

Abstract
We propose a general statistical framework for meta-analysis of gene- or region-based multimarker rare variant association tests in sequencing association studies. In genome-wide association studies, single-marker meta-analysis has been widely used to increase statistical power by combining results via regression coefficients and standard errors from different studies. In analysis of rare variants in sequencing studies, region-based multimarker tests are often used to increase power. We propose meta-analysis methods for commonly used gene- or region-based rare variants tests, such as burden tests and variance component tests. Because estimation of regression coefficients of individual rare variants is often unstable or not feasible, the proposed method avoids this difficulty by calculating score statistics instead that only require fitting the null model for each study and then aggregating these score statistics across studies. Our proposed meta-analysis rare variant association tests are conducted based on study-specific summary statistics, specifically score statistics for each variant and between-variant covariance-type (linkage disequilibrium) relationship statistics for each gene or region. The proposed methods are able to incorporate different levels of heterogeneity of genetic effects across studies and are applicable to meta-analysis of multiple ancestry groups. We show that the proposed methods are essentially as powerful as joint analysis by directly pooling individual level genotype data. We conduct extensive simulations to evaluate the performance of our methods by varying levels of heterogeneity across studies, and we apply the proposed methods to meta-analysis of rare variant effects in a multicohort study of the genetics of blood lipid levels.
Introduction
With the rapid advance of high-throughput sequencing technologies,1 rare variant association analysis is increasingly being conducted to identify genetic variants associated with complex traits. In recent years, significant effort has been devoted to develop powerful and efficient statistical methods to test for such associations. Because single-variant tests are underpowered to investigate rare variant effects unless sample sizes or effect sizes are large,2 region-based multimarker tests have been commonly used in an attempt to improve analysis power. For example, collapsing or burden tests summarize rare variant information within a gene or region into a single genetic score or genetic burden before performing association analysis.3–5 Variance component tests such as C-alpha6 and SKAT7 aggregate individual variant test statistics in a gene or region. Recently, unified tests that combine burden and variance component tests have been proposed.8,9
In genome-wide association studies (GWASs) of common variants, a single study is often underpowered to detect modest genetic effects.10 To overcome this limitation, meta-analysis is routinely used to analyze data across studies.11 Meta-analysis has several advantages over joint analysis of individual level data. Because meta-analysis uses study-specific summary statistics, it allows investigators to combine information across studies when individual-level data cannot be shared. Different studies often require specific sets of covariates, which can be difficult to accommodate in joint analysis. Finally, the summary statistic data files are much smaller than individual level data files, making for easier data transfer. For single-variant tests of common variants, it has been shown that meta-analysis can be essentially as powerful as joint analysis.12 Hundreds of trait-associated common variants have been discovered by meta-analysis.13–16
Detecting rare variant associations in sequencing studies probably will often require even larger sample sizes than common variant-oriented GWASs, making meta-analysis important for the identification of rare susceptibility alleles. However, little work has been done to develop meta-analysis methods for gene- or region-based multimarker tests. Although existing single-marker methods can be used for burden tests, no meta-analysis method exists for variance component and unified tests.
For single-marker tests of common variants from GWASs, meta-analysis typically analyzes regression coefficients and their standard errors across studies. However, because of low minor allele frequencies, estimated regression coefficients of rare variants in multimarker regression models are often unstable with very large variances, or regression models often fail to converge in the presence of many rare variants in a gene or region. Therefore, it is important to develop meta-analysis methods for rare variants that do not require estimating regression coefficients of rare variants.
In this paper, we propose a general framework for meta-analysis for gene- or region-based rare variant analysis for both continuous and binary traits. Unlike the traditional regression coefficient-based single-marker meta-analysis, a key advantage of the proposed method is that it aggregates score statistics, avoiding the need to estimate regression coefficients of rare variants. As variant component score tests that require fitting only the null model, the proposed methods are computationally efficient even for whole-genome analysis, and p values can be calculated analytically. Our meta-analysis framework uses gene-level summary statistics and is applicable to burden tests, variance component tests, and unified tests. The proposed approach can accommodate different levels of heterogeneity of genetic effects across studies,17 including between-ancestry heterogeneity,18 while achieving power similar to that of joint analysis. We evaluate the performance of the proposed methods through computer simulation and analysis of Metabochip array data for eight European cohorts to assess association of rare variants in lipoprotein lipase (LPL [MIM 609708]) gene with serum lipids.
Methods
Burden Tests, SKAT, and SKAT-O for a Single Study
Suppose one conducts a meta-analysis with K studies and performs a region- or gene-based analysis of rare variants. For the kth study, nk subjects are sequenced in a region that has mk variants. Let yki be the phenotype of the ith individual, let be a vector of mk genotypes (gkij = 0, 1, or 2) in the region, and let Xki be a vector of qk covariates including an intercept.
We consider the linear regression model
| (Equation 1) |
when phenotypes are continuous and the logistic regression model
| (Equation 2) |
when phenotypes are binary, where and are vectors of regression coefficients of covariate and genetic effects, respectively. The null hypothesis of no genetic association between variants in the region and the phenotype is H0: βk = 0.
Let be the score statistic of the jth variant in linear (for continuous traits) or logistic (binary traits) regression models, where is the estimated mean of yki under the null linear or logistic regression model, and for continuous traits and for binary traits with being estimated under the null linear model. For study k, the SKAT statistic for testing H0: βk = 0 is derived by assuming that the βkj has a distribution with mean 0 and variances and calculating a variance component score test for H0: τ = 0. The SKAT statistic7 is
where wkj is a weight for the variant j.
For example, Madsen and Browning (2009)4 proposed to up-weight rarer variants, where MAFj is the minor allele frequency of variant j. Note that they estimated MAFj by using only control samples in case-control studies. Wu et al. (2011)7 proposed the flexible beta density function wkj = Beta(MAFj, a1,a2) as weights with MAFj being estimated based on all study participants. The burden test statistic using the same weight first aggregates the rare variants in the region and then regresses the phenotype yki on the weighted total number of rare variants and can be written as
Recently, we9,19 proposed a unified approach that combines SKAT and the burden test into one framework as
a weighted average of the SKAT and burden test statistics. We demonstrated that ρ can be interpreted as a pair-wise correlation among the genetic effects coefficients βkj, that is, ρ = corr(βkj, βkj’) for j≠ j′.19 The asymptotic null distribution of Qk(ρ) is a mixture of chi-square distributions,20 and asymptotic p values can be obtained analytically by the Davies method,21,22 which approximates the inverse of the characteristic function.
Because in practice an optimal ρ is not known, we proposed an adaptive procedure SKAT-O to find an optimal ρ to maximize power.9 The SKAT-O test statistic T = min pk (ρ), where pk (ρ) is the p value for Qk(ρ). Hence, SKAT-O adaptively selects the optimal linear combination of SKAT and the burden test statistics. We derived the null distribution of the SKAT-O statistics; p values can be calculated by a computationally efficient one-dimensional numerical integration.19
Meta-analysis SKAT and SKAT-O
Input from Each Study
Single-variant meta-analysis can be conducted with study-specific summary statistics, such as the SNP estimated regression coefficient and its standard error. This same approach can be applied to meta-analysis by the burden test for rare variants, because burden tests collapse or summarize the rare variants within in a region into a single value. For a nonburden multimarker test, such as C-alpha or SKAT, meta-analysis can still be performed with summary statistics. However, additional summary statistics are necessary for calculating meta-analysis p values. For meta-analysis of variance component-based rare variant tests, such as SKAT, the required summary statistics for each study are the MAFs, the score statistics (Skj) for each variant, and the regional between-variant relationship matrix Φk.
Suppose Gk is an nk × mk genotype matrix, and is an nk × qk covariate matrix. The between-variant relationship matrix is
where is the projection matrix accounting for the fact that the effects of covariates Xk are estimated under the null model, for continuous traits (where I is an identity matrix), and for binary traits.
We show that if Xk has only an intercept term, then Φk is essentially the covariance matrix of the mk genetic markers up to a scale parameter. Suppose φkjl is the (j,l) element of Φk. When Xk has only an intercept,
where is the estimated variance of yki and is the mean of variant j. Therefore, Φk is a scaled sample covariance matrix of mk genetic markers up to the scale parameter .
Meta-analysis Assuming Homogeneous Genetic Effects across Studies
For simplicity, we here assume that all variants are observed in all K studies, so that m = m1 = … = mk. We relax this assumption later.
When genetic effects are homogeneous across studies, the effect size parameters β1 = β2 = … = βK, consistent with the assumption made for single-marker fixed-effect meta-analysis. Under this assumption, we use the following test statistic for meta-analysis:
| (Equation 3) |
When ρ = 0, Qhom-meta(0) corresponds to the meta-analysis version of SKAT, which first collapses the weighted score of the jth variant across studies and then aggregates the squared collapsed score statistics across the m variants within the region. When ρ = 1, Qhom-meta(1) corresponds to the meta-analysis burden test, which assumes that the effects of the variants in a region are the same for all the variants across studies. Qhom-meta-SKAT and Qmeta-Burden are identical to the joint analysis SKAT and burden test statistics assuming stratified covariate effects by study. In contrast to joint analysis, in which the same set of covariates needs to be used for all studies, the proposed meta-analysis allows study-specific covariates to be used.
Suppose Wk = diag[wk1, …, wkm] is an m × m diagonal matrix of marker-specific weights, where wkj will typically be a function of the MAF of SNP j. For example, wkj = Beta(MAFj, a1,a2)7 and (λ1, …, λm) are nonzero eigenvalues of Φρ = Lρ`(W1 Φ1 W1 + … + WK ΦK WK) Lρ, where Lρ is a Cholesky decomposition matrix of the compound symmetric matrix Rρ = (1- ρ) I + ρ11`, that is, Lρ Lρ` = Rρ. It can easily be shown that the asymptotic null distribution of Qhom-meta(ρ) is a mixture of chi-square distributions, , where are independent chi-square df = 1 random variables (Appendix B). Therefore, asymptotic p values can be calculated analytically by the Davies method.21,22
The homogeneous meta-analysis SKAT-O test statistic can be approximated by a simple grid search. We set a grid of D points 0 = ρ1 < ρ2 < … < ρD = 1, then Thom-meta = min [ p(ρ1), …, p(ρD) ], where p(ρ) is the p value of Qhom-meta(ρ). The asymptotic null distribution and the p value for Thom-meta can be obtained analytically (Appendix C).
Meta-analysis Assuming Heterogeneous Genetic Effects across Studies
Genetic effects may be heterogeneous across studies because of between-study heterogeneity, such as differences in ancestries. To allow for such heterogeneity, we modify Equation 3 with an assumption that effect sizes of markers in different studies are independent and follow a common distribution. The meta-analysis SKAT test statistic allowing for heterogeneity is
| (Equation 4) |
and the corresponding test statistic of the unified SKAT and burden test is
| (Equation 5) |
which is derived assuming that the regression coefficients βkj have correlation ρ for k ≠ k′ or j ≠ j′ (Appendix A). When ρ = 0, Qhet-meta(ρ) reduces to the heterogeneous meta-analysis SKAT statistic Qhet-meta-SKAT. We use the same burden test statistic for Qhom-meta(ρ) and Qhet-meta(ρ). The statistic Qhet-meta(ρ) can be viewed as an extension of the random effects model based meta-analysis for single variants.
In some meta-analyses, studies can be naturally grouped, for example based on ancestry. In this case, we might assume that genetic effects for the same ancestry group are homogeneous, whereas those for different ancestries are heterogeneous.18 Suppose K studies can be grouped into B ancestries and there are Kb studies in the bth ancestry group. The first K1 studies belong to the first ancestry group, the next K2 studies belong to the second ancestry group, and so forth. Let with . Then the heterogeneous meta-analysis SKAT test statistic can be written as
| (Equation 6) |
which first collapses the weighted score statistic of the jth marker across the studies in the same ancestry group and then aggregates the squared collapsed scores across ancestry groups and the m markers in the region. The corresponding unified test statistic has the same form as Equation 5 with Qhet-meta-SKAT in Equation 6 instead of Equation 4. Clearly Equation 4 is a special case of Equation 5 in which B = 1 and K1 = K. The generalized heterogeneous meta-analysis SKAT-O test statistic can be obtained as described in the previous section. Detailed derivations of the asymptotic null distributions for meta-analysis SKAT and SKAT-O are provided in Appendices B and C.
Weights for Variants and Dealing with Missing Variants
In rare variant analysis, MAF-based weighting schemes are often used to improve power.4,7 When genetic effects are assumed to be homogeneous across studies, the pooled estimates of MAFs can be used to construct the common weights for all studies. If studies are grouped based on ancestry, one can use ancestry-specific MAF-based weights. Further, we may test for association by using only a subset of variants selected based on bioinformatics analysis,23,24 for example by restricting analysis to nonsynonymous SNPs or constructing weights with Polyphen24 scores.
Some variants may be observed in only a subset of studies. If variant j was not observed in study k, we set Skj = 0 and φkjl = φklj = 0 for all l = 1, …, m, where φkjl is the (j,l) element of Φk. This corresponds to using a zero weight for the jth variant in the kth study (wkj = 0).
Numerical Simulations
To evaluate the proposed methods, we generated 10,000 European-like (EUR) and 10,000 admixed African-American-like (AA) haplotypes of length 200 kb by using the calibrated coalescent model.25 Because the average total exon length of a gene is ∼3 kb,26 we randomly selected a 3 kb region for each simulated data set and tested for association between the selected region and phenotypes.
To assess test calibration and estimate power, we considered six different scenarios (Table 1). Scenario 1 has three studies with the same set of covariates and scenarios 2 and 3 have three studies with different covariates. In scenarios 1 and 2, all studies were comprised of EUR samples; in scenario 3, the first two studies were comprised of EUR samples and the third study was comprised of AA samples. Scenarios 1–3 allow different sample sizes for different studies. Scenarios 4–6 are similar to scenarios 1–3 but assume the same sample sizes for all studies.
Table 1.
Simulation Study Settings
|
Sample Sizesa |
Covariatesb |
||||||
|---|---|---|---|---|---|---|---|
| Scenario | Pop.c | Study 1 | Study 2 | Study 3 | Study 1 | Study 2 | Study 3 |
| Different Cohort Sizes | |||||||
| 1 | EUR | 1,600 (800, 800) | 2,200 (1,000, 1,200) | 3,200 (1,800, 1,400) | (X1, X2) | (X1, X2) | (X1, X2) |
| 2 | EUR | 1,600 (800, 800) | 2,200 (1,000, 1,200) | 3,200 (1,800, 1,400) | (X1) | (X1, X2) | (X1, X2, X2) |
| 3 | EUR+AA | 1,600 (800, 800) | 2,200 (1,000, 1,200) | 3,200 (1,800, 1,400) | (X1) | (X1, X2) | (X1, X2, X2) |
| Equal Cohort Sizes | |||||||
| 4 | EUR | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | (X1, X2) | (X1, X2) | (X1, X2) |
| 5 | EUR | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | (X1) | (X1, X2) | (X1, X2, X2) |
| 6 | EUR+AA | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | 2,400 (1,000, 1,400) | (X1) | (X1, X2) | (X1, X2, X2) |
Total sample sizes, with numbers of cases and controls in the parentheses for simulation of binary traits.
Number of covariates in each study.
EUR refers to the scenario where all three studies had EUR samples. EUR + AA refers to the scenario where studies 1 and 2 had EUR samples and study 3 had AA samples.
We compared eight meta-analysis methods: SKAT-O and SKAT assuming homogeneous genetic effects across studies (Hom-Meta-SKAT-O and Hom-Meta-SKAT); SKAT-O and SKAT assuming heterogeneous genetic effects across studies (Het-Meta-SKAT-O and Het-Meta-SKAT); Fisher’s inverse chi-square27 and Stouffer’s Z score28 methods based on individual study SKAT-O p values (Meta-Fisher and Meta-Z); and fixed and random-effect model-based meta-analysis burden tests (Meta-Burden and Meta-Burden-RE). We used a grid search for in ρ values (0 ≤ ρ ≤ 1) for Hom-Meta-SKAT-O, Het-Meta-SKAT-O, Meta-Fisher, and Meta-Z analyses (Appendix C). For Meta-Z,30 Z scores of the kth study were computed as Zk = F−1(1 − pvaluek), where F is the standard normal cumulative distribution function and pvaluek is the p value of study k calculated by SKAT-O. The square roots of sample sizes were used as weights when Z scores were combined.29 Stouffer’s Meta-Z differs from the standard Z score method used in single-variant meta-analysis29,30 that uses the study-specific signs of the regression coefficients to compute Z scores. This standard single-marker-based Z score method cannot be directly generalized to multimarker tests because there is no obvious way to provide a sign for the joint multimarker effects. We used the inverse variance weighting for Meta-Burden and the REML estimation of variance component for Meta-Burden-RE via the metafor package.31
For all methods, we used beta(MAF; 1, 25) weights to up-weight rare variants (Figure S2 available online). We used MAF estimates pooled across all study cohorts to construct weights for Hom-Meta-SKAT and Hom-Meta-SKAT-O and used cohort-specific MAFs for Het-Meta-SKAT and Het-Meta-SKAT-O analyses, except for scenarios 3 and 6 where we used MAF estimates pooled within ancestry groups. Because the other methods cannot use common or ancestry-specific weights, we used cohort-specific weights.
Type I Error and Power Simulations
We generated continuous phenotypes according to the linear model
and binary phenotypes according to the logistic model
where Gki,causal is a genotype vector containing the causal variants and βk,causal is a vector of regression coefficients of genetic effects of the causal variants. X1k is a binary covariate taking values 0 and 1 each with probability 0.5, other covariates were continuous and distributed as standard normal, and qk indicates the number of covariates for study k (Table 1). For the binary trait simulations, we chose the intercept α0k to correspond to prevalence 0.01 or 0.10 when there was no genetic effect, and retrospective case-control phenotypes were generated, where the numbers of cases and controls are given in Table 1.
For type I error simulations, we set βk,causal = 0. To reduce the computational burden at stringent α levels, we first generated 50,000 genotype sets for randomly selected regions and then generated 500 phenotype sets for each genotype set. We set α = 2.5 × 10−6, 10−4, and 10−2, corresponding to genome-wide studies of 20,000 genes and candidate gene studies of 500 and 5 genes.
For power simulations, we assumed that 5%, 10%, 20%, or 50% of rare variants were causal. For each setting, we assumed either all causal SNPs were of positive effect or 80% were positive and 20% negative. Because it is possible that rarer variants have larger effect sizes, we modeled genetic marker regression coefficients as β = c|log10(MAF)|. For continuous traits, we set c = 0.25 when 20% of the rare variants were causal, which gave effect size |β| = 0.38 at MAF = 0.03. We used c = 0.475, c = 0.375, and c = 0.175, when 5%, 10%, and 50% of the rare variants were causal variants to compensate for the increased and decreased number of causal variants. For binary trait simulations, we used c = 0.35, when 20% of the rare variants were causal, which gave OR = 1.7 at MAF = 0.03. We used c = 0.6, c = 0.46, and c = 0.27, when 5%, 10%, and 50% of the rare variants were causal. For each simulation setting, we generated 1,000 data sets. Power was estimated as the fraction of p values less than α = 2.5 × 10−6, 10−4, and 10−2.
Rare Variant Association Meta-analysis of Lipid Traits in Eight European Cohorts
We analyzed data from eight studies collected in Northern and Western Europe: from Finland we used D2D 2007, METSIM, FUSION Stage 2, DPS, and DR’s EXTRA; from Norway, HUNT and Tromsø; and from Germany, DIAGEN (Table S3). Samples were collected according to protocols approved by local institutional review boards, and all individuals provided informed consent. DNA samples were genotyped on the Metabochip, a custom genotyping array that includes SNPs to fine map 257 known associations for cardiometabolic traits.32 Genotyping was performed at the Center for Inherited Disease Research (CIDR) and genotypes called with BeadStudio Genotyping Module, v.3.3.7. We excluded samples and SNPs with call rate < 98% and excluded one member of each of 970 first- and second-degree relative pairs identified via KING.33 For illustration, we selected LPL, a gene known to play a role in lipid biology. We identified seven UTR and protein-coding SNPs with MAF < 0.05 (Table S2) and analyzed them for association with HDL cholesterol, LDL cholesterol, and triglycerides (TG).
The two Norwegian cohorts were analyzed jointly, with a covariate for study origin; all other cohorts were analyzed individually. Age, age2, sex, and type 2 diabetes status were included as covariates, except for METSIM, which is comprised only of males. Because LPL was selected based on association with the common variant rs12678919 and we sought to identify independent association signals arising from rare variants in addition to common variants, we included rs12678919 genotype as a covariate. For each study, we performed inverse normal transformations on the residuals of a linear regression of raw (HDL and LDL) or log-transformed (TG) phenotypes on the covariates. Total sample sizes were 11,438; 10,619; and 11,004 for HDL, LDL, and TG, respectively.
Results
Type I Error Simulation Results
For these studies of 7,000 individuals, empirical type I error rates of the proposed Meta-SKAT and Meta-SKAT-O approaches are given in Table 2. The MAF spectrum of population allele frequencies in the simulated data (Figure S1) shows that the majority of variants were very rare. For example, 86% and 76% of variants in EUR had population MAF < 0.01 and < 0.001. Type I error rates were well controlled at significance levels α = 10−2 and 10−4 for both continuous and binary phenotypes. At α = 2.5 × 10−6, type I error rates were slightly inflated for continuous traits (with the exception of Het-Meta-SKAT-O) and slightly deflated for binary traits.
Table 2.
Type I Error Rate Estimates at Different α Levels via Simulation Studies under Scenarios 1–3 Based on 2.5 × 107 Simulations
| Scenarios | Level α | Hom-Meta-SKAT-O | Hom-Meta-SKAT | Het-Meta-SKAT-O | Het-Meta-SKAT |
|---|---|---|---|---|---|
| Continuous Trait | |||||
| Scenario 1 | 10−2 | 1.12 × 10−2 | 1.02 × 10−2 | 1.08 × 10−2 | 1.03 × 10−2 |
| 10−4 | 1.24 × 10−4 | 1.07 × 10−4 | 1.06 × 10−4 | 1.12 × 10−4 | |
| 2.5 × 10−6 | 3.16 × 10−6 | 3.12 × 10−6 | 2.00 × 10−6 | 3.16 × 10−6 | |
| Scenario 2 | 10−2 | 1.14 × 10−2 | 1.02 × 10−2 | 1.10 × 10−2 | 1.05 × 10−2 |
| 10−4 | 1.27 × 10−4 | 1.10 × 10−4 | 1.05 × 10−4 | 1.16 × 10−4 | |
| 2.5 × 10−6 | 3.32 × 10−6 | 2.84 × 10−6 | 2.20 × 10−6 | 3.32 × 10−6 | |
| Scenario 3 |
10−2 | 1.13 × 10−2 | 1.04 × 10−2 | 1.13 × 10−2 | 1.06 × 10−2 |
| 10−4 | 1.17 × 10−4 | 1.10 × 10−4 | 1.10 × 10−4 | 1.14 × 10−4 | |
| 2.5 × 10−6 | 2.76 × 10−6 | 3.44 × 10−6 | 2.52 × 10−6 | 3.00 × 10−6 | |
| Binary Trait | |||||
| Scenario 1 | 10−2 | 1.10 × 10−2 | 0.98 × 10−2 | 1.04 × 10−2 | 0.97 × 10−2 |
| 10−4 | 1.14 × 10−4 | 0.96 × 10−4 | 0.90 × 10−4 | 0.87 × 10−4 | |
| 2.5 × 10−6 | 2.56 × 10−6 | 2.24 × 10−6 | 1.52 × 10−6 | 1.88 × 10−6 | |
| Scenario 2 | 10−2 | 1.12 × 10−2 | 0.99 × 10−2 | 1.05 × 10−2 | 0.96 × 10−2 |
| 10−4 | 1.13 × 10−4 | 0.94 × 10−4 | 0.86 × 10−4 | 0.86 × 10−4 | |
| 2.5 × 10−6 | 2.64 × 10−6 | 2.00 × 10−6 | 1.68 × 10−6 | 2.04 × 10−6 | |
| Scenario 3 | 10−2 | 1.08 × 10−2 | 0.99 × 10−2 | 1.07 × 10−2 | 0.97 × 10−2 |
| 10−4 | 0.99 × 10−4 | 0.92 × 10−4 | 0.90 × 10−4 | 0.89 × 10−4 | |
| 2.5 × 10−6 | 1.80 × 10−6 | 1.68 × 10−6 | 1.24 × 10−6 | 1.56 × 10−6 | |
Each entry represents a type I error rate estimate calculated by the proportion of empirical p values smaller than the given level α.
Power Simulation Results
To investigate the effect on power of different degrees of heterogeneity of genetic effects across studies, we considered six scenarios (see Methods). The average numbers of observed variants ranged from 42 to 47 and the sum of causal allele MAFs from 0.003 to 0.07 (Table S1). Under scenario 3, EUR and AA cohorts shared on average 2.2%, 3.5%, 7%, and 17% of causal variants when 5%, 10%, 20%, and 50% of variants were causal.
We first compared power of the proposed meta-analysis methods with individual level data joint analysis. Figure 1 shows that for scenarios 1 and 4, the power of meta-analysis via Hom-Meta-SKAT and Hom-Meta-SKAT-O are nearly identical to those for joint analysis via SKAT and SKAT-O. We considered scenarios 1 and 4 only because the other scenarios allowed studies to have different sets of covariates, which creates difficulties in applying joint analysis.
Figure 1.
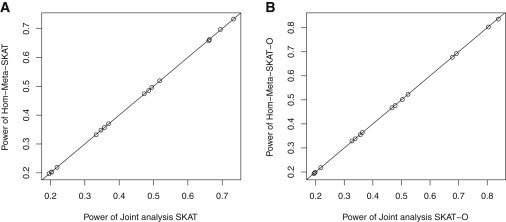
Comparisons of Meta and Joint Analysis of SKAT and SKAT-O
Sixteen dots represent sixteen combinations of scenarios (scenarios 1 and 4), the percentage of causal variants (5%, 10%, 20%, and 50%), and the percentage of risk-decreasing variants (0% and 20%). Empirical powers were obtained at α = 2.5 × 10−6.
(A) Comparison of Hom-Meta-SKAT and joint analysis SKAT.
(B) Comparison of Hom-Meta-SKAT-O and joint analysis SKAT-O.
Figure 2 compares the performance of the proposed and competing meta-analysis methods in a situation in which all causal variants were risk increasing. The performance of the methods varied depending on the degree of heterogeneity of genetic effects and the percentage of causal variants. When genetic effects were homogeneous across studies (scenario 1), the methods assuming homogeneous genetic effects (Hom-Meta-SKAT-O and Hom-Meta-SKAT) were more powerful than those assuming heterogeneous genetic effects (Hom-Meta-SKAT-O and Hom-Meta-SKAT), although the power loss via Het-Meta-SKAT-O was modest. When study cohorts shared about 50% of causal variants (scenario 2), both approaches had comparable power. When EUR and AA ancestry groups were more heterogeneous so that they shared only a small fraction of causal variants (scenario 3), the methods that accounted for ancestry-specific heterogeneity (Het-Meta-SKAT-O and Het-Meta-SKAT) were substantially more powerful than the methods assuming homogeneous genetic effects. Overall, Meta-Fisher had similar but slightly lower power than Het-Meta-SKAT-O. When 5%, 10%, and 20% of rare variants were causal, both Hom-Meta-SKAT and Het-Meta-SKAT had higher power than Meta-Burden and vice versa when the percentage of causal variants was 50%. In contrast, Hom-Meta-SKAT-O, Het-Meta-SKAT-O, and Meta-Fisher had robust power regardless of the percentage of causal variants, because they adaptively select the best test in the class of tests that are a linear combination of SKAT and burden test statistics.
Figure 2.
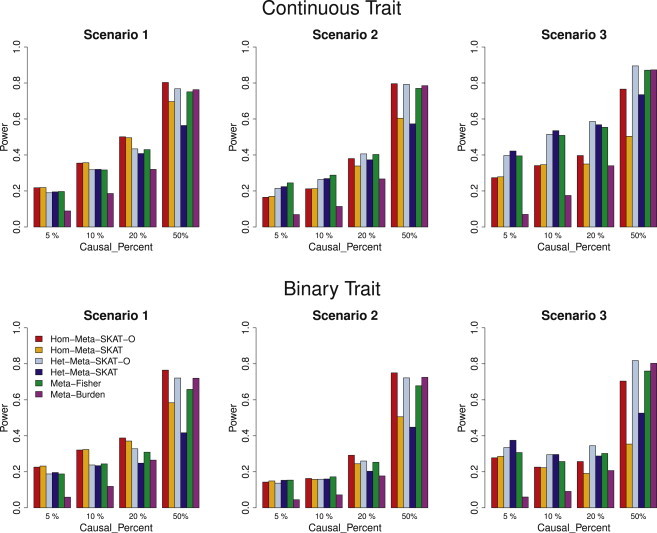
Power Comparisons of the Six Competing Methods when All Causal Variants Were Risk Increasing
Empirical power at α = 2.5 × 10−6 with different study cohort sizes (Table 1) when all causal variants in a region were risk increasing. Hom-Meta-SKAT and Hom-Meta-SKAT-O used the same weights for different studies calculated with the pooled MAF estimates and Meta-Fisher and Meta-Burden used the study-specific weights. In scenarios 1 and 2, Het-Meta-SKAT and Het-Meta-SKAT-O were conducted by assuming study-specific heterogeneity with study-specific weights. In scenario 3, Het-Meta-SKAT and Het-Meta-SKAT-O were conducted by assuming ancestry-specific heterogeneity with ancestry-specific weights. For each scenario, we considered three settings by randomly selecting 5%, 10%, 20%, and 50% of variants with MAF < 3% in a randomly selected 3 kb region as causal variants. For causal variants, we assumed that β = c|log10(MAF)|. Different c values were used for three different percentages of causal variants (see Methods). Therefore the power across the three settings (5%, 10%, 20%, and 50% of variants being causal) in each figure are not comparable.
Figure 3 compared the performance of the tests in the presence of both risk-increasing and risk-decreasing variants. As expected, the power for Meta-Burden was substantially reduced when 20% of causal variants were risk decreasing. The power of meta-analysis SKAT and SKAT-O methods were robust in this case, although powers decreased slightly compared to the case in which all causal variants were risk increasing (Figure 2). The conclusions on the relative performances between Hom-SKAT/SKAT-O and Het-SKAT/SKAT-O were similar to the case when all causal variants were risk increasing.
Figure 3.
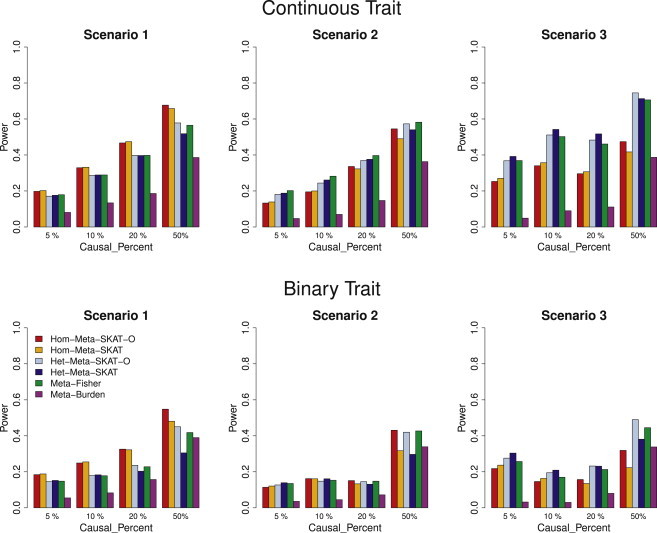
Power Comparisons of the Six Competing Methods when 20%/80% of Causal Variants Were Risk-Decreasing/Risk-Increasing
Empirical power at α = 2.5 × 10−6 with different study cohort sizes (Table 1) assuming 20% of the causal variants were risk decreasing and 80% of the causal variants were risk increasing. Hom-Meta-SKAT and Hom-Meta-SKAT-O used the same weights for different studies calculated via the pooled MAF estimates, and Meta-Fisher and Meta-Burden used the study-specific weights. In scenarios 1 and 2, Het-Meta-SKAT and Het-Meta-SKAT-O were conducted by assuming study-specific heterogeneity with study-specific weights. In scenario 3, Het-Meta-SKAT and Het-Meta-SKAT-O were conducted by assuming ancestry-specific heterogeneity with ancestry-specific weights. For each scenario, we considered three settings by randomly selecting 5%, 10%, 20%, and 50% of variants with MAF < 3% in a randomly selected 3 kb region as causal variants. For causal variants, we assumed that β = c|log10(MAF)|. Different c values were used for three different percentages of causal variants (see Methods). Therefore the power across the three settings (5%, 10%, 20%, and 50% of variants being causal) in each figure are not comparable.
Power results for scenarios 4–6 with equal sample sizes per group (Figures S3 and S4) and simulations assuming prevalence 0.10 rather than 0.01 (Figures S5 and S6) yielded the same conclusions regarding methods comparisons. We also compared power at α = 10−2 and 10−4 and results were the qualitatively the same (data not shown).
Power results for Meta-Z and Meta-Burden-RE for scenarios 1–3 (Figures S7 and S8) showed that Meta-Z was slightly more powerful than Meta-Fisher when the genetic effects were homogeneous (scenario 1) but less powerful than Meta-Fisher when the genetic effects were heterogeneous (scenarios 2 and 3). Meta-Burden-RE was substantially less powerful than the other methods.
In summary, the simulation results show that proper modeling of heterogeneity of genetic effects can increase the power of meta-analysis. Hom-Meta-SKAT-O was the most powerful and robust test when genetic effects were homogeneous, although the loss of power via Het-Meta-SKAT-O is modest. Het-Meta-SKAT-O was the most powerful and robust test when genetic effects were heterogeneous. The simulation results also show that the proposed methods are flexible in accounting for a range of heterogeneity of genetic effects between studies and are as powerful as joint analysis with all individual level data.
Rare Variant Association Meta-analysis of Lipid Traits in Eight European Cohorts
We applied our meta-analysis methods to analysis of the multicohort lipid data. We investigated the association between seven identified low-frequency and rare protein-coding and UTR variants in LPL with the three serum lipid phenotypes after adjusting for the common LPL variant rs12678919 previously shown to be associated with HDL and TG.13 For Het-Meta-SKAT and Het-Meta-SKAT-O, we assumed study-specific heterogeneous genetic effects by using Equation 4 (Table 3). Het-Meta-SKAT (p values = 6.5 × 10−5 and 1.5 × 10−5) and Het-Meta-SKAT-O (p values = 1.4 × 10−4 and 3.5 × 10−5) had the smallest p values for HDL and TG. The estimated optimal ρ of Het-Meta-SKAT-O for both HDL and TG was zero, corresponding to the SKAT test, which explains why Het-Meta-SKAT yielded smaller p values than Het-Meta-SKAT-O. None of the meta-analysis methods suggested strong association of LPL variants with LDL, consistent with the results for common variants in GWASs. Interestingly, the p values of Hom-Meta-SKAT and Hom-Meta-SKAT-O were substantially larger than the p values of Het-Meta-SKAT and Het-Meta-SKAT-O, consistent with the presence of heterogeneity of the genetic effects of LPL across the studies. Meta-Z had larger p values than Meta-Fisher, and all p values of Meta-Burden-RE were >0.05 (data not shown).
Table 3.
Analysis Results of the Multicohort Lipid Data for Testing the Effect of LPL on the Lipid Traits via Different Rare-Variant Meta-analysis Methods
| Trait | Hom-Meta-SKAT-O | Hom-Meta-SKAT | Het-Meta-SKAT-O | Het-Meta-SKAT | Meta-Fisher | Meta-Burden |
|---|---|---|---|---|---|---|
| HDL | 2.5 × 10−2 | 1.7 × 10−2 | 1.4 × 10−4 | 6.5 × 10−5 | 1.8 × 10−2 | 3.5 × 10−1 |
| LDL | 1.0 | 8.3 × 10−1 | 4.0 × 10−1 | 2.5 × 10−1 | 3.9 × 10−1 | 2.1 × 10−2 |
| TG | 5.3 × 10−3 | 3.7 × 10−3 | 3.5 × 10−5 | 1.5 × 10−5 | 6.0 × 10−4 | 7.7 × 10−2 |
Het-Meta-SKAT achieved the smallest p values for traits HDL and TG.
We also applied single-variant meta-analysis methods for the seven LPL SNVs individually (Table S2). The minimum p value among the seven SNPs was 5.5 × 10−3 for HDL and 1.3 × 10−4 for TG, which, even without correction for multiple comparisons, was substantially less significant than p values of Het-Meta-SKAT and Het-Meta-SKAT-O.
To estimate the trait variance explained by seven genetic variants in LPL, we computed adjusted R-squares by fitting standard linear regression, because the variants were not extremely rare on the Metabochip; all variants were present in at least two copies. When the seven rare variants were included in linear models, the adjusted R-squares were 0.32% and 0.44% for HDL and TG, after adjusting for covariates and SNV rs12678919. If all 20 exon and UTR SNVs were used, the adjusted R-squares were 0.88% and 1.21% for HDL and TG, respectively. These results show that only a small proportion of trait heritability was explained by the rare variants in LPL typed on the Metabochip.
Discussion
In this paper, we propose a statistical framework for meta-analysis of rare variant effects via burden tests, variance component tests, and unified tests that combine features of both. The framework is based on study-specific summary statistics for each region and is flexible enough to accommodate a range of heterogeneity of genetic effects across studies, including ancestry-specific heterogeneity for multiethnic studies. The power simulations and lipids data example demonstrate that power of the proposed meta-analysis framework is maximized when heterogeneity of genetic effects is properly modeled. From our simulation studies, we have found that Het-Meta-SKAT-O is reasonably robust to heterogeneity of genetic effects across studies; it is powerful when genetic effects are heterogeneous and loses little power when genetic effects are homogeneous across studies. The lipid data example shows that Het-Meta-SKAT-O works well in practice.
We note that the Hom-Meta-SKAT and Hom-Meta-SKAT-O test statistics assuming homogeneous genetic effects are essentially identical to joint analysis test statistics using all individual level data accounting for study-specific covariate effects, resulting in nearly identical power using meta-analysis and joint analysis. Our power simulations confirm this finding. Meta-analysis has several additional advantages over traditional individual data-based joint analysis. It avoids the need to share individual level data (requiring only summary statistics), allows one to adjust for different sets of covariates for different studies, and allows for heterogeneous genetic effects between studies. Liu et al. (2013)34 have recently independently developed meta-analysis burden and variance component tests that are analogous to our Meta-Burden and Hom-Meta-SKAT for continuous traits in addition to meta-analysis variable-threshold test.5
In contrast to joint analysis, which requires sharing of individual-level data, the proposed gene- or region-based multimarker tests require sharing of only summary statistics: single marker score statistics and between-variants relationship matrices that represent the linkage disequilibrium (LD) structure of the target regions. Although it is possible to estimate LD structure by using publicly available reference samples from the HapMap35 or 1000 Genomes32 Project for common variants,36 this approach is less effective for rare variant analysis, because these relationships may be more variable and because many rare variants may not even be observed in the reference samples. Future research is needed to determine the impact of using an external reference panel to estimate LD for rare variant meta-analysis.
Our Hom-Meta-SKAT and Hom-Meta-SKAT-O are multimarker score statistics analogous to that used in single-variant fixed-effects meta-analysis, and Het-Meta-SKAT and Het-Meta-SKAT-O are multimarker score statistics analogous to that used in single-variant random-effects meta-analysis.37 However, for a single marker, our Het-Meta-SKAT and Het-Meta-SKAT-O statistics do not reduce to that used in the traditional single-variant random effects model. Specifically, our null hypothesis assumes that study-specific population heterogeneous genetic effects are all zero, whereas the test used in the traditional single-variant random effect meta-analysis assumes that the overall mean is zero but allows study-specific effects to randomly vary around zero under the null. In this regard, our approach can be viewed as a multimarker extension of the modified single-variant random-effect model meta-analysis test proposed by Han and Eskin.37 The difference is that the score statistics are used instead of the likelihood ratio statistics and multiple markers are used instead of a single marker. Han and Eskin37 have shown that for single variants, the modified approach can have improved power over the traditional random effects model meta-analysis test and the fixed effect meta-analysis test in the presence of heterogeneous SNP effects. Our findings are consistent in this regard in multimarker settings.
One of the important features of the proposed meta-analysis framework is that it allows for flexible modeling of the degree of heterogeneity of genetic effects across studies. For multiethnic studies, the proposed framework allows for mixed homogeneous and heterogeneous genetic effects across different ancestry groups. By using simulation and real data analysis, we have shown that the power can be improved by properly modeling the heterogeneity of genetic effects. It is of future research interest to develop a test to examine whether gene-level effects are heterogeneous across studies.
In traditional single-variant meta-analysis of common variants, regression coefficients for the variant in each study are often used. Because sequencing studies consist of a large number of rare variants, estimation of the effects of rare variants by traditional regression methods is unstable with very large variances or often infeasible for both individual studies and meta-analysis when, for example, some rare variants are present in some studies but not all studies. An important advantage of our proposed meta-analysis methods is that they overcome this difficulty by using score statistics that do not require estimating regression coefficients of individual variants and only require fitting the null models. Our methods also allow p values to be calculated analytically, making our methods computationally fast even when analyzing data genome-wide. Our simulation study shows that our analytic p value calculations are quite accurate except when the nominal type I error is very low (α = 2.5 × 10−6), where the estimated type I error is slightly inflated for continuous traits and slightly deflated for binary traits. In future research, we plan to develop more accurate analytic p value calculations for very small type I error rates for the proposed methods, perhaps by using resampling methods, and to develop methods beyond traditional regression methods to estimate rare-variant effects in individual sequencing association studies and meta-analysis of multiple sequencing studies.
With rapid advances of sequencing technologies, more sequencing data from various existing large cohorts will be collected and more meta-analysis will be conducted in diverse populations. Our flexible meta-analysis framework provides an effective approach for rare-variant analysis across multiple and diverse studies.
Acknowledgments
This work was supported by grants K99 HL113264 (S.L.), R01 HG000376 (M.B.), and R37 CA076404 and P01 CA134294 (X.L.) from the National Institutes of Health.
Appendix A: Derivation of Qhet-meta(ρ)
Equations 1 and 2 can be written as the following generalized linear model with a canonical link function :
| (Equation A1) |
where is an identity function for continuous traits and a logistic function for binary traits. We assume that coefficients s are random variables with
| (Equation A2) |
for or . Equation A1 can be rewritten as
Let be a vector of nk phenotypes and be a vector of the estimated mean of yk under the null hypothesis of no association, H0: βk = 0. A score test statistic of is
| (Equation A3) |
where for continuous traits and for binary traits.
Next we derive Equation 5, which was obtained assuming B ancestry groups with the bth group consisting of Kb studies. We assume that when both and belong to the same ancestry. Otherwise, Equation A2 holds. Let be a vector of regression coefficients of the ancestry group b. Via a matrix notation, the generalized linear model is
The score test statistic of is
| (Equation A4) |
One can easily see that when B = 1, Equation A4 reduces to Qhom-meta in Equation 3.
Appendix B: Null Distribution of Qhom-meta(ρ) and Qhet-meta(ρ)
Because Equation 5 is identical to Equations 3 and 4 for B = 1 and B = K, respectively, we only need to derive the null distribution of Equation 5. Let and , and then asymptotically follows a multivariate Gaussian distribution with mean zero and variance . We further define and a block diagonal matrix
which is the covariance matrix of . By simple matrix algebra, we can show
| (Equation B1) |
It can be easily shown that Equation B1 asymptotically follows a mixture of chi-square distribution , where s are nonzero eigenvalues of and are independent and identically distrubuted chi-square random variables with one degree of freedom (df). An approximate p value of Qhet-meta (ρ) can be easily obtained by the Davies method.22
Appendix C: Null Distribution of Meta-SKAT-O
We only show the null distribution of Het-Meta-SKAT-O based on Equation 5, because Equation 5 is identical to Qhom-meta (ρ) when all studies belong to the same group. The test statistic of Het-Meta-SKAT-O is
| (Equation C1) |
where is a p value of Qhet-meta (ρ). The test statistic Thet-meta can be obtained by simple grid search across a range of : set a grid , then the test statistic . To obtain the null distribution of Thet-meta, we first define , , and Following the same logic in Lee et al.,19 we show that under the null hypothesis Qhet-meta (ρd) is asymptotically identical to
| (Equation C2) |
where are nonzero eigenvalues of , s are i.i.d random variables,, and satisfies the following conditions:
Because the correlation between and is zero, we can approximate Qhet-meta (ρ) as the mixture of two independent random variables wherein asymptotically follows a chi-square distribution with df = 1, and can be asymptotically approximated to a mixture of chi-square distributions with a proper adjustment. Now, the p value of Thet-meta can be quickly obtained by a one-dimensional numerical integration.
In a previous paper, we showed that the optimal , where is a proportion of causal variants.19 We also found that the unified test behaved similarly when ρ > 0.5. Hence, we used a grid of eight values of ρ = (0, 0.12, 0.22, 0.32, 0.42, 0.52, 0.5, 1) to perform the search of the optimal ρ in SKAT-O and Meta-SKAT-O in simulation studies and real data analysis. We also note that we modified the implementation of SKAT-O procedure to use the Davies method (rather than a moment matching method) to get a minimum p value over the grids of ρ values to improve the type I error control in the extreme tails of the distribution.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Meta-analysis SKAT and SKAT-O R-package (MetaSKAT), http://www.hsph.harvard.edu/xlin/software.html
References
- 1.Ansorge W.J. Next-generation DNA sequencing techniques. New Biotechnol. 2009;25:195–203. doi: 10.1016/j.nbt.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 2.Ladouceur M., Dastani Z., Aulchenko Y.S., Greenwood C.M.T., Richards J.B. The empirical power of rare variant association methods: results from sanger sequencing in 1,998 individuals. PLoS Genet. 2012;8:e1002496. doi: 10.1371/journal.pgen.1002496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li B., Leal S.M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madsen B.E., Browning S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009;5:e1000384. doi: 10.1371/journal.pgen.1000384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price A.L., Kryukov G.V., de Bakker P.I.W., Purcell S.M., Staples J., Wei L.J., Sunyaev S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010;86:832–838. doi: 10.1016/j.ajhg.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Neale B.M., Rivas M.A., Voight B.F., Altshuler D., Devlin B., Orho-Melander M., Kathiresan S., Purcell S.M., Roeder K., Daly M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011;7:e1001322. doi: 10.1371/journal.pgen.1001322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu M.C., Lee S., Cai T., Li Y., Boehnke M.C., Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Derkach A., Lawless J.F., Sun L. Robust and powerful tests for rare variants using Fisher’s method to combine evidence of association from two or more complementary tests. Genet. Epidemiol. 2013;37:110–121. doi: 10.1002/gepi.21689. [DOI] [PubMed] [Google Scholar]
- 9.Lee S., Emond M.J., Bamshad M.J., Barnes K.C., Rieder M.J., Nickerson D.A., Christiani D.C., Wurfel M.M., Lin X., NHLBI GO Exome Sequencing Project—ESP Lung Project Team Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012;91:224–237. doi: 10.1016/j.ajhg.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarthy M.I., Abecasis G.R., Cardon L.R., Goldstein D.B., Little J., Ioannidis J.P.A., Hirschhorn J.N. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat. Rev. Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 11.Zeggini E., Ioannidis J.P.A. Meta-analysis in genome-wide association studies. Pharmacogenomics. 2009;10:191–201. doi: 10.2217/14622416.10.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin D.Y., Zeng D. Meta-analysis of genome-wide association studies: no efficiency gain in using individual participant data. Genet. Epidemiol. 2010;34:60–66. doi: 10.1002/gepi.20435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teslovich T.M., Musunuru K., Smith A.V., Edmondson A.C., Stylianou I.M., Koseki M., Pirruccello J.P., Ripatti S., Chasman D.I., Willer C.J. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zeggini E., Scott L.J., Saxena R., Voight B.F., Marchini J.L., Hu T., de Bakker P.I.W., Abecasis G.R., Almgren P., Andersen G., Wellcome Trust Case Control Consortium Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat. Genet. 2008;40:638–645. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stahl E.A., Raychaudhuri S., Remmers E.F., Xie G., Eyre S., Thomson B.P., Li Y., Kurreeman F.A.S., Zhernakova A., Hinks A., BIRAC Consortium. YEAR Consortium Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010;42:508–514. doi: 10.1038/ng.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ioannidis J.P.A., Patsopoulos N.A., Evangelou E. Heterogeneity in meta-analyses of genome-wide association investigations. PLoS ONE. 2007;2:e841. doi: 10.1371/journal.pone.0000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morris A.P. Transethnic meta-analysis of genomewide association studies. Genet. Epidemiol. 2011;35:809–822. doi: 10.1002/gepi.20630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S., Wu M.C., Lin X. Optimal tests for rare variant effects in sequencing association studies. Biostatistics. 2012;13:762–775. doi: 10.1093/biostatistics/kxs014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang D., Lin X. Hypothesis testing in semiparametric additive mixed models. Biostatistics. 2003;4:57–74. doi: 10.1093/biostatistics/4.1.57. [DOI] [PubMed] [Google Scholar]
- 21.Duchesne P., Lafaye De Micheaux P. Computing the distribution of quadratic forms: Further comparisons between the Liu–Tang–Zhang approximation and exact methods. Comput. Stat. Data Anal. 2010;54:858–862. [Google Scholar]
- 22.Davies R.B. Algorithm AS 155: The distribution of a linear combination of χ 2 random variables. J. Royal Stat. Soc. C Applied Stat. 1980;29:323–333. [Google Scholar]
- 23.Kumar P., Henikoff S., Ng P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 24.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schaffner S.F., Foo C., Gabriel S., Reich D., Daly M.J., Altshuler D. Calibrating a coalescent simulation of human genome sequence variation. Genome Res. 2005;15:1576–1583. doi: 10.1101/gr.3709305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pruitt K.D., Tatusova T., Brown G.R., Maglott D.R. NCBI Reference Sequences (RefSeq): current status, new features and genome annotation policy. Nucleic Acids Res. 2012;40(Database issue):D130–D135. doi: 10.1093/nar/gkr1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher R.A., Genetiker S., Genetician S., Britain G., Généticien S. Oliver and Boyd; Edinburgh: 1970. Statistical Methods for Research Workers. [Google Scholar]
- 28.Stouffer, S.A., Suchman, E.A., DeVinney, L.C., Star, S.A., and Williams, R.M., Jr. (1949). The American soldier: adjustment during army life. In Studies in Social Psychology in World War II, Vol. 1.
- 29.Begum F., Ghosh D., Tseng G.C., Feingold E. Comprehensive literature review and statistical considerations for GWAS meta-analysis. Nucleic Acids Res. 2012;40:3777–3784. doi: 10.1093/nar/gkr1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu L., Sabo A., Neale B.M., Nagaswamy U., Stevens C., Lim E., Bodea C.A., Muzny D., Reid J.G., Banks E. Analysis of rare, exonic variation amongst subjects with autism spectrum disorders and population controls. PLoS Genet. 2013;9:e1003443. doi: 10.1371/journal.pgen.1003443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viechtbauer W. Conducting meta-analyses in R with the metafor package. J. Stat. Softw. 2010;36:1–48. [Google Scholar]
- 32.Altshuler D.M., Lander E.S., Ambrogio L., Bloom T., Cibulskis K., Fennell T.J., Gabriel S.B., Jaffe D.B., Shefler E., Sougnez C.L. A map of human genome variation from population scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manichaikul A., Mychaleckyj J.C., Rich S.S., Daly K., Sale M., Chen W.M. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–2873. doi: 10.1093/bioinformatics/btq559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu, D.J., Peloso, G.M., Zhan, X., Holmen, O., Zawitowski, M., Feng, S., Nikpay, M., Auer, P.L., Goel, A., Zhang, H., et al. (2013). Meta-analysis of gene level association tests. arXiv, arXiv:1305.1318, http://arXiv.org/abs/1305.1318 [DOI] [PMC free article] [PubMed]
- 35.Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., International HapMap Consortium A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang J., Ferreira T., Morris A.P., Medland S.E., Madden P.A.F., Heath A.C., Martin N.G., Montgomery G.W., Weedon M.N., Loos R.J., Genetic Investigation of ANthropometric Traits (GIANT) Consortium. DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 2012;44:369–375. doi: 10.1038/ng.2213. S1–S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han B., Eskin E. Random-effects model aimed at discovering associations in meta-analysis of genome-wide association studies. Am. J. Hum. Genet. 2011;88:586–598. doi: 10.1016/j.ajhg.2011.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.