

Summary
The capacity of the hematopoietic system to promptly respond to peripheral demands relies on adequate pools of progenitors able to transiently proliferate and differentiate in a regulated manner. However, little is known about factors that may restrain progenitor maturation to maintain their reservoirs. Conditional knockout mice for the Pbx1 proto-oncogene have a significant reduction in lineage-restricted progenitors in addition to a profound defect in hematopoietic stem cell (HSC) self-renewal. Through analysis of purified progenitor proliferation, differentiation capacity and transcriptional profiling, we demonstrate that Pbx1 regulates the lineage-specific output of multipotent and oligopotent progenitors. In the absence of Pbx1 multipotent progenitor (MPP) and common myeloid progenitor (CMP) pools are reduced due to aberrantly rapid myeloid maturation. This is associated with premature expression of myeloid differentiation genes and decreased maintenance of proto-oncogene transcriptional pathways, including reduced expression of Meis1, a Pbx1 dimerization partner, and its subordinate transcriptional program. Conversely, Pbx1 maintains the lymphoid differentiation potential of lymphoid-primed MPPs (LMPPs) and common lymphoid progenitors (CLPs), whose reduction in the absence of Pbx1 is associated with a defect in lymphoid priming that is also present in CMPs, which persistently express lymphoid and HSC genes underlying a previously unappreciated lineage promiscuity that is maintained by Pbx1. These results demonstrate a role for Pbx1 in restraining myeloid maturation while maintaining lymphoid potential to appropriately regulate progenitor reservoirs.
Key words: Pbx1, MPPs, CMPs, CLPs, Myeloid differentiation
Introduction
Hematopoiesis is sustained by hematopoietic stem cells (HSCs), which have the capacity to self-renew and differentiate into multiple blood cell lineages throughout the life of an individual. In addition to HSCs, the hematopoietic hierarchy is critically dependent on various progenitors, including transiently reconstituting multi-potent progenitors (MPPs), which in turn generate downstream progenitors characterized by progressively reduced self-renewal potentials and increasing lineage-restriction. Several transcription factors have been shown to have lineage-instructive roles in specific progenitors (reviewed in Iwasaki and Akashi, 2007; Orkin and Zon, 2008; Rothenberg, 2007), however little is known about factors that may restrain progenitor maturation, thus guaranteeing the constant presence of an adequate pool of undifferentiated cells ready for transient proliferative expansion to facilitate prompt responses to peripheral stresses such as infection or bleeding. This is especially important in the myeloid lineage, given the short life span of granulocytes and monocytes, and the lack of ‘memory’ cells in contrast to the lymphoid lineage. Whether the same factors regulating HSC self-renewal are also implicated in maintaining progenitor reservoirs has not been studied in detail. However, it is increasingly evident that lineage-restricted progenitors can serve as targets for oncogenic mutations that induce unlimited self-renewal and confer leukemia stem cell potential (Cano et al., 2008; Cleary, 2009; Krivtsov et al., 2006; Minami et al., 2008; Signer et al., 2010; Somervaille et al., 2009; Tremblay et al., 2010; Wojiski et al., 2009).
Pbx1 is a homeodomain transcription factor that forms hetero-oligomeric complexes with Hox, Meis and PKnox proteins to regulate developmental gene expression (Moens and Selleri, 2006). Postnatal hematopoiesis is profoundly perturbed in the absence of Pbx1, with severe reductions of HSCs and progenitors (Ficara et al., 2008), and an extreme self-renewal defect leading to non-functional stem cells. Pbx1 conditional knockout mice also exhibit significant reductions of common myeloid progenitors (CMPs) and common lymphoid progenitors (CLPs). Although lymphocytes are reduced, mature myeloid cell numbers are unaffected despite significant CMP reduction. This contrasts with several mouse models with mutations in other genes implicated in maintaining HSC self-renewal and/or cell cycle properties that have no phenotypes in the myeloid progenitor compartment (Galan-Caridad et al., 2007), or are characterized by myeloproliferative-like disease (Metcalf et al., 2006; Santaguida et al., 2009; Tothova et al., 2007; Viatour et al., 2008; Yilmaz et al., 2006) as a consequence of the HSC defect, strongly suggesting a specific role for Pbx1 in progenitors.
Here we demonstrate that Pbx1 restrains myeloid maturation in hematopoietic progenitors. Multiple genes and pathways are aberrantly regulated in Pbx1-deficient CMPs, with premature derepression of typical granulocyte and monocyte progenitor (GMP) transcripts, and downregulation of genes involved in malignant transformation. Pbx1 also sustains the persistent expression of HSC and B-lymphoid genes in CMPs, highlighting a lineage promiscuity that is maintained in committed progenitors by Pbx1.
Results
Multipotent progenitors differentially express Pbx1, which maintains their normal frequencies
Pbx1 expression was measured by real-time PCR in different subsets of prospectively isolated wild-type stem and progenitor cells within the hematopoietic hierarchy (Fig. 1A). For this purpose, MPPs were flow-sorted into three fractions according to Flk2 expression (Fig. 1A). Pbx1 transcripts were most abundant in HSCs, present at intermediate levels in MPPs with robust myeloid maturation potential (Flk2− MPPs and Flk2int MPPs) and CMPs, and at lower levels in lymphoid-primed MPPs (Flk2high, also known as LMPPs) (Månsson et al., 2007) and CLPs. This suggested that, in addition to HSCs, Pbx1 may serve important roles in MPPs and downstream progenitors. Markedly lower Pbx1 expression in CLPs compared to MPPs raised the possibility that the drastic reduction of CLPs observed in the absence of Pbx1 (Ficara et al., 2008 and Fig. 1B,C) might be the consequence of up-stream defects. Consistent with these observations, FACS analysis of MPP subsets revealed a significant decrease in the percentage and absolute number of LMPPs in mutant (Tie2Cre+.Pbx1−/f) mice compared to control (Tie2Cre−.Pbx1+/f) mice (Fig. 1E,F), supporting a role for Pbx1 in their maintenance. In addition, a consistent decrease in CMPs (Ficara et al., 2008 and Fig. 1C,D), and associated reduction of the absolute number of their Flk2int progenitors (Fig. 1F), was observed.
Fig. 1.

Evaluation of multi-potent progenitors in Pbx1-conditional knockout mice. (A) HSCs and progenitors indicated on the left were prospectively isolated by FACS from pooled BM of adult wild-type mice prior to RNA purification. The histogram shows Pbx1 transcript levels relative to HSCs as measured by real-time PCR (3–4 biological replicates, each performed in duplicate; *P<0.01). Expression levels of all progenitor subsets were significantly different from HSCs (P≤0.03). MPPs are defined as Lin−/cKithi/Scahi CD34+, divided into three fractions according to Flk2 expression: Flk2− MPPs, previously called short-term HSCs, Flk2-intermediate MPPs (Flk2int) and Flk2high MPPs or LMPPs. (B) Representative FACS analysis of CLPs from control (Tie2Cre−.Pbx1+/f) or mutant (Tie2Cre+.Pbx1−/f) mice. Contour plots on the left are referred to the Lin− gate. Percentages are relative to the parent gate. (C) Quantitative data from FACS analysis shown in B and D. Histograms represent the average CLP and CMP percentage within the BM of control or mutant mice (n = 14 and 15, respectively; *P<0.05). (D) Representative FACS analysis of CMPs and GMPs from control or mutant mice. Contour plots are referred to the Lin−cKit+Sca1− gate. Percentages are relative to the parent gate. (E) Representative FACS analysis is shown for MPPs from control or mutant mice, relative to the Lin−/cKithi/Scahi gate. (F) Histograms show the percentage of Flk2-negative, Flk2-intermediate and Flk2-high MPPs within the Lin−/cKithi/Scahi gate (left; qualitative plots are shown in E) or their absolute numbers per mouse (right); *P = 0.01, n = 8. Note that the mean fluorescence intensity was similar in mutants and controls, indicating that the reduction in progenitor subpopulations was not due to altered expression of the markers used to define them.
Myeloid differentiation from Pbx1-deficient MPPs is accelerated
To determine if Pbx1 regulates the ability of multipotent progenitors to differentiate into down-stream progeny, Flk2+ MPPs (including Flk2int MPPs and LMPPs) were prospectively isolated for in vitro analyses. In methylcellulose colony assays, MPPs from control and mutant mice gave rise to comparable numbers of colonies (supplementary material Fig. S1A). However, the relative proportion of erythroid and myeloid colonies was inverted (Fig. 2A left). Moreover, colonies were smaller, and yielded reduced numbers of total cells in pooled colonies at the end of the assay (Fig. 2A middle and right). FACS analysis of prospectively isolated MPPs over several days in liquid culture demonstrated that Pbx1-deficient cells upregulated expression of myeloid markers Mac-1 and Gr-1, however they were prone to do it with faster kinetics compared to wild-type cells (Fig. 2B). Moreover, mutant MPPs extinguished their cKit+Sca1+ phenotypes earlier compared to controls (supplementary material Fig. S1B). Thus, accelerated maturation in mutants suggested that Pbx1 may normally restrain MPP differentiation along the myeloid lineage.
Fig. 2.

Differentiation ability of Pbx1-null multi-potent progenitors. (A) Methylcellulose colony assay of prospectively isolated Flk2+ MPPs. Erythroid and myeloid colonies were counted 9 days after plating. The histogram on the left represents the average of four independent assays, each performed in duplicate (*P≤0.04). Representative myeloid colonies (CFU-GM) are shown in the middle panels. The histogram on the right represents pooled colonies from three independent experiments. (B) Time-course FACS analysis (representative of four) was performed on prospectively isolated MPPs from control or mutant mice, cultured in the presence of serum and cytokines for 3 or 6 days. Contour plots show events within the live/forward and side light scatter gate, and the average percentage of Mac-1+ cells is represented in the histograms on the right. (C) Prospectively isolated LMPPs from control or mutant mice were transplanted into wild-type recipients. FACS plots show representative analysis of spleen (left, gated on CD19+ cells) and thymus (right, gated on CD45+ cells) from a mouse transplanted 3 weeks earlier with Pbx1-null cells. The histogram depicts the average of four experiments (4–7 independent donors/group, *P = 0.05). (D) Limiting dilution analysis (representative of two) of the frequency of control or mutant LMPPs able to develop into CD19+ B cells in co-culture with GFP-OP9 stromal cells (R2 = 0.91 and 0.98, respectively). (E) Frequency of wells negative for the presence of myeloid cells when 10 LMPPs per well were seeded in the OP9 B-cell differentiation assay.
Pbx1 maintains lymphoid differentiation potential of LMPPs
Prospectively isolated LMPPs were assessed for their ability to differentiate into lymphoid progeny in vitro (supplementary material Fig. S1C) and in vivo (Fig. 2C). FACS analysis 11 days after plating of limiting numbers of LMPPs in co-culture with OP9 stromal cells revealed that Pbx1-null cells differentiated into B220+CD19+ B cells in vitro (supplementary material Fig. S1C). However, unlike myeloid differentiation, FACS analysis shortly after plating did not reveal premature lymphoid differentiation by Pbx1-null LMPPs. When transplanted into sub-lethally irradiated recipient mice, Pbx1-deficient LMPPs competed with host lymphoid progenitor cells and differentiated into lymphocytes, as shown by the presence of donor-derived B cells in the spleen and T cells in the thymus three weeks after transplant (Fig. 2C), however lymphoid outputs from Pbx1-null LMPPs were lower compared to wild-type, suggesting that Pbx1 normally maintains lymphoid potential. This was further investigated in limiting dilution in vitro B-cell differentiation assays from control or Pbx1-null LMPPs to highlight potential defects not detectable with bulk cultures. This showed a markedly lower frequency of B cell progenitors from Pbx1-null LMPPs compared to control LMPPs (Fig. 2D), in accordance with the lower in vivo B cell output after transplant. In contrast, in vitro myeloid output from Pbx1-null LMPPs was increased (Fig. 2E). Thus, Pbx1 regulates the relative lineage output of MPPs by restraining myeloid differentiation and maintaining lymphoid differentiation.
Pbx1-deficient CMPs display altered proliferation kinetics
To determine whether intrinsic defects may contribute to the reduced number of CMPs and CLPs in the absence of Pbx1, we quantified the in vivo steady-state cycling activity of lineage-restricted progenitors. Mice were injected with BrdU and two hours later FACS analysis of CLP and CMP revealed no significant decreases in BrdU incorporation indicating that the observed reductions in adult mutant mice were not due to a decreased capacity to proliferate (Fig. 3A). Rather, there was a tendency toward higher BrdU incorporation in mutant CMPs, although it did not reach statistical significance (P = 0.07). Furthermore, annexin V analysis excluded increased apoptosis as a major cause underlying CMP and CLP reduction (not shown).
Fig. 3.
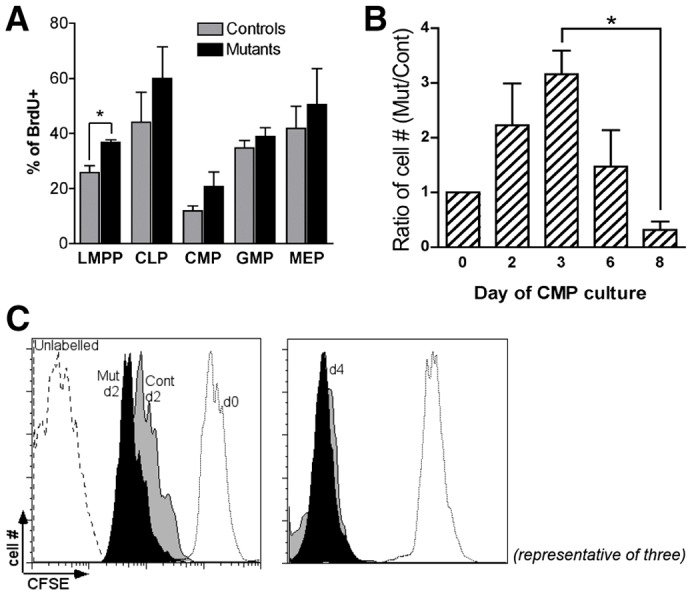
Altered progenitor proliferation. (A) Bar graph shows the percentage of BrdU+ cells in vivo within the indicated progenitor compartments (two independent experiments; average of three mutants and four controls, *P<0.02). (B) Prospectively isolated CMPs (2000–10,000 cells) were placed in liquid culture in the presence of cytokines. The graph shows the ratio of Pbx1-null cell number versus control at different time points (n = 3, *P = 0.003). (C) Prospectively isolated CMPs were labeled with CFSE immediately after sorting and cultured for 2 or 4 days. FACS analysis was performed immediately after labeling (d0), or at the indicated time points after plating. Histograms are representative of two independent experiments (n = 3).
Proliferation of prospectively isolated CMPs and GMPs was then studied in liquid culture in response to growth factor stimulation. Unexpectedly, Pbx1-null CMPs displayed a higher proliferation rate compared to wild-type in the first few days of culture. However, their number increased at a much lower rate compared to wild-type at later time points, so that the ratio of mutant CMPs versus wild-type was inverted at day 8 (Fig. 3B). Of note, there was no evidence of increased cell death as a major underlying cause of the observed phenotype at these time points (not shown). The in vitro proliferation capacity of mutant GMPs, on the other hand, did not differ from normal controls (not shown). The short-term higher proliferative response to cytokine stimulation in mutant CMPs was confirmed by monitoring dilution of the CFSE dye after 2 days of liquid culture (Fig. 3C). Thus, the reduced numbers of post-natal Pbx1-deficient committed progenitors in vivo are not due to an intrinsic lack of proliferation, but rather to its aberrantly rapid extinction in the myeloid lineage.
Pbx1-deficient CMPs differentiate prematurely
Prospectively isolated CMPs were placed in liquid culture to assess whether the increased proliferation was associated with aberrant differentiation capacity. Within 1 day of culture, a higher proportion of Pbx1-null CMPs had acquired Mac-1 expression compared to controls (Fig. 4A). At day 3, cultures initiated by mutant CMPs contained a smaller proportion of immature c-Kit+ Mac-1− cells, and a higher fraction of c-Kit− Mac-1+ cells, demonstrating premature differentiation. Analysis of c-Kit+ Mac-1− gated cells showed that mutant CMPs lost their immature phenotype more rapidly than their wild-type counterparts (Fig. 4A,B). Analysis of GMPs showed a similar but milder trend (supplementary material Fig. S1D). Concomitant FACS analysis for CFSE and Mac-1 after 2 days of culture showed that cells that had acquired Mac-1 had also diluted CFSE (supplementary material Fig. S1E) indicating that the observed premature differentiation by Pbx1-null CMPs was not due to an absence of proliferation.
Fig. 4.

Premature differentiation of Pbx1-null lineage-restricted progenitors. (A) FACS analysis (representative of five) is shown for the progeny of CMPs prospectively isolated from control or mutant mice. Dot plots on the right are relative to the c-Kit+Mac-1− gate at day 3. (B) Histograms summarize data shown in A, day 3 (n = 5, *P<0.05). (C) Methylcellulose colony assay from prospectively isolated CMPs. The top and bottom histograms represent the average of four and two independent assays, respectively, each performed in duplicate (*P≤0.02). Once scored, colonies were pooled, counted (bottom histogram), cytospun and stained with May-Grunwald and Giemsa. Arrowheads on cytospin images indicate blast-like, not terminally differentiated cells (number of blast-like cells per field was 11.5±1.25 and 1.2±0.57 in control and mutant, respectively; n = 10, P<0.0001). Pictures were taken using a Nikon ECLIPSE E1000M microscope equipped with SPOT advanced software, using a 20× magnification. (D) Limiting dilution analysis (representative of two) of the frequency of control or mutant CLPs able to develop into CD19+ B cells, as shown in Fig. 2D (R2 = 0.92 and 0.96, respectively). (E) Bar graph shows average % of donor-derived cells within B lymphocytes in bone marrow (BM) or spleen (SP), and within T cells in the thymus (TH), 1 week after transplant of control or mutant CLPs (three recipient mice analyzed per group; two different donors).
In colony-forming-cell assays, CMPs from mutant and control mice gave rise to similar numbers of colonies (supplementary material Fig. S1A). Nevertheless, there was a significant reduction in the relative proportion of erythroid colonies with a concomitant increase of myeloid colonies in cultures initiated by mutant CMPs (Fig. 4C top left). Moreover, a lower proportion of undifferentiated, ‘blast-like’ cells in cytospin preparations from pooled Pbx1-null colonies was observed compared to controls (Fig. 4C right). In addition, colonies from Pbx1-null CMPs tended to be smaller compared to wild-type (not shown), resulting in a lower number of total cells (Fig. 4C, bottom left), in agreement with results in liquid culture at day 8 (Fig. 3B). Overall, these data indicate that accelerated maturation to the next stage in the hematopoietic hierarchy in the absence of sufficient self-renewal, with a preferential skewing towards the granulocytic-monocytic vs the erythroid lineage, likely contributes to the observed CMP reduction in the absence of Pbx1.
To study differentiation capacity toward B cells by mutant lymphoid progenitors, prospectively isolated CLPs from control or mutant mice were co-cultured with OP9 cells and FACS analysis was performed at two time points (supplementary material Fig. S1F). Pbx1-null CLPs differentiated as well as control CLPs into B220+CD19+ cells in vitro. However, a lower frequency of functional CLPs from Pbx1-null mice was observed in vitro in limiting dilution B-cell differentiation assays (Fig. 4D). Prospectively isolated CLPs were also transplanted into sub-lethally irradiated wild-type recipients. Transplanted mice were sacrificed 1 or 3 weeks later to monitor the presence of donor-derived B cells in bone marrow (BM) and spleen, and of T cells in the thymus. Pbx1-null CLPs differentiated into mature lymphocytes in vivo, although the degree of chimerism in the B cell lineage tended to be lower in mice transplanted with mutant CLPs compared to controls at both time points (Fig. 4E and not shown). Thus, both myeloid and lymphoid progenitors differentiate into mature progeny in the absence of Pbx1, but with a remarkably higher efficiency in the myeloid lineage versus decreased efficiency in the lymphoid lineage.
GMP gene activation in Pbx1-deficient CMPs
Transcriptional profiling was performed to define Pbx1-dependent genes and pathways responsible for the CMP premature maturation phenotype. To provide a baseline for comparison, we initially interrogated the normal CMP-to-GMP transition, which revealed 705 (592 non-redundant) differentially expressed transcripts (282 downregulated in GMPs = typical CMP genes, including Pbx1, and 423 upregulated in GMPs = typical GMP genes) (supplementary material Table S1; supplementary material Fig. S2A). Their classification based on gene ontology (GO; supplementary material Fig. S3B) underscored a substantial increase in metabolic activity as CMPs progress towards the GMP stage, with a concomitant reduction of transcripts coding for DNA-binding proteins/transcription factors, as expected with an increase in specialization (supplementary material Fig. S3A).
Comparison of Pbx1 mutant versus control CMPs revealed 329 (305 non-redundant) differentially expressed transcripts (207 downregulated, and 122 upregulated in the absence of Pbx1) (supplementary material Tables S2, S3; supplementary material Fig. S2A). Comparison of control versus mutant GMPs revealed fewer (129) differentially regulated transcripts, in accordance with their milder phenotype, and with the lower (although detectable) expression level of Pbx1 in wild-type GMPs relative to wild-type CMPs (see later) and to MPPs (supplementary material Fig. S2C), suggesting that Pbx1 does not play a crucial role in GMPs. Expression of genes encoding surface markers used for isolation of CMPs and GMPs, respectively, were not perturbed in mutants.
Gene set enrichment analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005) was employed to compare the rank-ordered dataset of mutant versus wild-type CMP transcripts against the gene set defining normal CMP-to-GMP transition. Transcripts downregulated in mutant CMPs were highly enriched for normal CMP-specific genes (Fig. 5A), whereas transcripts upregulated in mutant CMPs correlated with normal GMP genes. Accordingly, 40% of the 305 non-redundant transcripts differentially expressed in mutant versus wild-type CMPs by SAM (statistical analysis of microarray) analysis were also differentially regulated in the normal CMP-to-GMP transition (Fig. 5B). Principal component analysis (PCA) confirmed that Pbx1-null CMPs tended to be located between normal GMPs and CMPs (supplementary material Fig. S2B). In support of this notion, classification of genes upregulated in the absence of Pbx1 based on GO almost completely overlapped with that of normal GMPs (supplementary material Fig. S3D). Thus, in the absence of Pbx1, phenotypically defined CMPs prematurely express a subset of genes typical of their downstream GMP progeny.
Fig. 5.

Pbx1 deficiency leads to derepression of GMP genes in CMPs. (A) GSEA plots show enrichment of the gene set characteristic of the normal CMP-to-GMP transition in the Pbx1-null CMP transcriptome. Transcripts downregulated in Pbx1-null CMPs positively correlate with typical CMP genes (left panel, FDR q-value 0), and negatively correlate with typical GMP genes (right panel, FDR q-value 0). (B) Venn diagram shows overlapping gene expression among Pbx1-null CMPs and the normal CMP-to-GMP transition. (C) Bar graphs shows the number of gene sets belonging to different functional groups that are enriched in Pbx1-null CMP downregulated (in blue) and upregulated (in red) transcripts. Gene sets with FDR q-value <0.2 and Nominal P≤0.05 were considered significantly enriched. (D) Heat map shows the expression of the top 31 differentially regulated transcripts between control and mutant CMPs, whose level of expression changed at least threefold between the two groups. Red arrows indicate genes that were also downregulated in Pbx1-null HSCs (Ficara et al., 2008).
GSEA was then employed to compare the rank-ordered lists of mutant and normal CMP and GMP transcripts against published gene sets. The transcripts downregulated in Pbx1-null CMPs were notably enriched for several gene sets (supplementary material Table S4; Fig. 5C) downregulated during differentiation of myeloid or other cell types, providing further support to the notion that Pbx1 normally restrains myeloid differentiation. Unexpectedly, several lymphoid gene sets were downregulated in mutant CMPs, suggesting decreased lineage promiscuity and strong myeloid-commitment in Pbx1-null progenitors. Transcripts downregulated in Pbx1-null CMPs were also enriched for HSC genes, indicating that Pbx1 normally maintains at least part of the HSC signature that is persistently expressed in committed progenitors.
Pbx1 maintains proto-oncogenic pathways in myeloid progenitors
The most numerous group of annotated gene sets downregulated in mutant CMPs was associated with various malignancies, including myeloid leukemias, underscoring the fact that Pbx1 may normally sustain pathways frequently perturbed in oncogenesis. In support of this hypothesis, a search of the GEO and GSEA microarray databases revealed that Pbx1 is highly expressed in a subset of human cancers (supplementary material Fig. S4A–F; supplementary material Table S4), and overexpressed compared to corresponding normal tissues in mouse and human breast cancer (supplementary material Fig. S4G,H), but downregulated in response to treatment (supplementary material Fig. S4I).
Thus, multiple genes and pathways are aberrantly regulated in Pbx1-null CMPs, with premature derepression of typical GMP transcripts, and downregulation of genes involved in malignant transformation.
Meis1 is part of the Pbx1-dependent transcriptional program
Among the cancer/transformation gene sets identified by GSEA, the two with the highest enrichment score and the lowest false discovery rate (FDR; 0.00) and q-value consisted of Meis1 target genes in transformed cell lines (supplementary material Table S4). Meis1 forms a transcriptional complex with Pbx1 and is one of the few genes downregulated in Pbx1-null CMPs that are also downregulated in Pbx1-null HSCs (Fig. 5D and Ficara et al., 2008), suggesting that Pbx1 might directly or indirectly regulate expression of its dimerization partner.
qPCR analysis on prospectively isolated wild-type CMPs and GMPs showed that Pbx1 and Meis1 were downregulated to similar extents during the CMP-to-GMP transition, as expected if Pbx1 is transcriptionally up-stream of Meis1 (Fig. 6A). To further test the latter hypothesis, cKit+ cells (enriched in myeloid progenitors) were transduced with a retroviral vector expressing either GFP alone (MOCK) or Pbx1 and GFP (PBX1). Pbx1 and Meis1 overexpression was measured by real-time PCR before (day 0, day 1) and after (day 3, in GFP+ cells) transduction (Fig. 6B). In mock-transduced cells, both transcripts were downregulated upon culture, as expected since at day 3 most cells had differentiated (c-Kit− Mac-1+ phenotype). However, Meis1 expression was upregulated in Pbx1 transduced cells, in accordance with the hypothesis that Meis1 is transcriptionally downstream of Pbx1, directly or indirectly. Flt3, a known Meis1 target gene, displayed a similar expression profile (supplementary material Fig. S5). Nevertheless, forced expression of Meis1 was unable to rescue the null phenotype in absence of Pbx1 (data not shown) consistent with their function as a heterodimer. Thus, Meis1 is both a downstream effector as well as a dimerization partner in the Pbx1-dependent transcriptional program of hematopoietic progenitors.
Fig. 6.
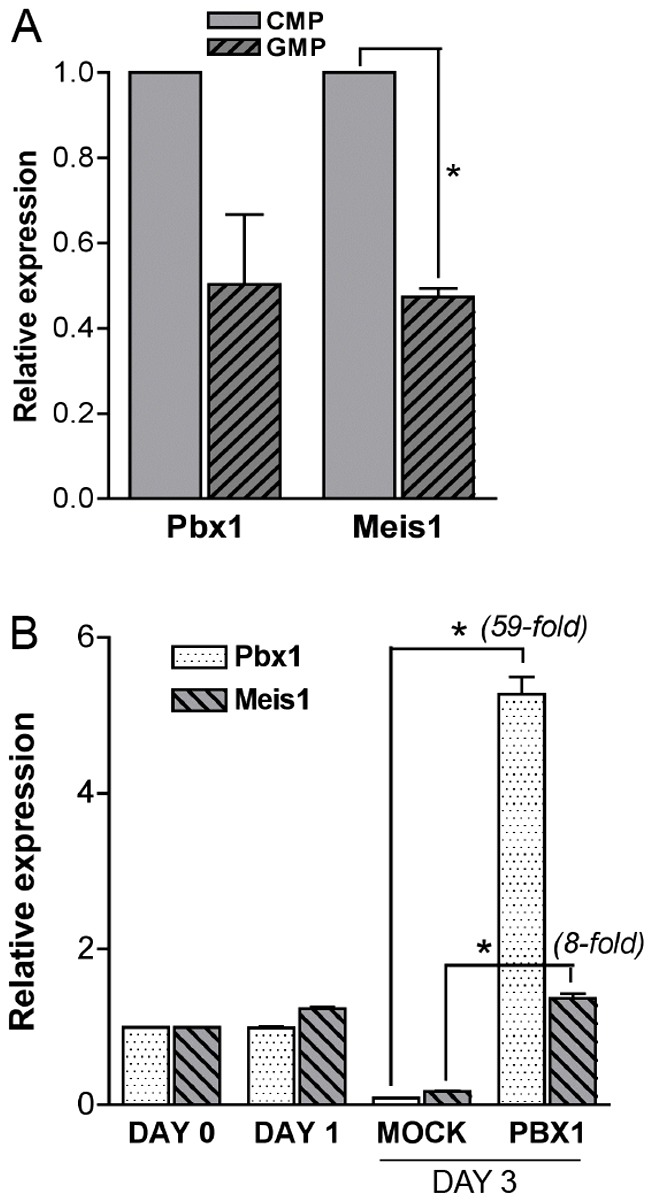
Meis1 is transcriptionally downstream of Pbx1. (A) Bar graph shows Pbx1 and Meis1 transcript levels in GMPs relative to CMPs measured by real-time PCR on progenitors freshly isolated from wild-type BM cells (*P≤0.002). (B) Bar graph shows Pbx1 and Meis1 transcript levels measured by real-time PCR either on freshly isolated wild-type c-Kit+ cells (day 0), or from cultured cells before (day 1) or after (day 3) transduction with a retroviral vector encoding Pbx1 (*P≤0.0007).
Discussion
In this study we demonstrate a role for Pbx1 in sustaining expression of transcriptional programs that restrain myeloid maturation in hematopoietic progenitors and are frequently perturbed in human cancers. Pbx1 also sustains the persistent expression of HSC and B-lymphoid genes in CMPs, highlighting a lineage promiscuity that is maintained by Pbx1. These data support the conclusion that Pbx1 serves dual roles in hematopoietic progenitors to temporally restrict myeloid differentiation, and maintain lymphoid potential, thus being crucial for homeostasis of the hematopoietic system.
Our study provides new insight into the role of Pbx1 as a brake on cellular differentiation and a regulator of the balance between self-renewal versus maturation. Previous studies have attributed the hypoplasia or aplasia of multiple organ systems associated with embryonic deficiency of Pbx1 to a progenitor proliferation defect (Brendolan et al., 2005; Manley et al., 2004; Schnabel et al., 2003; Selleri et al., 2001). In contrast, our studies based on analyses of hematopoietic progenitors at a single cell resolution showed increased short-term proliferation that was prematurely extinguished as progenitors (MPP and CMP) rapidly progressed to the next stage of differentiation. As a consequence, total cell numbers generated in culture and in vivo were reduced but the proliferative index was not significantly perturbed. Accelerated myeloid maturation was confirmed in vivo by gene expression profiles of freshly isolated Pbx1-deficient CMPs, which displayed premature derepression of GMP genes and enrichment for differentiation gene sets. Accelerated maturation has also been observed in embryonic chondrocytes (Selleri et al., 2001), which undergo premature ossification in Pbx1-knockout embryos. Taken together, these studies suggest that the primary defect associated with Pbx1 deficiency is not reduced proliferation per se, but accelerated maturation that shortens the temporal phase of proliferative expansion for progenitors resulting in hypoplasia or cytopenia.
In addition to functioning as a brake on myeloid differentiation, Pbx1 also maintains lymphoid potential in hematopoietic progenitors. In the absence of Pbx1, the earliest progenitors with skewed or restricted lymphoid differentiation capacity (LMPPs and CLPs, respectively) are numerically reduced and functionally compromised with decreased lymphoid outputs in culture and transplant experiments. This likely reflects a substantial role for Pbx1 in maintaining lymphoid gene expression in progenitors consistent with previous studies showing that lymphoid gene expression in HSCs (so-called lymphoid priming) is compromised in the absence of Pbx1 (Ficara et al., 2008). In the current studies, persistent expression of lymphoid genes in wild-type CMPs enabled our demonstration that Pbx1 maintains their lineage inappropriate expression. The functional significance for promiscuous expression of the lymphoid signature in wild-type CMPs is unclear although it may underlie the well-documented lineage switching potential of myeloid progenitors (Anderson et al., 2007; Boeckx et al., 2004; Chiang and Monroe, 1999; Yu et al., 2003). Nevertheless, its perturbation in Pbx1 mutants provides evidence of a substantial role for Pbx1 in maintaining lymphoid gene expression in a variety of progenitors, as well as HSCs, which may account for the major reductions in LMPP, CLP and B cell progenitors of Pbx1-deficient mice.
Our study provides a rationale to investigate whether Meis1 is a direct downstream target of Pbx1, and as a possible mediator of Pbx1 function in CMPs. Meis1 is generally considered a typical HSC gene based on its high expression in the most immature primitive hematopoietic populations, and downregulation upon differentiation (Argiropoulos and Humphries, 2007; Pineault et al., 2002). In this study we confirm that the expression of Pbx1 and Meis1 is decreased in myeloid oligopotent progenitors compared to HSCs, but nevertheless functions to limit their otherwise rapid maturation. In accordance with our conclusion that Meis1 might be a major mediator of Pbx1 in restraining myeloid differentiation, knockdown of Meis1 in murine leukemia cells increases monocytic differentiation (Kumar et al., 2009). Meis1 is also a critical downstream mediator of the MLL oncogenic program, functioning as an essential regulator of leukemia stem cell (LSC) potential (Wong et al., 2007), and our data suggest that its reported function in maintaining self-renewal of LSCs might partly be due to a role in preventing differentiation. Meis1 regulation by Pbx1 is unexpected considering that it is a Pbx1 dimerization partner (Chang et al., 1997), although is consistent with the recently reported compensatory increase in Pbx1 levels in response to loss of Meis1 (Unnisa et al., 2012). However, Pbx1 directly regulates Hox11 in spleen progenitor cells during spleen ontogeny, and Hox11, in turn, regulates its own promoter in complex with Pbx1 (Brendolan et al., 2005). Therefore, Pbx1 participates in complex cross-regulatory circuits with its DNA binding partners.
Our bioinformatics analyses indicated that Pbx1 maintains expression of transcriptional pathways associated with oncogenesis suggesting a proto-oncogenic function for Pbx1 in various lineages, including CMPs. This observation was confirmed upon removal of the annotated cell cycle genes from the analysis (not shown), indicating that the proto-oncogenic signature is not a consequence of the perturbation of CMP proliferation properties. The same observation was also corroborated upon application of more stringent criteria for accepting gene sets as enriched for (not shown). Genes associated with malignancies that are downstream of Pbx1 in CMPs include Mpl, Pf4, Angpt1, Mef2c, Flt3, Fyn, Bcl2, Syk, and Abl1, among others. Despite these hierarchical relationships, retroviral-mediated forced expression of wild-type Pbx1 was not sufficient to induce leukemia in hematopoietic progenitors (data not shown) likely reflecting that its oncogenic activation requires protein fusion as originally observed in B-cell precursor acute lymphoblastic leukemia (Kamps et al., 1990; Nourse et al., 1990), which alters its transcriptional properties. Nevertheless, wild-type Pbx1 is a component of crucial transcriptional cascades initiated by oncoproteins in leukemia pathogenesis (Eklund, 2007; Wong et al., 2007), and our studies demonstrate that Pbx1 in turn regulates transcriptional pathways that normally oppose differentiation and are perturbed in a variety of malignancies.
Materials and Methods
Mice
Tie2Cre+.Pbx1−/f mutant mice have been described previously (Ficara et al., 2008). Congenic Ly5.1 mice, purchased from Jackson Laboratories (Bar Harbor, ME, USA), were maintained in the Stanford animal facility and used as recipients in transplantation experiments. All experiments were performed with the approval of and in accordance with Stanford's and Istituto Clinico Humanitas Administrative Panel on Laboratory Animal Care.
Analysis and isolation of hematopoietic progenitor cells
BM cell suspensions were obtained by crushing of multiple bones as described (Ficara et al., 2008). Progenitors were purified based on the absence of lineage markers using a Lineage Cell Depletion kit (Miltenyi Biotec, Auburn, CA, USA) and an automated cell separator (AutoMACS, Miltenyi Biotec), and then subjected to cell surface staining prior to FACS analysis or sorting. Lineage-depleted BM was further deprived of residual Lin+ cells after incubation with APC-Cy7 or PE-conjugated Streptavidin (eBioscience, San Diego, CA, USA) and monoclonal antibodies (mAbs) to additional T cell markers not present in the cocktail (CD3, 4, 8, BD Pharmingen, San Jose, CA, USA). In the present study the following subpopulations were analyzed: HSCs (Lin−/cKithi/Scahi/CD34−/Flk2−); MPPs at different stages of maturation (Lin−/CD127−/cKithi/Scahi/CD34+/Flk2− or Flk2int or Flk2high); CLPs (Lin−/CD127+/Flk2high/cKitint/Scaint); CMPs (Lin−/cKit+/Sca−/CD34+/FcgRII/IIIint); GMPs (Lin−/cKit+/Sca−/CD34+/FcgRII/IIIhigh). The following fluorochrome-conjugated mAbs were purchased from eBioscience as conjugates to APC, Cy5-PE, Cy7-PE, or PE: cKit (2B8), Sca1 (D7), CD127 (A7R34), CD16/32 (93), CD135 (AF2 10.1). FITC-conjugated CD34 (RAM34) was purchased from BD Pharmingen. CLPs were isolated using procedures described previously (Karsunky et al., 2008). Cell sorts were performed using a FACS Vantage (BD Biosciences, San Jose, CA, USA) equipped with Diva software (BD, Franklin Lakes, New Jersey, USA), and data were analyzed using FlowJo (Tree Star, Ashland, OR, USA). For in vitro B cell differentiation assays, lineage-depleted BM cells were prepared with a lineage-depletion kit (Stem Cell Technologies, Vancouver, BC, Canada) and the automated Robosep cell separator, prior to cell surface staining and sorting. Cell sorts were performed using a FACS ARIA (BD Biosciences).
Real-time quantitative PCR
HSCs and progenitors were sorted from wild-type C57BL/6 mice or from GFP+ cells prior to RNA extraction with Trizol (Invitrogen, Carlsbad, CA, USA). cDNA was prepared using Superscript First-Strand Synthesis System for RT-PCR (Invitrogen) and then subjected to real-time PCR using Taqman probes (Applied Biosystems, Foster City, CA, USA).
Cell culture
For colony-forming unit assays, sorted hematopoietic progenitors were seeded into methylcellulose-containing medium (methoCult 3234; Stem Cell Technologies) in the presence of SCF, Flt3 ligand (20 ng/ml each), IL-6, IL-3, GM-CSF, TPO (10 ng/ml each) and Epo (3 U/ml), and colonies were scored at day 7. MPPs, CMPs and GMPs were cultured in IMDM supplemented with 10% FBS (Hyclone, Logan, UT, USA) and SCF (20 ng/ml), IL-6, IL-3 (10 ng/ml each), in U-bottomed 96-well plates, and analyzed by FACS 3 or 6 days later upon staining with Abs to Mac1/CD11b (M1/70, BD Pharmingen), Gr1 (RB6-8C5, eBioscience), and/or Abs defined above. LMPPs and CLPs were co-cultured with OP9 stromal cells in Mem-alpha supplemented with heat-inactivated 10% FBS, 50 mM β-mercaptoethanol, and SCF, Flt3L (20 ng/ml each), and IL-7 (10 ng/ml), and analyzed by FACS 4 or 10–11 days later upon staining with Abs to B220 (RA3-6B2, eBioscience) and CD19 (1D3, BD Pharmingen). For limiting dilution assays, OP9 cells were seeded in flat-bottomed 96-well plates, and different amounts of sorted LMPPs or CLPs from pools of three mice were added to each well (10 to 96 wells/condition). Medium was half-replaced every third day, and at day 10 cells from each well were analyzed by FACS to assess the presence of B220+CD19+CD11b−Gr1− B cells.
Transplantation assays
Transplants of LMPPs or CLPs (2000–4500 cells) from mutant or control mice (Ly5.2) were performed by retro-orbital injection of sublethally irradiated Ly5.1 mice (450 cGy). Spleens and thymuses were dissected from transplanted animals 1 or 3 weeks later as described (Karsunky et al., 2008). Donor and recipients were distinguished using mAbs to Ly5.2/CD45.2 (104) and Ly5.1/CD45.1 (A20).
Cell cycle analysis
Mice received a single intraperitoneal injection of 5-bromodeoxyuridine (BrdU) 2 hours prior to sacrifice. Analysis of BrdU incorporation in BM progenitors was performed using the FITC BrdU Flow Kit (BD Pharmingen) according to the manufacturer's protocol. Concomitant use of the anti-BrdU mAb and Abs to cell surface markers allowed detection of proliferating cells within the different progenitor subsets after lineage-depletion (LMPPs and CLPs) or cKit positive-selection (CMP, GMP, MEP). CFSE labeling (Invitrogen) was performed according to manufacturer's instructions.
Statistical analysis
Significance of differences was determined by two-tailed Student's t-test. Error bars in bar graphs indicate s.e.m.
Microarray and bioinformatics analyses
BM cells were obtained from multiple bones of individual 3- to 5-week-old Tie2Cre+.Pbx1−/f or Tie2Cre+.Pbx1+/f mice, and maintained on ice when possible through all procedures. CMPs and GMPs were sorted as described above into RNAlater (Ambion – Life Technologie, Grand Island, NY, USA). RNA was purified using Trizol followed by RNeasy MinElute Cleanup Kit (Qiagen, Valencia, CA, USA), then subjected to amplification with the Affymetrix 3′ IVT Express kit (Affymetrix, Santa Clara, CA, USA). Microarray experiments were performed in the Stanford PAN Facility using Affymetrix 430-2.0 arrays. Arrays were scanned with a Gene Chip Scanner 3000 (Affymetrix) running GCOS 1.1.1 software. Microarray data were normalized with Expression Console software (Affymetrix), using RMA algorithms, then further normalized with dCHip (Li and Wong, 2001). Low signals (below 60) were filtered out using the PreprocessDataset module in GenePattern (http://www.broad.mit.edu/cancer/software/genepattern/). Hierarchical clustering of microarray samples (Eisen et al., 1998) was used to identify outliers. Differentially expressed genes in CMP or GMP were identified using Significance Analysis of Microarrays software (Tusher et al., 2001). Genes with the false-discovery rate below 13% were considered significant genes. Microarray raw data are available for download at Gene Expression Omnibus (http://ncbi.nlm.nih.gov/geo, Accession Number GSE30028). Principal component analysis (PCA) was carried out on all genes analyzed to assign the general variability in the data to a reduced set of variables called principal components (Jolliffe, 2002).
DNA constructs and virus production
Human PBX1B cDNA was subcloned into the pMYs-ires-GFP retroviral vector (a gift from T. Kitamura) into the XhoI sites. Retrovirus supernatant was prepared using standard techniques.
Supplementary Material
Acknowledgments
We thank M. Ambrus and C. Nicolas for technical assistance, T. Serwald for useful suggestions regarding CLP prospective isolation and culture and for the GFP+ OP9 cell line, A.G. Rolink for the GFP− OP9 cell line, C. Buracchi for assistance during cell sorts, and T. Kitamura for the pMYs-ires-GFP vector.
Footnotes
Author contributions
F.F. designed and performed experiments, interpreted data, and wrote the manuscript. L.C., M.I. and K.S.S. performed experiments and interpreted data. C.L. and L.Z. conducted bioinformatics analyses. M.L.C. directed the research, oversaw data analysis, and edited the manuscript.
Funding
We acknowledge support from the Children's Health Initiative of the Packard Foundation and the National Institutes of Health (NIH) [grant number CA90735 to M.L.C.]. This work was also supported by an American Society of Hematology (ASH) Scholar Award [to F.F.]; and by a Marie Curie International Reintegration Grant [grant number PIRG6-GA-2009-256452 to F.F.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.125435/-/DC1
References
- Anderson K., Rusterholz C., Månsson R., Jensen C. T., Bacos K., Zandi S., Sasaki Y., Nerlov C., Sigvardsson M., Jacobsen S. E. (2007). Ectopic expression of PAX5 promotes maintenance of biphenotypic myeloid progenitors coexpressing myeloid and B-cell lineage-associated genes. Blood 109, 3697–3705 10.1182/blood-2006-05-026021 [DOI] [PubMed] [Google Scholar]
- Argiropoulos B., Humphries R. K. (2007). Hox genes in hematopoiesis and leukemogenesis. Oncogene 26, 6766–6776 10.1038/sj.onc.1210760 [DOI] [PubMed] [Google Scholar]
- Boeckx N., van der Velden V. H., Boogaerts M., Hagemeijer A., Vandenberghe P., van Dongen J. J. (2004). An inv(16)(p13q22) positive acute myeloid leukaemia relapsing as acute precursor B-cell lymphoblastic leukaemia. Haematologica 89, ECR28. [PubMed] [Google Scholar]
- Brendolan A., Ferretti E., Salsi V., Moses K., Quaggin S., Blasi F., Cleary M. L., Selleri L. (2005). A Pbx1-dependent genetic and transcriptional network regulates spleen ontogeny. Development 132, 3113–3126 10.1242/dev.01884 [DOI] [PubMed] [Google Scholar]
- Cano F., Drynan L. F., Pannell R., Rabbitts T. H. (2008). Leukaemia lineage specification caused by cell-specific Mll-Enl translocations. Oncogene 27, 1945–1950 10.1038/sj.onc.1210818 [DOI] [PubMed] [Google Scholar]
- Chang C. P., Jacobs Y., Nakamura T., Jenkins N. A., Copeland N. G., Cleary M. L. (1997). Meis proteins are major in vivo DNA binding partners for wild-type but not chimeric Pbx proteins. Mol. Cell. Biol. 17, 5679–5687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang M. Y., Monroe J. G. (1999). BSAP/Pax5A expression blocks survival and expansion of early myeloid cells implicating its involvement in maintaining commitment to the B-lymphocyte lineage. Blood 94, 3621–3632 [PubMed] [Google Scholar]
- Cleary M. L. (2009). Regulating the leukaemia stem cell. Best Pract. Res. Clin. Haematol. 22, 483–487 10.1016/j.beha.2009.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen M. B., Spellman P. T., Brown P. O., Botstein D. (1998). Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. USA 95, 14863–14868 10.1073/pnas.95.25.14863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eklund E. A. (2007). The role of HOX genes in malignant myeloid disease. Curr. Opin. Hematol. 14, 85–89 10.1097/MOH.0b013e32801684b6 [DOI] [PubMed] [Google Scholar]
- Ficara F., Murphy M. J., Lin M., Cleary M. L. (2008). Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell 2, 484–496 10.1016/j.stem.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galan-Caridad J. M., Harel S., Arenzana T. L., Hou Z. E., Doetsch F. K., Mirny L. A., Reizis B. (2007). Zfx controls the self-renewal of embryonic and hematopoietic stem cells. Cell 129, 345–357 10.1016/j.cell.2007.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki H., Akashi K. (2007). Myeloid lineage commitment from the hematopoietic stem cell. Immunity 26, 726–740 10.1016/j.immuni.2007.06.004 [DOI] [PubMed] [Google Scholar]
- Jolliffe I. T. (2002). Principal Component Analysis New York, NY: Springer-Verlag [Google Scholar]
- Kamps M. P., Murre C., Sun X. H., Baltimore D. (1990). A new homeobox gene contributes the DNA binding domain of the t(1;19) translocation protein in pre-B ALL. Cell 60, 547–555 10.1016/0092-8674(90)90658-2 [DOI] [PubMed] [Google Scholar]
- Karsunky H., Inlay M. A., Serwold T., Bhattacharya D., Weissman I. L. (2008). Flk2+ common lymphoid progenitors possess equivalent differentiation potential for the B and T lineages. Blood 111, 5562–5570 10.1182/blood-2007-11-126219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov A. V., Twomey D., Feng Z., Stubbs M. C., Wang Y., Faber J., Levine J. E., Wang J., Hahn W. C., Gilliland D. G. et al. (2006). Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature 442, 818–822 10.1038/nature04980 [DOI] [PubMed] [Google Scholar]
- Kumar A. R., Li Q., Hudson W. A., Chen W., Sam T., Yao Q., Lund E. A., Wu B., Kowal B. J., Kersey J. H. (2009). A role for MEIS1 in MLL-fusion gene leukemia. Blood 113, 1756–1758 10.1182/blood-2008-06-163287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Wong W. H. (2001). Model-based analysis of oligonucleotide arrays: expression index computation and outlier detection. Proc. Natl. Acad. Sci. USA 98, 31–36 10.1073/pnas.98.1.31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley N. R., Selleri L., Brendolan A., Gordon J., Cleary M. L. (2004). Abnormalities of caudal pharyngeal pouch development in Pbx1 knockout mice mimic loss of Hox3 paralogs. Dev. Biol. 276, 301–312 10.1016/j.ydbio.2004.08.030 [DOI] [PubMed] [Google Scholar]
- Månsson R., Hultquist A., Luc S., Yang L., Anderson K., Kharazi S., Al-Hashmi S., Liuba K., Thorén L., Adolfsson J. et al. (2007). Molecular evidence for hierarchical transcriptional lineage priming in fetal and adult stem cells and multipotent progenitors. Immunity 26, 407–419 10.1016/j.immuni.2007.02.013 [DOI] [PubMed] [Google Scholar]
- Metcalf D., Dakic A., Mifsud S., Di Rago L., Wu L., Nutt S. (2006). Inactivation of PU.1 in adult mice leads to the development of myeloid leukemia. Proc. Natl. Acad. Sci. USA 103, 1486–1491 10.1073/pnas.0510616103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minami Y., Stuart S. A., Ikawa T., Jiang Y., Banno A., Hunton I. C., Young D. J., Naoe T., Murre C., Jamieson C. H. et al. (2008). BCR-ABL-transformed GMP as myeloid leukemic stem cells. Proc. Natl. Acad. Sci. USA 105, 17967–17972 10.1073/pnas.0808303105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens C. B., Selleri L. (2006). Hox cofactors in vertebrate development. Dev. Biol. 291, 193–206 10.1016/j.ydbio.2005.10.032 [DOI] [PubMed] [Google Scholar]
- Mootha V. K., Lindgren C. M., Eriksson K. F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E. et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 10.1038/ng1180 [DOI] [PubMed] [Google Scholar]
- Nourse J., Mellentin J. D., Galili N., Wilkinson J., Stanbridge E., Smith S. D., Cleary M. L. (1990). Chromosomal translocation t(1;19) results in synthesis of a homeobox fusion mRNA that codes for a potential chimeric transcription factor. Cell 60, 535–545 10.1016/0092-8674(90)90657-Z [DOI] [PubMed] [Google Scholar]
- Orkin S. H., Zon L. I. (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644 10.1016/j.cell.2008.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineault N., Helgason C. D., Lawrence H. J., Humphries R. K. (2002). Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Exp. Hematol. 30, 49–57 10.1016/S0301-472X(01)00757-3 [DOI] [PubMed] [Google Scholar]
- Rothenberg E. V. (2007). Negotiation of the T lineage fate decision by transcription-factor interplay and microenvironmental signals. Immunity 26, 690–702 10.1016/j.immuni.2007.06.005 [DOI] [PubMed] [Google Scholar]
- Santaguida M., Schepers K., King B., Sabnis A. J., Forsberg E. C., Attema J. L., Braun B. S., Passegué E. (2009). JunB protects against myeloid malignancies by limiting hematopoietic stem cell proliferation and differentiation without affecting self-renewal. Cancer Cell 15, 341–352 10.1016/j.ccr.2009.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel C. A., Selleri L., Cleary M. L. (2003). Pbx1 is essential for adrenal development and urogenital differentiation. Genesis 37, 123–130 10.1002/gene.10235 [DOI] [PubMed] [Google Scholar]
- Selleri L., Depew M. J., Jacobs Y., Chanda S. K., Tsang K. Y., Cheah K. S., Rubenstein J. L., O'Gorman S., Cleary M. L. (2001). Requirement for Pbx1 in skeletal patterning and programming chondrocyte proliferation and differentiation. Development 128, 3543–3557 [DOI] [PubMed] [Google Scholar]
- Signer R. A., Montecino-Rodriguez E., Witte O. N., Dorshkind K. (2010). Immature B-cell progenitors survive oncogenic stress and efficiently initiate Ph+ B-acute lymphoblastic leukemia. Blood 116, 2522–2530 10.1182/blood-2010-01-264093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somervaille T. C., Matheny C. J., Spencer G. J., Iwasaki M., Rinn J. L., Witten D. M., Chang H. Y., Shurtleff S. A., Downing J. R., Cleary M. L. (2009). Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell 4, 129–140 10.1016/j.stem.2008.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S. et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothova Z., Kollipara R., Huntly B. J., Lee B. H., Castrillon D. H., Cullen D. E., McDowell E. P., Lazo-Kallanian S., Williams I. R., Sears C. et al. (2007). FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell 128, 325–339 10.1016/j.cell.2007.01.003 [DOI] [PubMed] [Google Scholar]
- Tremblay M., Tremblay C. S., Herblot S., Aplan P. D., Hébert J., Perreault C., Hoang T. (2010). Modeling T-cell acute lymphoblastic leukemia induced by the SCL and LMO1 oncogenes. Genes Dev. 24, 1093–1105 10.1101/gad.1897910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V. G., Tibshirani R., Chu G. (2001). Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 98, 5116–5121 10.1073/pnas.091062498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unnisa Z., Clark J. P., Roychoudhury J., Thomas E., Tessarollo L., Copeland N. G., Jenkins N. A., Grimes H. L., Kumar A. R. (2012). Meis1 preserves hematopoietic stem cells in mice by limiting oxidative stress. Blood 120, 4973–4981 10.1182/blood-2012-06-435800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P., Somervaille T. C., Venkatasubrahmanyam S., Kogan S., McLaughlin M. E., Weissman I. L., Butte A. J., Passegué E., Sage J. (2008). Hematopoietic stem cell quiescence is maintained by compound contributions of the retinoblastoma gene family. Cell Stem Cell 3, 416–428 10.1016/j.stem.2008.07.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojiski S., Guibal F. C., Kindler T., Lee B. H., Jesneck J. L., Fabian A., Tenen D. G., Gilliland D. G. (2009). PML-RARalpha initiates leukemia by conferring properties of self-renewal to committed promyelocytic progenitors. Leukemia 23, 1462–1471 10.1038/leu.2009.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong P., Iwasaki M., Somervaille T. C., So C. W., Cleary M. L. (2007). Meis1 is an essential and rate-limiting regulator of MLL leukemia stem cell potential. Genes Dev. 21, 2762–2774 10.1101/gad.1602107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yilmaz O. H., Valdez R., Theisen B. K., Guo W., Ferguson D. O., Wu H., Morrison S. J. (2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia-initiating cells. Nature 441, 475–482 10.1038/nature04703 [DOI] [PubMed] [Google Scholar]
- Yu D., Allman D., Goldschmidt M. H., Atchison M. L., Monroe J. G., Thomas-Tikhonenko A. (2003). Oscillation between B-lymphoid and myeloid lineages in Myc-induced hematopoietic tumors following spontaneous silencing/reactivation of the EBF/Pax5 pathway. Blood 101, 1950–1955 10.1182/blood-2002-06-1797 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.