

Abstract
Intestinal tumors from mice and humans can have a polyclonal origin. Statistical analyses indicate that the best explanation for this source of intratumoral heterogeneity is the presence of interactions among multiple progenitors. We sought to better understand the nature of these interactions. An initial progenitor could recruit others by facilitating the transformation of one or more neighboring cells. Alternatively, two progenitors that are independently initiated could simply cooperate to form a single tumor. These possibilities were tested by analyzing tumors from aggregation chimeras that were generated by fusing together embryos with unequal predispositions to tumor development. Strikingly, numerous polyclonal tumors were observed even when one genetic component was highly, if not completely, resistant to spontaneous tumorigenesis in the intestine. Moreover, the observed number of polyclonal tumors could be explained by the facilitated transformation of a single neighbor within 144 μm of an initial progenitor. These findings strongly support recruitment instead of cooperation. Thus, it is conceivable that these interactions are necessary for tumors to thrive, so blocking them might be a highly effective method for preventing the formation of tumors in the intestine and other tissues.
Keywords: colon cancer, spatial statistics, clonal interactions, mouse model
Tumors are often heterogeneous with respect to several distinguishable properties, including differentiation state, proliferation rate, metastatic potential, and therapeutic response. Two models to explain intratumoral heterogeneity have been proposed. The clonal evolution model asserts that different subclones arise from a single progenitor as a consequence of molecular changes followed by selection for dissimilar microenvironments within a tumor (1). By contrast, the cancer stem cell model contends that a small population of stem cells originating from a single progenitor is responsible for tumor maintenance but the progeny can differentiate in several diverse ways (1). A key assumption in both models is that tumors are derived from a single progenitor.
Evidence is steadily accruing that intestinal tumors are often polyclonal rather than monoclonal (2). Merritt et al. (3) demonstrated that hereditary tumors in the mouse intestine are often derived from multiple progenitors. In this study, aggregation chimeras were generated by fusing embryos carrying the Min allele of the Adenomatous polyposis coli gene (ApcMin/+) to embryos carrying Min and the Rosa26 lineage marker (ApcMin/+ R26+). Clonal structure was assessed in histologic sections of tumors stained for the lineage marker. A significant number (8%) of early adenomas were heterotypic, being composed of cells from the two different embryos. Using a similar approach, Thliveris et al. (4) demonstrated that carcinogen-induced tumors in mice are also derived from multiple progenitors. In both studies, the intestines consisted of small blue and white patches. This chimeric pattern increases the power to detect polyclonality because a heterotypic tumor forming on a border between the two colors is clearly polyclonal, whereas a homotypic tumor could be polyclonal as the result of being derived from two progenitors with the same R26 status or else monoclonal. The findings from the Merritt and Thliveris studies are consistent with those of other investigators demonstrating that hereditary and sporadic colorectal tumors in humans are often polyclonal (5, 6). Therefore, multiple progenitors contributing to a single tumor are an additional source of intratumoral heterogeneity.
Although evidence supports the existence of polyclonality, this phenomenon could have been merely a consequence of random collision between independently derived tumors instead of necessary clonal interactions. In the Merritt study, the aggregation chimeras developed far too many tumors to rule out random collision. To distinguish between the possible explanations for polyclonality, Thliveris et al. (7) generated aggregation chimeras that developed relatively few intestinal tumors. They found that the percentage of heterotypic tumors was still high (20%), even though the multiplicity of tumors was very low. This observation when combined with statistical analyses ruled out random collision and favored clonal interactions. In the Thliveris study (7), tumor phenotypes were linked with image data describing the pattern of chimerism to estimate the range of clonal interactions. They found that interactions occurring between progenitors in neighboring crypts (i.e., 40–120 μm apart) were sufficient to account for the percentage of heterotypic tumors that was observed. Thus, polyclonality could be explained by multiple progenitors interacting over a very short distance.
The details of clonal interactions during the initial stages of tumorigenesis remain unknown. One possibility is some form of recruitment in which a single progenitor, following the loss of Apc activity, subsequently facilitates the neoplastic transformation of one or more neighboring cells. Alternatively, multiple independently derived progenitors arising in close proximity might effectively cooperate and gain a selective growth advantage over an isolated progenitor. Although prior studies of tumor clonality were unable to distinguish between recruitment and cooperation, the two models predict different frequencies of heterotypic tumors in aggregation chimeras formed from embryos that have unequal susceptibilities to tumorigenesis. On the basis of this realization, we characterized clonal interactions by generating and analyzing two types of aggregation chimeras: C57BL/6 (B6) ApcMin/+ ↔ Apc1638N/+ R26+ and B6 ApcMin/+ ↔ Apc+/+ R26+, because B6 ApcMin/+ mice spontaneously develop many more intestinal tumors than either B6 Apc1638N/+ mice or B6 Apc+/+ mice (8).
Results
We reasoned that recruitment and cooperation could be distinguished by generating aggregation chimeras from embryos with unequal tumor susceptibilities (Fig. 1). If polyclonal tumors form as a consequence of recruitment, the number will depend on how many progenitors lie on the border between patches that are derived from the different embryos and the ability of the susceptible tissue to facilitate the transformation of the more resistant tissue. This number should be relatively high because progenitors arising from cells highly susceptible to neoplastic transformation (white) are common. If polyclonal tumors form as a consequence of cooperation, the number will depend on the probability that two or more progenitors are juxtaposed to each other. This number should be relatively low because progenitors arising from cells that are resistant (blue) to neoplastic transformation are rare. Conceptually, these two distinct models explaining polyclonality should be distinguishable.
Fig. 1.

Different models could explain interactions leading to the formation of polyclonal tumors. Aggregation chimeras were generated by fusing an embryo that is relatively susceptible to the formation of intestinal tumors (white) to an embryo that is relatively resistant (blue). (A) The number of polyclonal tumors that are heterotypic with a mixture of white and blue (red star) will be high if the more susceptible white progenitors are able to recruit the more resistant neighboring blue cells (Left), but the number will be low if one of the white progenitor needs to be juxtaposed to a rare blue progenitor (Right). (B) Statistical modeling validated this conceptualization. The percentage of tumors that were predicted to be homotypic white (white lines), homotypic blue (blue lines), or a mixture of white and blue (red lines) was plotted vs. the initiation differential, which is the ratio of tumor susceptibilities, using average image statistics for all measured chimeric patterns and using different tumor formation models (each optimized in its parameters): full recruitment with interactions of 67 μm (Left, solid lines); partial recruitment with interactions over 144 μm (Left, dashed lines); and cooperation with interactions over 5,004 μm (Right). The models make distinguishable predictions when the initiation differential is far from one (e.g., 0.15 or less). The dark gray band ranges from 0.01 to 0.02, which is the probable initiation differential between Apc1638N/+ and ApcMin/+. In contrast, when the ratio is 1 [i.e., both embryos are equally susceptible to the formation of intestinal tumors as in previous studies (3, 4, 7)], the two models are indistinguishable.
To advance our reasoning, we constructed statistical models that predict the frequency of heterotypic tumors under recruitment and cooperation in chimeras that were formed from embyros with unequal susceptibilities to neoplastic transformation. Calculations leveraged information in the complex chimeric patterns revealed through images of the intestine. These complex chimeric patterns exhibited statistical regularities, for example in terms of proportions of a given color, or typical distances between points of opposite color (Table 1). A small distance between points of opposite color corresponds to the notion of small patch size; the precise way in which the chimeric properties predict heterotypic tumors depends on whether we model recruitment or cooperation as the generative mechanism. For recruitment, an ApcMin/+ cell at a random position is transformed into its neoplastic counterpart after the loss of Apc activity, and this initial progenitor then facilitates the transformation of neighboring cells. This model is simplified by assuming all neighboring cells can be recruited to form a single tumor derived from several progenitors. In the recruitment calculations, the patch-size information is conveyed by the distribution of the distance from a point to the nearest point of the opposite color. For cooperation, multiple transformed cells arising from independent events in which Apc is inactivated and in close proximity form a single tumor because the interactions provide a selective advantage. Here, the patch-size information is conveyed by properties of locally averaged chimeric images. The details of both statistical models are provided in Methods, SI Methods, and Fig. S1. A key finding is that these models make significantly different predictions about the probability for a tumor to be heterotypic, especially in the case in which chimeras are formed from embyros with unequal tumor susceptibilities (Fig. 1B). For example, when one embryo is 100 times more likely to develop a tumor than the other [i.e., the initiation differential (β/α) is 0.01], the heterotypic frequency is roughly 20% under recruitment and less than 1% under cooperation. Recruitment and cooperation are clearly distinguishable by analyzing aggregation chimeras that are generated from embryos with unequal tumor susceptibilities.
Table 1.
Clonal structure of tumors in the aggregation chimeras
| No. of tumors |
|||||||||||
| Type | Mouse ID | Section | % Blue | Median distance to blue, μm | Median distance to white, μm | Total | White | Blue | Heterotypic | Not scorable | Not studied |
| ApcMin/+ ↔ Apc1638N/+ R26+ | 77 | 1 | 42 | 78 | 52 | 7 | 0 | 0 | 0 | 1 | 6 |
| 2 | 40 | 68 | 43 | 14 | 0 | 0 | 0 | 2 | 12 | ||
| 3 | 46 | 59 | 48 | 41 | 0 | 0 | 0 | 2 | 39 | ||
| 4 | 36 | 78 | 42 | 59 | 0 | 0 | 1 | 1 | 57 | ||
| C | 25 | 108 | 49 | 5 | 0 | 0 | 1 | 0 | 4 | ||
| 124 | 1 | 50 | 60 | 54 | 7 | 1 | 0 | 1 | 5 | 0 | |
| 2 | 46 | 57 | 48 | 11 | 7 | 1 | 1 | 2 | 0 | ||
| 3 | 30 | 59 | 31 | 31 | 29 | 0 | 0 | 2 | 0 | ||
| 4 | 30 | 78 | 38 | 30 | 28 | 0 | 1 | 1 | 0 | ||
| C | 51 | 43 | 57 | 11 | 11 | 0 | 0 | 0 | 0 | ||
| 130 | 1 | 54 | 60 | 70 | 10 | 0 | 0 | 0 | 2 | 8 | |
| 2 | 57 | 42 | 51 | 13 | 2 | 0 | 0 | 3 | 8 | ||
| 3 | 50 | 60 | 64 | 62 | 4 | 0 | 0 | 4 | 54 | ||
| 4 | 52 | 54 | 57 | 27 | 1 | 0 | 0 | 1 | 25 | ||
| C | 59 | 59 | 78 | 12 | 1 | 0 | 1 | 2 | 8 | ||
| 136 | 1 | 38 | 88 | 57 | 7 | 2 | 0 | 1 | 1 | 3 | |
| 2 | 38 | 87 | 60 | 9 | 0 | 0 | 0 | 0 | 9 | ||
| 3 | 28 | 86 | 42 | 53 | 8 | 0 | 0 | 1 | 44 | ||
| 4 | 28 | 73 | 31 | 45 | 0 | 0 | 0 | 0 | 45 | ||
| C | 18 | 167 | 51 | 20 | 3 | 0 | 0 | 0 | 17 | ||
| Total | 474 | 97 | 1 | 7 | 30 | 339 | |||||
| ApcMin/+ ↔ Apc+/+ R26+ | 88 | 1 | 78 | 48 | 115 | 8 | 4 | 0 | 2 | 2 | 0 |
| 2 | 61 | 54 | 68 | 16 | 6 | 0 | 0 | 10 | 0 | ||
| 3 | 58 | 54 | 59 | 4 | 4 | 0 | 0 | 0 | 0 | ||
| 4 | 54 | 34 | 42 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| C | 50 | 57 | 54 | 1 | 0 | 0 | 0 | 1 | 0 | ||
| 111 | 1 | 48 | 78 | 66 | 4 | 1 | 0 | 1 | 2 | 0 | |
| 2 | 35 | 92 | 51 | 3 | 0 | 0 | 1 | 2 | 0 | ||
| 3 | 30 | 97 | 42 | 7 | 6 | 0 | 0 | 1 | 0 | ||
| 4 | 50 | 57 | 54 | 3 | 2 | 0 | 0 | 1 | 0 | ||
| C | 42 | 60 | 42 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| 120 | 1 | 57 | 72 | 102 | 2 | 0 | 0 | 2 | 0 | 0 | |
| 2 | 42 | 85 | 68 | 2 | 0 | 0 | 1 | 1 | 0 | ||
| 3 | 34 | 76 | 43 | 2 | 0 | 0 | 1 | 1 | 0 | ||
| 4 | 41 | 66 | 51 | 2 | 2 | 0 | 0 | 0 | 0 | ||
| C | 45 | 72 | 61 | 0 | 0 | 0 | 0 | 0 | 0 | ||
| Total | 54 | 25 | 0 | 8 | 21 | 0 | |||||
Sections 1–4 refer to quarters of the small intestine numbered from proximal to distal; section C refers to the colon. In all aggregation chimeras, some tumors were not scorable with complete certainty because of poor fixation, sectioning, or staining. In others, tumor multiplicity was very high so it was impractical to analyze every tumor. Because all tumors were essentially a mixture of blue and white cells and their clonal structure cannot be predicted from wholemounts, a subset of tumors from throughout the intestine was chosen randomly for histological analysis. Aggregation chimeras 77, 124, 130, 136, 88, 111, and 120 were killed at 121, 101, 94, 79, 115, 108, and 122 d of age, respectively.
We fused B6 ApcMin/+ embryos to B6 Apc1638N/+ R26+ embryos because B6 ApcMin/+ mice (n = 228) develop on average 95 ± 53 intestinal tumors, whereas B6 Apc1638N/+mice (n = 94) develop on average 0.98 ± 1.17 tumors in our mouse colony. The resulting aggregation chimeras were killed when moribund at ages ranging from 79 to 121 d (Fig. 2 and Table 1). The intestinal tract was removed, divided into five equal segments, splayed open, stained to identify cells carrying the R26 lineage marker, and photographed; images were digitized for statistical analysis in which the chimeric pattern was characterized (Fig. 2 A and B and Table 1). The fixed samples were scored for intestinal tumors using a dissecting microscope; an average of 118 ± 19 was observed (Table 1). Many tumors were excised, embedded in paraffin, sectioned, and assessed by two pathologists to determine phenotype (Fig. 2D). Seven heterotypic tumors consisting of blue and white neoplastic cells with nuclear β-catenin were observed (Table 1). To further contrast recruitment with cooperation as distinct models for clonal interactions, we also fused ApcMin/+ embryos to Apc+/+ R26+ embryos because Apc+/+ mice have never been observed to develop intestinal tumors spontaneously. The resulting aggregation chimeras were killed when moribund at ages ranging from 108 to 122 d (Fig. 3 and Table 1). These mice developed on average 18 ± 10 intestinal tumors. Of the 54 tumors excised, 8 were heterotypic and 25 homotypic white (Table 1 and Fig. 4). In both types of aggregation chimeras, polyclonal tumors are relatively common in aggregation chimeras that are generated from embryos with unequal tumor susceptibilities.
Fig. 2.

Tumors from ApcMin/+ ↔ Apc1638N/+ R26+ aggregation chimeras can have a polyclonal origin. The mice were generated and killed when moribund. The intestinal tract was removed and stained with X-Gal. ApcMin/+ cells are white, and Apc1638N/+ cells carrying R26+ are blue. (A and B) The intestine was photographed (A) and digitized (B) for statistical analysis. (C) Tumors (wholemount) were then excised, embedded in paraffin, and sectioned. (D) Sections were stained with hematoxylin and eosin to determine whether a tumor was composed of white, blue, or a mixture of white and blue neoplastic cells. (Scale bar, 200 μm.)
Fig. 3.

Tumors from ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras can have a polyclonal origin. (A and B) The intestines from these chimeras are a patchwork of ApcMin/+ cells (white) and Apc+/+ R26+ cells (blue) as evidenced in this representative image, which was photographed (A) and digitized (B). (C and D) Several tumors were heterotypic, being composed of both cell types as indicated in the wholemount and verified by histology. (Scale bar, 200 μm.)
Fig. 4.
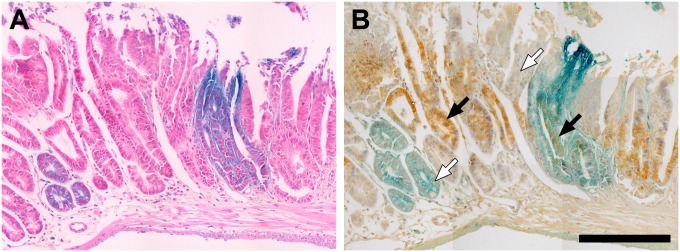
Polyclonal tumors are composed of blue and white neoplastic cells. (A and B) To confirm the impression of two pathologists based on hematoxylin and eosin stained sections (A), immunohistochemistry was performed to assess the localization of β-catenin (B). Blue and white cells within the the tumor clearly have β-catenin localized to the nucleus (black arrows), which is a marker for neoplastic transformation, whereas histologically normal cells of either color do not (white arrows). (Note that 4A and 4B are composites to create the full images.)
The probability that a tumor is heterotypic, homotypic blue, or homotypic white depends on the pattern of chimerism surrounding the initiation point and the range of interactions, regardless of the model being tested. These probabilities were calculated for all five segments of the intestinal tract in ApcMin/+ ↔ Apc1638N/+ R26+ and ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras, considering recruitment in which all neighbors are transformed, partial recruitment in which some neighbors are transformed, and cooperation (Fig. 5). The values were compared with the observed rate of heterotypic tumors. Both recruitment models significantly outperformed the cooperation model. This finding supports the notion that polyclonal tumors arise because an initial progenitor following the loss of Apc activity facilitates the transformation of one or more neighboring cells.
Fig. 5.

The probablity that a tumor is heterotypic, homotypic blue, or homotypic white was determined using three different models. (A and B) Probabilities were calculated for each of the 20 intestinal sections from ApcMin/+ ↔ Apc1638N/+ R26+ aggregation chimeras (A) and each of the 15 intestinal sections from ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras (B). The values for heterotypic and homotypic white that were obtained considering recruitment in which all neighbors are transformed (white triangles), partial recruitment in which some neighbors are transformed (gray squares), and cooperation (black circles) were compared with the observed rate of heterotypic tumors (black diamonds with white centers). Some sections have a higher probability of forming heterotypic tumors (upper left quadrant) than others (lower right quadrant) because of the chimeric pattern. The probability that a tumor is homotypic blue is extremely low (Apc1638N/+) or essentially zero (Apc+/+) for all models, so these values are not shown.
The range of interactions for recruitment that are necessary to explain polyclonality was estimated. The maximum log likelihood was calculated at different interaction distances and with different numbers of partners (Fig. S2). A distance of 25 μm resulted in the best fit if all neighbors were recruited; the distance increased to 144 μm if only a single neighbor was recruited. Note that the distance between two neighboring crypts is only approximately 50 μm. Thus, recruitment over a very short distance easily accounts for the observed number of polyclonal tumors in ApcMin/+ ↔ Apc1638N/+ R26+ and ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras.
Discussion
Evidence is steadily accruing that indicates that several tumor types can be polyclonal as a consequence of being derived from multiple progenitors and not merely the emergence of distinct subclones during tumor evolution. We sought to better understand the interactions among progenitors in the intestine. Aggregation chimeras were generated by fusing together embryos with unequal tumor susceptibilities to create a biological model that allows us to distinguish between recruitment and cooperation. Statistical analyses that combined tumor phenotype with the pattern of chimerism indicated that recruitment is the best explanation for polyclonality. In fact, the formation of a polyclonal tumor in this experimental system can be explained by recruitment of only a single neighboring cell within two to three crypts of the initial progenitor.
Tumors were analyzed from two sets of aggregation chimeras. In ApcMin/+ ↔ Apc1638N/+ R26+ aggregation chimeras, the expected number of polyclonal tumors was 14 under recruitment and 1 under cooperation. These numbers were calculated by knowing the percentage of the chimera that was derived from each embryo, the multiplicity of intestinal tumors, the phenotype of scorable tumors, and the statistical models for recruitment and cooperation (Fig. 1 and Table 1). The observed number was seven. Similarily, in ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras, the expected numbers of polyclonal tumors was four under recruitment and zero under cooperation because Apc+/+ mice never spontaneously develop intestinal tumors. Eight were observed. This finding was unexpected: a previous study found no polyclonal tumors in ApcMin/+ ↔ Apc+/+ aggregation chimeras (3, 9). Our study differed from the previous study in important ways. One key difference is the way in which aggregation chimeras were constructed. In our study, the Apc+/+ embryo always carried the R26 lineage marker because it is easier to detect blue cells in a predominanlty white mass than it is to detect white cells in a predominantly blue mass. In the previous studies, the embryo carrying the lineage marker varied from chimera to chimera. Another key difference is how the intestines and tumors were analyzed. In our study the entire intestinal tract was removed, stained, and scored, and then all of the tumors were isolated, embedded in paraffin, and sectioned through and through for pathological assessment. In the previous studies only a third of the intestinal tract was removed, and then tumors were sampled from areas in which blue tissue and white tissue were juxtaposed. Finally, in our study, sections from each tumor were examined by two pathologists. Our findings with two sets of aggregation chimeras indicate that an initial progenitor is able to recruit a nearby wild-type partner.
How does an initial progenitor recruit neighboring cells? A number of different mechanisms are possible. The loss of Apc activity in the initial progenitor could trigger the loss of Apc activity in neighboring cells. An ApcMin/+ cell could lose the wild-type copy of Apc by point mutation or somatic recombination (10) and be transformed into its neoplastic counterpart. This initial progenitor and its immediate descendants might express mitogenic factors that increase the rate of cellular proliferation in neighboring cells. Rapid proliferation might result in spontaneous mutations in Apc and consequent loss of activity and neoplastic transformation. Kuraguchi et al. (11) found that intestinal tumors from Apc1638N/+ mice lacking DNA mismatch activity often carried two distinct somatic mutations in Apc. In addition, Thirlwell et al. (6) found that tumors from patients afflicted with familial adenomatous polyposis often carried two distinct somatic mutations. Thus, recruitment could be mediated through additional genetic events, particularly in the context of hereditary cancers. However, if recruitment involved only Apc mutations, the number of polyclonal tumors should be higher in ApcMin/+ ↔ Apc1638N/+ R26+ aggregation chimeras, in which only two hits are required for the development of polyclonal tumors, than in ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras, in which three are required. However, the number of polyclonal tumors was comparable in ApcMin/+ ↔ Apc1638N/+ R26+ and ApcMin/+ ↔ Apc+/+ R26+ aggregation chimeras even though in the first set both embryos carry a germ-line Apc mutation and in the second set only one embryo carries a germ-line Apc mutation. Analyzing the status of Apc in polyclonal tumors in this study is extremely challenging given the amount of tissue that is available and the condition of the tissue after X-gal staining, which is harsh, involving two fixation steps and an overnight incubation at 37 °C.
Another possible mechanism for recruitment is paracrine oncogenic signaling. The initial ApcMin/+ progenitor after the loss of Apc activity and its immediate progeny could produce signaling factors that facilitate transformation of neighboring cells that are responsive to the signal. For example, secreted Wnt molecules could lead to the translocation of β-catenin to the nucleus in neighboring cells that are expressing Frizzled receptors. β-Catenin is clearly localized to the nucleus in neoplastic cells that are derived from the ApcMin/+, Apc1638N/+, and even the Apc+/+ lineages (Fig. 4). Thus, recruitment might involve signaling instead of additional genetic events in certain biological contexts. Thirlwell et al. (6) have demonstrated that some sporadic colon cancers were polyclonal, consisting of dysplastic crypts that carry mutations in Apc and those that do not.
Several lines of investigation support the notion that recruitment could be mediated by Wnt molecules. Neoplastic cells in which β-catenin is localized to the nucleus protrude out from the normal crypt structure (12). This change in position would place an initial progenitor and its immediate progeny in close proximity to neighboring cells such that secreted factors could elicit changes in signaling. Once β-catenin has translocated to the nucleus, it stimulates the expression of numerous genes, including Wnt3A (13). Several Wnt signaling molecules are transforming factors in vitro and in vivo (14–16). Epithelial cells expressing Wnt1 can transform other epithelial cells. Mammary tumors that are induced by a virus in GR mice are usually polyclonal with two or more mutually interdependent cell populations, but only one population expresses activated Wnt1 (15). Similarly, stromal cells expressing Wnt1 can transform epithelial cells. Fibroblasts expressing Wnt1 elicit a morphological transformation of neighboring mammary epithelial cells in coculture experiments when the neighboring cells are responsive to the signal (16). Recently, other investigators have suggested that Wnt signaling is a marker of colon cancer stem cells (CSC) and that it is regulated by the microenvironment (17). Tumor cells that have lost the capacity to form tumors can be reprogrammed to express CSC markers including CD133 and regain tumorigenic capacity when stimulated by myofibroblast-derived factors that activate Wnt signaling (17). Thus, recruitment might involve interactions among epithelial cells as well as interactions among epithelial and stromal cells that are mediated through molecules that affect Wnt signaling.
This study has limitations. The statistical models described are extreme simplifications of the dynamic mechanisms truly at work. As has been found in so many other domains, the use of such simplified models provides a language to discuss primary factors affecting the system (18) and puts a sharper focus on the information content of the available data. We also recognize that staining to detect the R26 lineage marker severely impairs the type of molecular analyses that can be performed.
This study extends our understanding of the clonal origin of intestinal tumors. An initial progenitor recruits neighboring cells within a very short distance; molecules mediating this interaction might affect Wnt signaling. The ability of one cell to facilitate the transformation of another cell has been observed with cancer-associated fibroblasts (18). Understanding recruitment will likely impact prevention strategies. For example, if the initial progenitor that transforms neighboring cells is responsive to some mitogenic factor, drugs that target that factor might prevent tumors from forming or becoming established and growing. A number of known naturally occurring factors down-regulate Wnt signaling including Dickkopf-1 (19). New models are now becoming available to identify the cells involved and the molecules that mediate clonal interactions that give rise to polyclonal tumors.
Methods
Mouse Strains and Generation of Aggregation Chimeras.
All animal studies were conducted under protocols approved by the Institutional Animal Care and Use Committee at the University of Wisconsin-Madison, following the guidelines of the American Association for the Assessment and Accreditation of Laboratory Animal Care. C57BL/6J (B6) mice heterozygous for ApcMin were from the laboratory stock of William F. Dove at the University of Wisconsin. B6 and B6 mice heterozygous for the Rosa26 transgene expressing LacZ were obtained from The Jackson Laboratory. B6 mice heterozygous for Apc1638N were obtained from the National Cancer Institute Repository. Stocks of these strains were maintained by continually backcrossing to B6 mice (The Jackson Laboratory) that were imported every fifth generation. Offspring were screened for ApcMin, Rosa26, and Apc1638N using PCR assays of DNA isolated from toe clips (7, 20).
Aggregation chimeras were generated by fusing together embryos from two crosses: B6 Apc+/+ × B6 ApcMin/+ and B6 Apc+/+ × B6 Apc1638N/+ Rosa26+. Ear snips were also taken from each animal and stained with 5-bromo-4-chloro-indolyl-β-d-galactopyranoside (X-Gal) (United States Biological) to ascertain chimerism for β-galactosidase activity.
Tumor Phenotyping.
Wholemounts of the intestines were prepared, stained with X-gal, photographed and digitized. Tumors were isolated, embedded in paraffin, serially sectioned in toto, and counterstained with hematoxylin and eosin. The phenotype of each tumor was determined by two pathologists independently of each other. Some slides were stained with β-catenin to confirm heterotypic tumors were composed of blue and white neoplastic cells. Details are in Supporting Information or were described previously (7).
Statistical Analyses.
Treating the intestinal epithelium as a finite planar region, we consider positions x on this surface in an aggegation chimera, and we let c(x) be the indicator function that defines the chimeric pattern. Say, c(x) = 1 indicates the tissue is blue at x and c(x) = 0 indicates the tissue is white, as evidenced by image data (Fig. S1). Let d(x,y) denote the distance along the intestinal surface between points x and y, and further let D(x, δ) denote a disk on the surface centered at x and having radius δ; that is, D(x, δ) = {y: d(x,y) ≤ δ }. Allowing different rates of tumor initiation in the contributing lineages, we treat the probability density of initiation events as f(x) = α + (β − α)c(x), where the different tumor susceptibilities are α (white tissue) and β (blue tissue). The initiation differential ρ = β/α is relevant in comparing the probabilities of the different tumor phenotypes in different models.
Cooperation model.
The central concept is that multiple progenitors initiated independently but in close proximity have a selective advantage over an isolated progenitor. A simple mathematical expression of this idea involves two initiation events at random locations X and Y, distributed independently and possibly nonuniformly according to f(x) as above. We consider a tumor forming only if the distance d(X,Y) is less than some value δ. The tumor is homotypic blue if c(X) = c(Y) = 1; it is homotypic white if c(X) = c(Y) = 0, and otherwise it is heterotypic. In the cooperation model, tumor phenotype probabilities under these assumptions are:
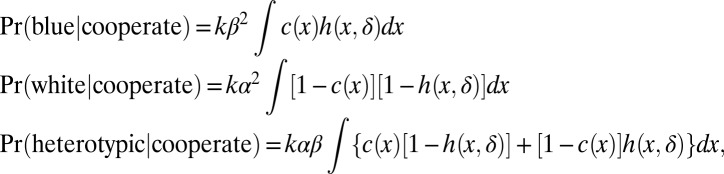 |
where k is a normalizing constant, and the function h(x,δ), shown in Fig. S1, is a smoothed version of a chimeric pattern c(x); that is, h(x,δ) = (1/(πδ2)) ∫ 1[d(x,y) ≤ δ] c(y) dy. Probabilities are computed using parameter settings and image characteristics derived from smoothed chimeric pattern images. Additional details are provided in SI Methods.
Recruitment model.
An elementary recruitment model entails a single initiation event at a random position X, followed by neoplastic transformation of all cells in the disk D(X, δ). The tumor initiated at X is homotypic blue if c(y) = 1 for all y in the disk [i.e., all cells within a given distance are blue, homotypic white if all c(y) = 0, and otherwise heterotypic]. The rationale is simply that to be homotypic blue, the initiation point X must occur in blue tissue and also be sufficiently far from white tissue to avoid recruitment of that opposite type. In the original version of this model that was proposed by Thliveris et al. (7), the initiation point X is uniformly distributed over the surface, but uniformity is an unnecessary restriction and can be usefully extended to the density f(x) (above) when we consider chimeras formed from two genetic backgrounds that differ in tumor susceptibility. Probabilities of the three tumor phenotypes have integral representations:
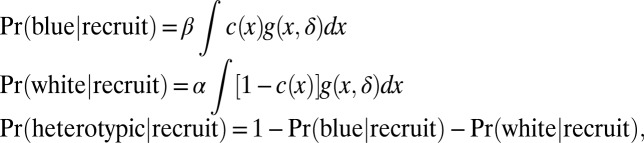 |
where integrals are over the planar intestinal surface, and g(x,δ) indicates that position x is more than δ units from any cell of the opposite lineage; that is, g(x, δ) = 1 if c(y) = c(x) for all y in D(x, δ), otherwise g(x, δ) = 0. Fixing parameters α, β, and δ, phenotype probabilities are computable in a given intestinal region using characteristics of the chimeric patterns recorded in the image data. Each binary image is converted to a distance-map image, which records at each pixel the distance to the nearest pixel of the opposite color. The empirical distribution of these distances, separated for blue and white source pixels, determines the integrals and thus the phenotype probabilities. We used the R package EBImage to compute these distance maps (21). Additional details are provided in SI Methods.
The heterotypic rate Pr(heterotypic|recruit) increases with disk radius δ, because larger disks are more likely to include both lineages. Thliveris et al. (7) estimated that a value for δ of 40–120 μm was sufficiently large to explain the observed percentage of heterotypic tumors in seven aggregation chimeras. For various values of the initiation differentials ρ, the probability of each tumor phenotype is shown (Fig. 1B and Fig. S1). In this elementary recruitment model, the rate of heterotypic tumors is affected very slightly by the initiation differential ρ.
By comparison, a key observation concerns the behavior of Pr(heterotypic|cooperate) for initiation differentials ρ that are far from unity. Under cooperation, it becomes highly improbable to see a heterotypic tumor when the two tissue types exhibit substantially different initiation rates (Fig. 1B and Fig. S1). The present analyses quantify predictions from recruitment vs. cooperation calculations to support statistical testing generated in the proposed experiments.
Partial recruitment.
We considered a variation of the recruitment model in which only a select number of neighboring cells were transformed. Specifically, we allowed a single initiation event at random position X following density f(x), as in the recruitment model. Next some number of partners (ν), sampled uniformly at random in the disk D(X, δ), are recruited to form a tumor whose phenotype is homotypic if c(X) matches that of all of the partners, otherwise it is heterotypic. The phenotype probabilities involve smoothing images rather than taking distance maps, as in the cooperation model, and they take the following specific forms, which are fully derived in the SI Methods.
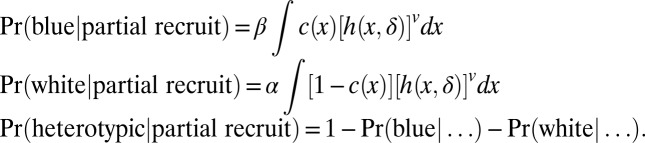 |
It is readily confirmed that these probabilities converge to the full-recruitment probabilities as ν increases.
Likelihood computations.
The phenotype of each tumor (homotypic blue, homotypic white, or heterotypic) was treated as the realization of a random variable with trinomial probabilities determined by the specific model on test as well as by image characteristics of the intestinal region harboring the tumor. Intestines were segmented into five regions for this purpose. Initiation rate parameters α and β were fixed at values estimated from the multiplicity of tumors in parental strains; ApcMin/+ mice develop on average 100 tumors, whereas Apc1638N/+ mice develop on average 1 tumor, and Apc+/+ mice develop none. With PHENOi, the phenotype of tumor i, the log likelihood in a given model MODEL is:
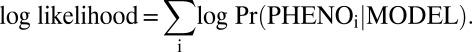 |
Free parameters in each MODEL, including disk diameter and the number of recruited partners, were fixed by maximizing this log likelihood.
Source Code.
R scripts to perform all computations are given in Dataset S1. Scripts were generated to calculate distance maps, smooth images, compute phenotype probabilities, and calculate likelihoods.
Supplementary Material
Acknowledgments
We thank Ella Ward and Jane Weeks in Experimental Pathology at the University of Wisconsin (UW) Carbone Cancer Center for technical assistance, members of the Laboratory Animal Research staff for their conscientious care of animals involved in this and other studies, and Drs. William F. Dove, Norman Drinkwater, Mark Reichelderfer, and William Schelman for critical review of the manuscript. This study was supported by funding from National Cancer Institute Grants R01 CA123438 (to R.B.H.), R01 CA063677 (to W. F. Dove), P30 CA014520 (UW Carbone Cancer Center Core Grant), P50 CA095103 (Gastrointestinal Specialized Program of Research Excellence Grant, Vanderbilt-Ingram Cancer Center), K08 CA84146 (to A.T.T.), T32 CA009614 (to D.A.D.), and T32 CA009135 (to C.D.Z.), and by start-up funds from the UW Division of Gastroenterology and Hepatology, the UW Department of Medicine, and the UW School of Medicine and Public Health (to R.B.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. P.P. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303064110/-/DCSupplemental.
References
- 1.Pietras A. Cancer stem cells in tumor heterogeneity. Adv Cancer Res. 2011;112:255–281. doi: 10.1016/B978-0-12-387688-1.00009-0. [DOI] [PubMed] [Google Scholar]
- 2.Parsons BL. Many different tumor types have polyclonal tumor origin: Evidence and implications. Mutat Res. 2008;659(3):232–247. doi: 10.1016/j.mrrev.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 3.Merritt AJ, Gould KA, Dove WF. Polyclonal structure of intestinal adenomas in ApcMin/+ mice with concomitant loss of Apc+ from all tumor lineages. Proc Natl Acad Sci USA. 1997;94(25):13927–13931. doi: 10.1073/pnas.94.25.13927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thliveris AT, et al. Clonal structure of carcinogen-induced intestinal tumors in mice. Cancer Prev Res (Phila) 2011;4(6):916–923. doi: 10.1158/1940-6207.CAPR-11-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Novelli MR, et al. Polyclonal origin of colonic adenomas in an XO/XY patient with FAP. Science. 1996;272(5265):1187–1190. doi: 10.1126/science.272.5265.1187. [DOI] [PubMed] [Google Scholar]
- 6.Thirlwell C, et al. Clonality assessment and clonal ordering of individual neoplastic crypts shows polyclonality of colorectal adenomas. Gastroenterology. 2010;138(4):1441–1454. doi: 10.1053/j.gastro.2010.01.033. 1454.e1-7. [DOI] [PubMed] [Google Scholar]
- 7.Thliveris AT, et al. Polyclonality of familial murine adenomas: Analyses of mouse chimeras with low tumor multiplicity suggest short-range interactions. Proc Natl Acad Sci USA. 2005;102(19):6960–6965. doi: 10.1073/pnas.0502662102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uronis JM, Threadgill DW. Murine models of colorectal cancer. Mamm Genome. 2009;20(5):261–268. doi: 10.1007/s00335-009-9186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gould KA, Dove WF. Localized gene action controlling intestinal neoplasia in mice. Proc Natl Acad Sci USA. 1997;94(11):5848–5853. doi: 10.1073/pnas.94.11.5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haigis KM, Dove WF. A Robertsonian translocation suppresses a somatic recombination pathway to loss of heterozygosity. Nat Genet. 2003;33(1):33–39. doi: 10.1038/ng1055. [DOI] [PubMed] [Google Scholar]
- 11.Kuraguchi M, et al. The distinct spectra of tumor-associated Apc mutations in mismatch repair-deficient Apc1638N mice define the roles of MSH3 and MSH6 in DNA repair and intestinal tumorigenesis. Cancer Res. 2001;61(21):7934–7942. [PubMed] [Google Scholar]
- 12.Roberts RB, et al. Importance of epidermal growth factor receptor signaling in establishment of adenomas and maintenance of carcinomas during intestinal tumorigenesis. Proc Natl Acad Sci USA. 2002;99(3):1521–1526. doi: 10.1073/pnas.032678499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Z, et al. Secreted frizzled related protein 2 protects cells from apoptosis by blocking the effect of canonical Wnt3a. J Mol Cell Cardiol. 2009;46(3):370–377. doi: 10.1016/j.yjmcc.2008.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimizu H, et al. Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. 1997;8(12):1349–1358. [PubMed] [Google Scholar]
- 15.Mester J, Wagenaar E, Sluyser M, Nusse R. Activation of int-1 and int-2 mammary oncogenes in hormone-dependent and -independent mammary tumors of GR mice. J Virol. 1987;61(4):1073–1078. doi: 10.1128/jvi.61.4.1073-1078.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jue SF, Bradley RS, Rudnicki JA, Varmus HE, Brown AM. The mouse Wnt-1 gene can act via a paracrine mechanism in transformation of mammary epithelial cells. Mol Cell Biol. 1992;12(1):321–328. doi: 10.1128/mcb.12.1.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vermeulen L, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 18.Franco OE, Shaw AK, Strand DW, Hayward SW. Cancer associated fibroblasts in cancer pathogenesis. Semin Cell Dev Biol. 2010;21(1):33–39. doi: 10.1016/j.semcdb.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.González-Sancho JM, et al. The Wnt antagonist DICKKOPF-1 gene is a downstream target of beta-catenin/TCF and is downregulated in human colon cancer. Oncogene. 2005;24(6):1098–1103. doi: 10.1038/sj.onc.1208303. [DOI] [PubMed] [Google Scholar]
- 20.Fodde R, et al. A targeted chain-termination mutation in the mouse Apc gene results in multiple intestinal tumors. Proc Natl Acad Sci USA. 1994;91(19):8969–8973. doi: 10.1073/pnas.91.19.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sklyar O, Pau G, Smith M, Huber W. EBImage: Image processing toolbox for R. R package version 3.8.0. 2012 Available at http://www.bioconductor.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.