MIM 611223), PIK3R2 (MIM 603157), and PIK3CA cause disorders characterized by overgrowth (megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome [MIM 603387]).9,10 In addition, somatic mutations of various genes encoding components within this pathway, including PIK3R1, PIK3CA, and AKT3, have been associated with cancer development.10
We investigated two independent families (Table 1) affected by autosomal-dominant SHORT syndrome (short stature, hyperextensibility of joints, ocular depression, Rieger anomaly, and teething delay [MIM 269880]) and also displaying partial lipodystrophy, low body mass index (BMI), and a progeroid face. The study subjects provided written informed consent, and the regional ethics committee approved the study. Statistical significance was calculated with SPSS software and a Mann-Whitney U nonparametric test comparing affected subjects and controls. In family 1,11 four affected subjects in three generations had infancy-onset partial lipodystrophy, low BMI, and dysmorphic features, including a progeroid face (Figures 1A and 1B and Table S1, available online). Affected subjects had low birth weight, short stature, teething delay, and Rieger anomaly, but no joint hyperextensibility. Diabetes developed between 22 and 55 years of age. Available family members were followed up 38 years after the initial description (Table 1). Subjects II-2 and III-2 were treated with insulin, had poor metabolic control, and were very lean (BMI 15.1 and 17.7 kg/m2, respectively). We obtained blood samples after an overnight fast and measured serum levels of lipids, cholesterols, and leptins, which were within the normal range, whereas adiponectin levels were in the upper limits of the normal range (Table 1). Computed-tomography (CT) scans revealed extremely scarce amounts of intra-abdominal and subcutaneous fat in the flanks and slightly scarce subcutaneous fat in the buttocks and thighs (Figure 2). There was no hepatic steatosis.
Table 1.
Clinical and Biochemical Characteristics
| Characteristic |
Family 1 |
Family 2 |
Controlsa |
||||||
|---|---|---|---|---|---|---|---|---|---|
| II-1 | II-2 | II-3 | III-1 | III-2 | II-1 | III-1 | III-2 | n = 16 | |
| Genotype | NM | NM | NN | NN | NM | NM | NN | NM | NN |
| Gender | female | female | male | male | male | female | male | male | 7 males and 9 females |
| Current age (years) | deceased | 79 | 65 | 45 | 42 | 53 | 20 | 15 | 38 ± 9 |
| BMI (kg/m2)b | 22.5 | 15.1 | 25.0 | NA | 17.7 | 17.1 | NA | 13.9 | 21.5 ± 1.7 |
| Partial lipodystrophyc | yes | yes | no | no | yes | yes | no | yes | no |
| Stature (cm) | short (152) | short (152) | tall (192) | normal | short (163) | short (153) | normal | short (161) | normal (175 ± 10) |
| Progeroid faced | yes | yes | no | no | yes | yes | no | yes | no |
| Rieger anomaly | yes | yes | no | no | yes | yes | no | yes | no |
| Diabetes | yes | yes | yes | no | yes | no | no | no | no |
| Insulin treatment (U/kg/day) | NA | 1.4 | varies | no | 1.6 | no | no | no | no |
| Vascular complications | NA | microalbuminuria, neuropathy | yes | no | no | no | no | no | no |
| Fasting glucose (4.0–6.0 mmol/l)e | 5.1 | 12.1 | 14.1 | NA | 23.2 | 5.7 | 5.0 | 5.5 | 5.0 ± 0.3 |
| HbA1c (4.0%–6.4%) | NA | 9.9 | 10.7 | NA | 10.6 | 5.5 | 5.0 | 5.8 | 5.1 ± 0.3 |
| Total cholesterol (3.9–7.8 mmol/l) | NA | 5.5 | 4.6 | NA | 6.2 | 5.0 | 2.8 | 2.7 | 4.6 ± 0.9 |
| HDL cholesterol (0.8–2.1 mmol/l) | NA | 2.0 | 1.0 | NA | 1.5 | 2.2 | 1.1 | 1.5 | 1.9 ± 0.6 |
| LDL cholesterol (1.8–5.7 mmol/l) | NA | 3.4 | 3.2 | NA | 4.4 | 2.7 | 1.5 | 0.9 | 2.8 ± 0.8 |
| Triglycerides (0.45–2.60 mmol/l) | NA | 0.93 | 1.46 | NA | 1.46 | 0.98 | 0.80 | 0.68 | 0.7 ± 0.2 |
| Leptin (pmol/l)f | NA | 285 | NA | NA | 389 | 1,121 | 68 | 61 | 138 ± 36 (males) and 388 ± 248 (females) |
| Adiponectin (150–746 nmol/l) | NA | 560 | NA | NA | 469 | 491 | 439 | 307 | 361 ± 174 |
The following abbreviations are used: NN, normal PIK3R1 genotype; NM, heterozygous PIK3R1 mutation c.1945C>T (p.Arg649Trp); NA, not available or not applicable; HDL, high-density lipoprotein; and LDL, low-density lipoprotein.
We recruited BMI and age-matched controls from the staff at the Haukeland University Hospital.
Reference interval is 18–25 kg/m2.
Mainly affecting the face, flanks, and buttocks.
Midface hypoplasia, wide and deep-set eyes, triangular face, and hypotrichosis.
To convert the values for glucose into mg/dl, multiply by 18.056.
Reference values are <465 pmol/l for males and <930 pmol/l for females.
Figure 1.
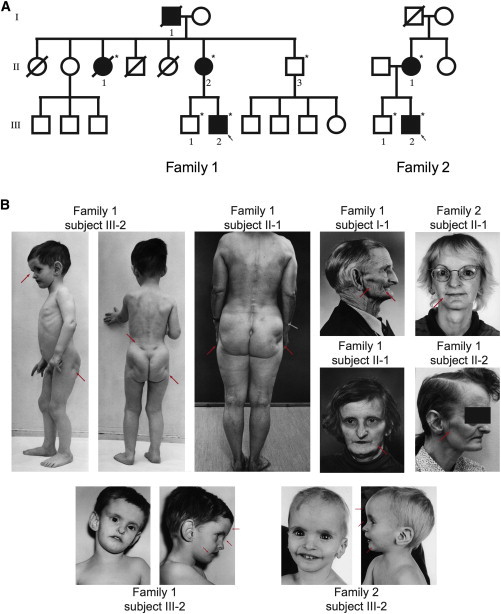
Pedigrees and Photos of Affected Family Members
(A) Pedigrees of Norwegian family 1 and German family 2. Filled symbols represent affected family members, and open symbols represent healthy individuals. DNA from subject I-1 of family 1 was unavailable for testing. The five other affected family members were investigated and had the mutation. Healthy subjects II-3 and III-1 of family 1 and subject III-1 of family 2 did not have the mutation. Circular symbols represent females, and squares represent males. Arrows represent the probands of each family. Stars indicate those individuals from whom DNA was available.
(B) The photos reveal the key clinical signs. Partial lipodystrophy is especially noticeable in the face and the buttocks, and dysmorphic features, including a progeroid face with hypotrichosis (thin hair), frontal bossing, wide and deep-set eyes, thin ala nasi, a beaked nose, downturned lateral corners of the mouth, and large ears, are evident. Red arrows point to the typical features. Photos are printed with permission from John Wiley & Sons11 and Wolters Kluwer Health.12
Figure 2.
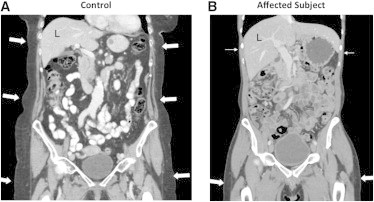
CT Scans
Coronal reconstruction of contrast-enhanced CT scans of the abdomen and proximal lower extremities of subject III-2 of family 1 (B) and a 79-year-old healthy female control (A). In the affected person (B), the amount of subcutaneous fat (arrows) is strikingly sparse in the upper abdomen and lower thorax (closed arrows), whereas it seems normal in the buttocks and thighs (filled arrows). In the control (A), there is proportional distribution of subcutaneous fat (arrows) in the thorax, abdomen, and pelvis. The affected individual had normal liver attenuation (62 versus 8 Hounsfield units in a subject with fatty liver), indicating no signs of hepatic steatosis (not shown). The CT scans were performed according to standard procedures.
In family 2,12 two affected subjects with strikingly similar features to those of affected individuals in family 1 had infancy-onset partial lipodystrophy, low BMI, and progeroid facial features (Figures 1A and 1B and Table S1). They had low birth weight, short stature, teething delay, and Rieger anomaly, but no joint hyperextensibility. The mother exhibited impaired glucose tolerance and mildly elevated postprandial serum insulin. At the follow-up examination, subjects II-1 and III-2 had normal fasting glucose and A1c values (Table 1). Serum lipids were within the normal range, whereas adiponectin levels were in the upper limits of the normal range (Table 1).
To establish the genetic cause of the disease, we performed whole-exome sequencing of DNA from two affected members (subjects II-1 and III-2) and one unaffected member (subject III-1) of family 1. Whole-exome sequencing, read mapping, and variant calling were performed as described in Johansson et al.13 On average, we obtained 39× median coverage, and 88% of the exome was covered at least 8×. We identified an average of 14,253 exonic variants per individual after initial quality control (Table S2). We used a fully penetrant autosomal-dominant model and removed variants that were synonymous, noncoding, or identified in 52 Norwegian control exomes or that had an allele frequency > 0.5% in 1000 Genomes (Table S2). After filtering, 182 exonic variants were classified as rare. Filtering based on an autosomal-dominant model reduced the number of variants to 22 (Table S3). We searched for direct or indirect associations between our 22 candidates and 12 genes involved in insulin signaling or lipodystrophy by performing a protein-based network analysis (STRING). Only one candidate gene, PIK3R1, clustered directly with insulin signaling genes (PIK3CA, IRS1, IRS2, and INSR) and indirectly with lipodystrophy genes (LMNA, BSCL2, AGPAT2, and PPARG) (Figure S1). PIK3R1 had the heterozygous mutation c.1945C>T (RefSeq accession number NM_181523.2), predicted to cause the substitution of arginine with tryptophan at amino acid position 649 (p.Arg649Trp) (RefSeq NP_852664.1).
Sanger resequencing (Figure S2A and S2B) confirmed that all affected members, but not the healthy members (subjects II-3 and III-1), of family 1 carried the c.1945C>T mutation. To exclude that this mutation was common, we sequenced 340 Norwegian controls and found that none of them carried the mutation. Amplification of genomic DNA was carried out with primers 5′-TTTGTCCTGGGAGGTTGCACTGGA-3′ and 5′-AAGGCAGGCACTGCCACTTCCAT-3′, yielding a PCR product of 692 bp. We performed Sanger sequencing with the same primers.
To investigate whether the mutant allele was transcribed, we extracted RNA from cultured skin fibroblasts with the RNeasy Kit (QIAGEN) and subsequently performed cDNA synthesis (OneStep RT-PCR, QIAGEN). A PCR product of 189 bp was amplified from the cDNA with primers 5′-AACACTGAACAATATTCACTGGTGG-3′ and 5′-TTCGCCGTCCACTACAGAGC-3′. Sanger sequencing was then performed with the same primers on an ABI 3730 capillary sequencer (Applied Biosystems). Sequencing the PIK3R1 cDNA isolated from fibroblasts confirmed that the mutated allele was transcribed (Figure S2B).
PIK3R1 encodes the PI3K p85α regulatory subunit, which binds to the catalytic subunit p110 to form the holoenzyme.14 The arginine residue at position 649 is highly conserved. This amino acid is located in the Src homology 2 (SH2) domain and is predicted to be involved in direct binding to phosphorylated substrates including receptor tyrosine kinases and adaptor proteins, such as IRS-1 (Figures 3A and 3B). The mutation (c.1945C>T) was scored by four different bioinformatics programs as follows: PolyPhen-2, “probably damaging” (HumDiv score 1.000, HumVar score 1.000); SIFT, “damaging” (0.00); Mutation Taster, “moderately radical” (by Grantham Matrix, 101); and Align GVGD, “less likely” (class C0).
Figure 3.
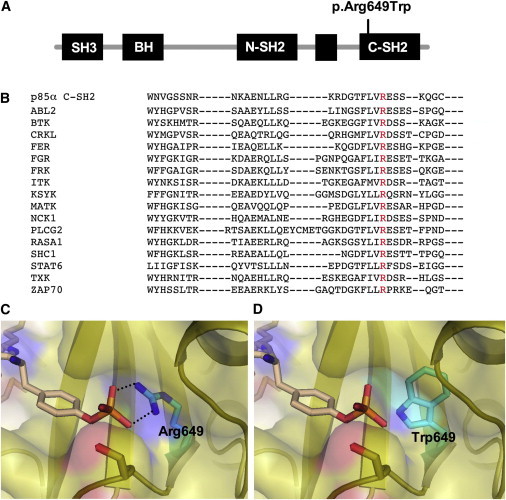
Overview of the p85 Protein with the p.Arg649Trp Substitution and SH2 Sequences of Various Proteins Related to p85
(A) Schematic illustration of p85α, the different domains, and the position of the p.Arg649Trp substitution.
(B) Multiple-sequence alignment of SH2 domains from the indicated human proteins was performed with the Clustal Omega sequence-alignment tool and demonstrated absolute conservation of the arginine residue (red color).
(C and D) Illustration that the p.Arg649Trp-containing protein impairs binding with the phosphotyrosine phosphate moiety. The panels show the substrate-binding pocket of the p85α SH2 domain (Protein Data Bank ID 1H90) with important residues for recognition of the phosphotyrosine of a platelet-derived growth-factor-receptor peptide (pYMPMS).
(C) Arg649 in the wild-type (WT) p85α SH2 domain is critical for the formation of a bidentate interaction with the phosphotyrosine phosphate. Represented by black dotted lines, the distances between the amino group of Arg649 and the phosphate group of pTyr are 2.7 Å (top) and 2.8 Å (bottom).
(D) Introduction of the p.Arg649Trp substitution causes loss of this key interaction for substrate recognition.
Sanger sequencing of DNA from two affected members of family 2 demonstrated an identical PIK3R1 mutation to that of family 1. The mutation was a C>T transition occurring in the context of a CpG dinucleotide. CpGs are potential methylation sites and, as such, are susceptible to mutations because a methylated cytosine can spontaneously deaminate to thymine. This might explain the recurrent nature of the PIK3R1 mutation reported here. We performed Genome-Wide Human SNP Array 6.0 (Affymetrix) genotyping of both probands and their affected parents (child-parent duos) and excluded a recent common ancestry of the families by PLINK analysis. We also used SNPs in the PIK3R1 region to search for contrary homozygosity between the two families. This analysis allowed us to confidently exclude sharing for all but a 41 kb region in which all SNPs surrounding the mutation were uninformative: 67,573,663–67,615,528 bp on chromosome 5 (Human Genome Database build 19; the mutation is located at 67,592,129). Haplotype reconstruction in the region surrounding PIK3R1 showed that the mutations reside on two different haplotype backgrounds (Figure S4). Therefore, they stem from two independent mutational events.
To understand the functional role of the mutation, we made a structural model (PyMOL) of the C-terminal SH2 domain of p85α with arginine (normal) and tryptophan (substituted) residues at position 649 in the phosphopeptide binding pocket in the presence of a platelet-derived growth-factor-receptor phosphopeptide (Figures 3C and 3D). The normal arginine residue participates in a bivalent interaction (2.7 and 2.8 Å) with the oxygen atoms of the phosphotyrosine residue, leading to enhanced contact with the phosphopeptide within the binding pocket. The altered tryptophan residue disrupts this interaction, which could lead to reduced affinity for the phosphopeptide (Figure 3D).
To test this hypothesis directly, we assessed PI3K activation and downstream insulin signaling in fibroblasts prepared from punch biopsies of the skin from a normal (subject II-3) and an affected (subject II-2) family member. Human primary fibroblasts were cultured in Dulbecco's modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (Life Technologies). Cells were serum starved in DMEM supplemented with 0.1% BSA for 3 hr and treated in the absence or presence of 10 nM insulin (Sigma Aldrich) for the indicated time points. Cells were lysed with 1× RIPA lysis buffer (EMD Millipore), and lysates were subjected to SDS-PAGE and subsequently immunoblotted with the indicated antibodies. For PI3K-activation assays, immunoprecipitations with the indicated antibodies were performed overnight in buffer A containing 25 mM Tris-HCl (pH 7.4), 2 mM Na3VO4, 10 mM NaF, 10 mM Na4P2O7, 1 mM EGTA, 1 mM EDTA, and 1% Nonidet P-40 supplemented with protease and phosphatase inhibitor cocktails (Sigma Aldrich). Immune complexes were captured by incubation with Protein A/G Plus Agarose (Santa Cruz Biotechnology) for 1 hr at 4°C. Immunoprecipitates were subsequently washed three times in buffer A containing 500 mM NaCl and two times in PI3K reaction buffer (20 mM Tris-HCl [pH 7.4], 100 mM NaCl, and 0.5 mM EGTA) and suspended in 50 μl of PI3K reaction buffer containing 0.1 mg/ml phosphatidylinositol (Avanti Polar Lipids). The reactions were performed for 30 min at room temperature, and phosphorylated lipids were separated by thin-layer chromatography as described.15
In normal fibroblasts, insulin stimulation led to a robust ∼4-fold increase in phosphotyrosine-associated PI3K activity, whereas in the mutant fibroblasts, only a ∼2-fold increase was observed (Figure 4A). Analysis of PI3K activity in IRS-1 immunoprecipitates revealed a parallel reduction in insulin-stimulated activity in mutant fibroblasts, although this did not quite reach statistical significance (Figure 4B). The observed trend for a reduction in IRS-1-associated PI3K activity was not due to decreased levels of p85α or efficiency of IRS-1 immunoprecipitation in mutant fibroblasts (Figure S3A and S3B). In normal fibroblasts, insulin stimulation led to a robust increase in the activation of downstream effectors of the PI3K pathway, including AKT and GSK-3β (Figure 4C). In contrast, insulin stimulation of fibroblasts from affected individuals revealed that phosphorylation of AKT and GSK-3β was 53% and 12% lower, respectively, than that of normal fibroblasts. This defect in PI3K-dependent signaling was not due to a decrease in the protein levels of components in this pathway, given that levels of p85α, p110, and GSK-3β were unaltered in mutant fibroblasts.
Figure 4.
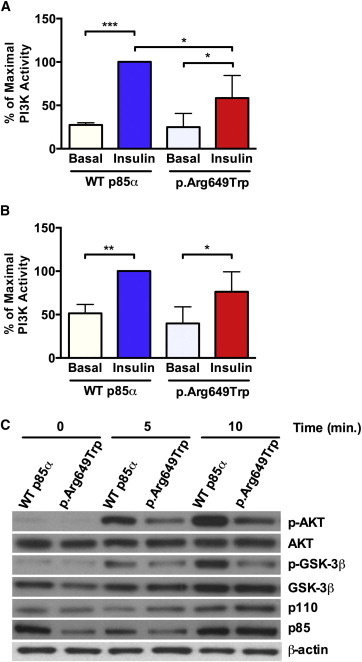
The p85α p.Arg649Trp Substitution Leads to Impaired Insulin Signaling in Primary Human Fibroblasts
(A and B) PI3K activity was evaluated in (A) anti-phosphotyrosine (PY) or (B) anti-IRS-1 immunoprecipitates in WT (normal) and p.Arg649Trp human fibroblast cell lines before and after insulin stimulation (10 mM) for 10 min. Data are expressed as a percentage of maximal stimulation, and p values were calculated by ordinary one-way ANOVA testing (∗p < 0.05, ∗∗∗p < 0.001) (n = 3, data are represented as mean ± SEM).
(C) Immunoblot analysis of lysates obtained from WT (normal) and p.Arg649Trp fibroblasts before and after insulin stimulation was performed with the indicated antibodies. Representative immunoblots are shown.
To further explore the impact of the PIK3R1 mutation, we reconstituted preadipocytes made deficient for both p85α and p85β by genetic knockout (controls)16 with constructs for Arg649 (normal) or Trp649 (substitution) p85α by retroviral-mediated gene transduction. PIK3R1 mutation c.1945C>T was introduced into a human p85α cDNA clone (Thermo Scientific) with the use of the QuickChange Lightening Site-Directed Mutagenesis Kit (Agilent Technologies). Resultant clones were sequenced for verification that the desired mutation had been introduced and were subsequently cloned into the expression plasmid pBabe (Addgene). Reconstituted preadipocytes were cultured in DMEM supplemented with 10% FBS, penicillin, and streptomycin. Cells were serum starved in DMEM supplemented with 0.1% BSA for 6 hr prior to insulin stimulation. A PI3K assay was performed as described above.
In the control cells lacking p85α and p85β, insulin elicited a ∼2-fold increase in phosphotyrosine-associated PI3K activity at 5 and 10 min, indicating that some signaling was possible through alternative pathways (Figure 5A). As expected, this was augmented (∼5-fold) when reconstituted with normal p85α. Interestingly, compared with pBabe controls, cells reconstituted with Trp649 p85α showed a statistically significant (p < 0.05) increase in basal activity, but this activity was not further stimulated by insulin. Assessment of IRS-1-associated activity revealed a significant (p < 0.01), insulin-dependent increase in normal p85α-reconstituted cell lines, but not in mutant cells (Figure 5B). Protein levels and immunoprecipitation efficiency were unaltered between the cell lines examined either before or after insulin stimulation, suggesting that any changes in IRS-1-associated activity were due to recruitment of the PI3K enzyme to IRS-1 (Figure S3C and S3D). Insulin-dependent activation of downstream signals, such as AKT and GSK-3β, was barely detectable in control knockout cells, whereas a robust activation was observed in normal p85α-reconstituted cells (Figures 5C and 5D). Again, compared to pBabe controls, p85α-reconstituted cells showed a statistically significant (p < 0.01), ∼15-fold increase in basal activation, which was not further stimulated by insulin; this is consistent with results obtained for PI3K activation (Figure 5D).
Figure 5.
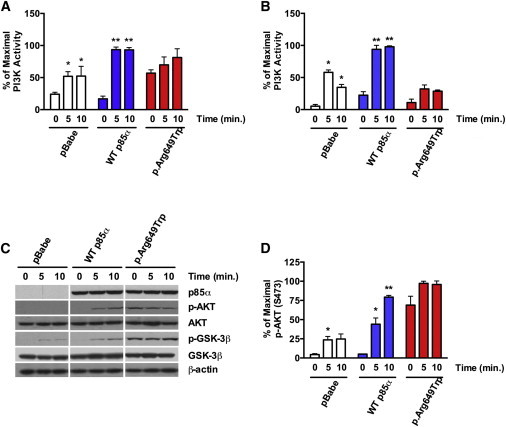
Assessment of PI3K Pathway Activation in WT and p.Arg649Trp-Reconstituted Cell Lines
(A and B) PI3K activity was measured in (A) anti-phosphotyrosine or (B) anti-IRS-1 immunoprecipitates before and after insulin stimulation (10 mM) for 10 min.
(C) Immunoblot analysis using the indicated antibodies was performed on cell lysates obtained from cell lines at the indicated time points after insulin stimulation (10 mM). Representative immunoblots are shown.
(D) Quantification of AKT phosphorylation (S473) was performed with ImageJ. Data are expressed as a percentage of maximal stimulation, and p values were calculated by ordinary one-way ANOVA testing (∗p < 0.05, ∗∗p < 0.01) (n = 3, data are represented as mean ± SEM).
There are multiple inherited clinical syndromes characterized by lipodystrophy, i.e., abnormal distribution of subcutaneous adipose tissue.17,18 Recessive forms can exhibit generalized (Berardinelli-Seip syndrome [MIM 269700]) or partial loss of fat because of mutations in AGPAT2 (MIM 608594), BSCL2 (MIM 269700), CAV1 (MIM 601047), or PTRF (MIM 613327).19 Autosomal-dominant forms are either generalized (Hutchinson-Gilford syndrome [MIM 176670]) as a result of LMNA mutations or partial as a result of mutations in LMNA (Dunnigan syndrome [MIM 151660]), PPARG (MIM 604367), AKT2 (MIM 164731), or PLIN1 (MIM 613877). The disease mechanism of these lipodystrophies mainly involves disturbances in adipocyte differentiation and metabolism, which is reflected in disturbances in serum levels of lipids, leptin, and adiponectin.
The phenotype of our affected individuals seems compatible with that of SHORT syndrome20, in which partial lipodystrophy and facial dysmorphic features are part of the phenotype as well. Some individuals also had insulin resistance and/or diabetes. Certain features develop relatively late in life. This, as well as other modifier genes, might be the cause of the varying clinical presentation described in case reports. Until now, the genetic cause of SHORT syndrome has been unknown. By finding that a PIK3R1 mutation, resulting in the substitution of an amino acid in the SH2 domain, can cause this disease, we have identified a cause of SHORT syndrome and a pathway associated with partial lipodystrophy and growth retardation.
SH2 domains function as regulatory modules of intracellular-signal-transduction pathways by virtue of their high-affinity interaction with tyrosine-phosphorylated peptides.21,22 The majority of disease-causing alterations of the SH2 domain affect highly conserved residues.23 Alignment of 120 human SH2 domains reveals that the arginine residue at position 649 of p85α is conserved in 98% of these motifs. This arginine is predicted to participate in the recognition and binding of tyrosine-phosphorylated substrates.24 When arginine is substituted with tryptophan, structural modeling suggests that p85α loses the interaction with its substrate and predicts a reduced affinity for phosphopeptides. This is supported by our studies in fibroblasts from individuals harboring the PIK3R1 mutation; they showed a decrease in phosphotyrosine and IRS1-associated PI3K activity and a decrease in downstream activation of AKT and GSK-3β in response to insulin.
The p85α SH2 domains also negatively regulate basal enzyme activity through allosteric, inhibitory interactions with the catalytic subunit.25 This has been largely ascribed to the N-terminal SH2 domain. However, reconstituted preadipocytes with the altered p85α exhibited increased basal activity of PI3K and a commensurate increase in the activation of AKT and GSK-3β, supporting an important role for the C-terminal SH2 domain in both basal and insulin-stimulated activation of p110. In contrast to individuals with many other lipodystrophy forms, the individuals studied here exhibited no or very mild insulin resistance and showed near normal levels of lipids, leptin, and adiponectin. This might be due to insulin-independent PI3K activation,26 explaining the elevated basal PI3K levels found in mutant fibroblasts and in preadipocytes reconstituted with Trp649 p85α. Loss of adipose tissue in the affected individuals was confined mainly to the face, flanks, and buttocks. The normal leptin levels in our affected subjects indicate that leptin-substitution therapy in this condition is not warranted. Whether the observed short stature is due to impaired insulin signaling remains to be explored. It is, however, interesting to note that birth weights were low in affected family members, and impaired insulin signaling might therefore be the cause of the intrauterine and postnatal growth retardation. Affected subjects displayed a spectrum of phenotypes that seem developmental in origin and seem to be caused by impaired PI3K signaling rather than by a secondary effect due to the development of insulin resistance. In contrast to family 2, three affected members of family 1 had diabetes. It is noteworthy that family 1 subject II-3, who did not carry the mutation, had diabetes and signs of insulin resistance. Thus, diabetes and possibly insulin resistance might at least partly be due to other susceptibility factors present within this family.
In conclusion, by identifying a genetic cause of SHORT syndrome, we have revealed a mechanism for partial lipodystrophy and growth retardation, adding expanded possibilities for diagnosis and targeted treatment in these diseases.
Acknowledgments
We thank the families for participation, as well as Stig Å. Eide and Louise Grevle for technical assistance, Francisco S. Roque and Inge Jonassen for support with computational analysis, and the University of Bergen Audiovisual Section for help with the photographs. The study was supported by the Research Council of Norway, the University of Bergen, the K.G. Jebsen Foundation, Helse Vest, Innovest, and the European Research Council (to P.R.N.), the Nils Normans Foundation (to S.J.), and National Institutes of Health grant DK55545 (to C.R.K.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Align GVGD, http://agvgd.iarc.fr/agvgd_input.php
Clustal Omega, http://www.ebi.ac.uk/Tools/msa/clustalo/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
MutationTaster, http://www.mutationtaster.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2
PyMOL, http://www.pymol.org
SIFT, http://sift.jcvi.org
STRING, http://string-db.org/
References
- 1.Aubin D., Gagnon A., Sorisky A. Phosphoinositide 3-kinase is required for human adipocyte differentiation in culture. Int J Obes (Lond) 2005;29:1006–1009. doi: 10.1038/sj.ijo.0802961. [DOI] [PubMed] [Google Scholar]
- 2.Engelman J.A., Luo J., Cantley L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat. Rev. Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 3.Ueki K., Fruman D.A., Yballe C.M., Fasshauer M., Klein J., Asano T., Cantley L.C., Kahn C.R. Positive and negative roles of p85 alpha and p85 beta regulatory subunits of phosphoinositide 3-kinase in insulin signaling. J. Biol. Chem. 2003;278:48453–48466. doi: 10.1074/jbc.M305602200. [DOI] [PubMed] [Google Scholar]
- 4.Miki H., Yamauchi T., Suzuki R., Komeda K., Tsuchida A., Kubota N., Terauchi Y., Kamon J., Kaburagi Y., Matsui J. Essential role of insulin receptor substrate 1 (IRS-1) and IRS-2 in adipocyte differentiation. Mol. Cell. Biol. 2001;21:2521–2532. doi: 10.1128/MCB.21.7.2521-2532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Taniguchi C.M., Emanuelli B., Kahn C.R. Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 6.Yu W., Chen Z., Zhang J., Zhang L., Ke H., Huang L., Peng Y., Zhang X., Li S., Lahn B.T., Xiang A.P. Critical role of phosphoinositide 3-kinase cascade in adipogenesis of human mesenchymal stem cells. Mol. Cell. Biochem. 2008;310:11–18. doi: 10.1007/s11010-007-9661-9. [DOI] [PubMed] [Google Scholar]
- 7.Pal A., Barber T.M., Van de Bunt M., Rudge S.A., Zhang Q., Lachlan K.L., Cooper N.S., Linden H., Levy J.C., Wakelam M.J. PTEN mutations as a cause of constitutive insulin sensitivity and obesity. N. Engl. J. Med. 2012;367:1002–1011. doi: 10.1056/NEJMoa1113966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hussain K., Challis B., Rocha N., Payne F., Minic M., Thompson A., Daly A., Scott C., Harris J., Smillie B.J. An activating mutation of AKT2 and human hypoglycemia. Science. 2011;334:474. doi: 10.1126/science.1210878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rivière J.B., Mirzaa G.M., O’Roak B.J., Beddaoui M., Alcantara D., Conway R.L., St-Onge J., Schwartzentruber J.A., Gripp K.W., Nikkel S.M., Finding of Rare Disease Genes (FORGE) Canada Consortium De novo germline and postzygotic mutations in AKT3, PIK3R2 and PIK3CA cause a spectrum of related megalencephaly syndromes. Nat. Genet. 2012;44:934–940. doi: 10.1038/ng.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rodon J., Dienstmann R., Serra V., Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10:143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 11.Aarskog D., Ose L., Pande H., Eide N. Autosomal dominant partial lipodystrophy associated with Rieger anomaly, short stature, and insulinopenic diabetes. Am. J. Med. Genet. 1983;15:29–38. doi: 10.1002/ajmg.1320150104. [DOI] [PubMed] [Google Scholar]
- 12.Koenig R., Brendel L., Fuchs S. SHORT syndrome. Clin. Dysmorphol. 2003;12:45–49. doi: 10.1097/00019605-200301000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Johansson S., Irgens H., Chudasama K.K., Molnes J., Aerts J., Roque F.S., Jonassen I., Levy S., Lima K., Knappskog P.M. Exome sequencing and genetic testing for MODY. PLoS ONE. 2012;7:e38050. doi: 10.1371/journal.pone.0038050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winnay J.N., Boucher J., Mori M.A., Ueki K., Kahn C.R. A regulatory subunit of phosphoinositide 3-kinase increases the nuclear accumulation of X-box-binding protein-1 to modulate the unfolded protein response. Nat. Med. 2010;16:438–445. doi: 10.1038/nm.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueki K., Algenstaedt P., Mauvais-Jarvis F., Kahn C.R. Positive and negative regulation of phosphoinositide 3-kinase-dependent signaling pathways by three different gene products of the p85alpha regulatory subunit. Mol. Cell. Biol. 2000;20:8035–8046. doi: 10.1128/mcb.20.21.8035-8046.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klein J., Fasshauer M., Ito M., Lowell B.B., Benito M., Kahn C.R. beta(3)-adrenergic stimulation differentially inhibits insulin signaling and decreases insulin-induced glucose uptake in brown adipocytes. J. Biol. Chem. 1999;274:34795–34802. doi: 10.1074/jbc.274.49.34795. [DOI] [PubMed] [Google Scholar]
- 17.Garg A. Acquired and inherited lipodystrophies. N. Engl. J. Med. 2004;350:1220–1234. doi: 10.1056/NEJMra025261. [DOI] [PubMed] [Google Scholar]
- 18.Fiorenza C.G., Chou S.H., Mantzoros C.S. Lipodystrophy: pathophysiology and advances in treatment. Nat Rev Endocrinol. 2011;7:137–150. doi: 10.1038/nrendo.2010.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Garg A. Clinical review#: Lipodystrophies: genetic and acquired body fat disorders. J. Clin. Endocrinol. Metab. 2011;96:3313–3325. doi: 10.1210/jc.2011-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gorlin R.J., Cervenka J., Moller K., Horrobin M., Witkop C.J., Jr. Malformation syndromes. A selected miscellany. Birth Defects Orig. Artic. Ser. 1975;11:39–50. [PubMed] [Google Scholar]
- 21.Marengere L.E., Pawson T. Structure and function of SH2 domains. J. Cell Sci. Suppl. 1994;18:97–104. doi: 10.1242/jcs.1994.supplement_18.14. [DOI] [PubMed] [Google Scholar]
- 22.Songyang Z., Shoelson S.E., Chaudhuri M., Gish G., Pawson T., Haser W.G., King F., Roberts T., Ratnofsky S., Lechleider R.J. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 23.Lappalainen I., Thusberg J., Shen B., Vihinen M. Genome wide analysis of pathogenic SH2 domain mutations. Proteins. 2008;72:779–792. doi: 10.1002/prot.21970. [DOI] [PubMed] [Google Scholar]
- 24.Waksman G., Shoelson S.E., Pant N., Cowburn D., Kuriyan J. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell. 1993;72:779–790. doi: 10.1016/0092-8674(93)90405-f. [DOI] [PubMed] [Google Scholar]
- 25.Dupuis J., Langenberg C., Prokopenko I., Saxena R., Soranzo N., Jackson A.U., Wheeler E., Glazer N.L., Bouatia-Naji N., Gloyn A.L., DIAGRAM Consortium. GIANT Consortium. Global BPgen Consortium. Anders Hamsten on behalf of Procardis Consortium. MAGIC investigators New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pereira R.I., Draznin B. Inhibition of the phosphatidylinositol 3′-kinase signaling pathway leads to decreased insulin-stimulated adiponectin secretion from 3T3-L1 adipocytes. Metabolism. 2005;54:1636–1643. doi: 10.1016/j.metabol.2005.07.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.