

Background: Understanding the coupling of O2 reduction to proton pumping by CcO requires detection of reaction intermediates.
Results: We have detected two oxoferryl intermediates at the PM oxidation state.
Conclusion: The H-bonding properties of the proximal heme a3 His ligand control the strength of the oxoferryl species.
Significance: The role of His-411, Thr-389, Gly-386, and Asp-399 residues in the proton pumping P→F transition is outlined.
Keywords: Bioenergetics, Computation, Cytochrome Oxidase, Heme, Raman Spectroscopy
Abstract
Identification of the intermediates and determination of their structures in the reduction of dioxygen to water by cytochrome c oxidase (CcO) are particularly important to understanding both O2 activation and proton pumping by the enzyme. In this work, we report the products of the rapid reaction of O2 with the mixed valence form (CuA2+, heme a3+, heme a32+-CuB1+) of the enzyme. The resonance Raman results show the formation of two ferryl-oxo species with characteristic Fe(IV)=O stretching modes at 790 and 804 cm−1 at the peroxy oxidation level (PM). Density functional theory calculations show that the protein environment of the proximal H-bonded His-411 determines the strength of the distal Fe(IV)=O bond. In contrast to previous proposals, the PM intermediate is also formed in the reaction of Y167F with O2. These results suggest that in the fully reduced enzyme, the proton pumping νFe(IV)=O = 804 cm−1 to νFe(IV)=O = 790 cm−1 transition (P→F, where P is peroxy and F is ferryl) is triggered not only by electron transfer from heme a to heme a3 but also by the formation of the H-bonded form of the His-411-Fe(IV)=O conformer in the proximal site of heme a3. The implications of these results with respect to the role of an O=Fe(IV)-His-411-H-bonded form to the ring A propionate of heme a3-Asp-399-H2O site and, thus, to the exit/output proton channel (H2O) pool during the proton pumping P→F transition are discussed. We propose that the environment proximal to the heme a3 controls the spectroscopic properties of the ferryl intermediates in cytochrome oxidases.
Introduction
Dioxygen activation and reduction by the respiratory enzyme cytochrome c oxidase (CcO)3 are of fundamental importance in bioenergetics and cell respiration (1–26). The enzyme uses four redox-active centers, CuA, heme a, heme a3, and CuB, to sustain mitochondrial electron transport by reducing O2 to H2O. This catalytic strategy provides an effective means by which to couple the free energy available in late oxygen intermediates to the proton pumping of the enzyme (27–31). Establishing the structures of the intermediates subsequent to O=O bond cleavage is essential to understanding the linkage of these events with proton transfer. The O=O bond cleavage mechanism by CcO and the structures of the intermediates have been a matter of considerable debate (11–29).
Resonance Raman (RR) spectroscopy has been considered as a reliable technique for identifying the structure of the intermediate species (11–29), and time-resolved resonance Raman has been used to monitor the kinetics of the formation and decay of the various intermediates (18, 19, 24, 25). This way, the relationship between the electron transfer events and the formation of each intermediate, which are required to clarify the molecular mechanisms of the O2 reduction, was monitored in the ns-ms time scale. Some of the structures were inferred based on observed kinetic changes, and others were proposed based on spectroscopic properties. Since the characterization of the P (607 nm) intermediate in the reduction of O2 to H2O by CcO as a ferryl-oxo species, several structures and reaction mechanisms have been proposed (20–29). There is a consensus now that the O–O bond cleavage takes place when the binuclear heme Fe-CuB center is reduced, and recently, it has been suggested that the additional electron, which is needed to produce the so-called P (607 nm), Fe(IV)=O intermediate, is provided by either a tyrosine or a tryptophan residue (29–31). The determination of the structure(s) of the final product of the reaction of MV-CcO with O2, which is linked to the proton pumping function of the enzyme, may resolve controversial aspects of the O2 mechanism. We have studied the reaction of mixed valence MV-CcO (CuA2+, heme a3+, heme a32+, CuB1+) from Paracoccus denitrificans with O2 by RR. Our results show, for the first time, the presence of two ferryl-oxo intermediates with characteristic frequencies at 790 and 804 cm−1 at the same oxidation state of the heme Fe-CuB center. We attribute the 14 cm−1 frequency difference between the ferryl-oxo species to variations in the basicity (H-bonded versus non-H-bonded conformers) of the His-411 ligand proximal to the heme a3 (Fig. 1), induced at the point of the O–O bond cleavage process. Density functional theory calculations showed that the H-bonded properties of the proximal His-411 control the strength the Fe(IV)=O bond, and the strength of the H-bonding is controlled by the distance between the heme Fe(IV)=O and CuB. In addition, the DFT data excluded the formation of a His-Fe(V)=O species or that of a heme macrocycle π cation radical Fe(IV)=O structure for the PM (804 cm−1) intermediate.
FIGURE 1.

Schematic presentation of the binuclear heme a3-CuB center and the protonation states of Asp-399/ring A propionate (prop-A) of CcO from P. denitrificans.
EXPERIMENTAL PROCEDURES
Cytochrome c oxidase was purified from P. denitrificans according to published procedures (27, 28, 31). The enzyme was concentrated to 150 μm in Tris buffer containing 0.1% dodecyl β-d-maltoside at pH 7.5 and stored in liquid nitrogen until use. The enzyme was diluted to 5 and 50 μm for the optical and resonance Raman experiments, respectively. The MV-CcO was prepared by anaerobic incubation of the oxidized enzyme with carbon monoxide (CO). Resonance Raman spectra were acquired as described elsewhere (27, 28). The excitation wavelength was 428.7 nm, provided by a diode laser (Melles Griot). The incident power was 1 milliwatt. Optical absorbance spectra were recorded with a PerkinElmer Life Sciences Lambda 20 UV-visible spectrometer.
Density Functional Theory (DFT)-based Geometry Optimizations and Frequency Calculations were performed for all models by the TURBOMOLE software package on the same level of theory to derive vibrational frequencies (32). A force module of TURBOMOLE used calculated analytically harmonic vibrational frequencies within the Hartree–Fock (HF) or (RI)DFT methods for closed shell and spin-unrestricted open shell systems. RI was only used partially, which means that the resulting Hessian is only a (very good) approximation to exact second derivatives of the (RI)DFT energy expression. A standard force constant calculation predicted all allowed and forbidden vibrational transitions.
In our DFT studies, we used a CuB(II) and a Fea3(IV) site, giving a (+1) overall charge. A doublet state was assumed for the binuclear ferryl Fe(IV)-Cu(II) models. It should be noted that a quartet state leads to a bridged Fe-O-CuB species following a histidine dissociation. For each structure considered, a full geometry optimization was performed using the density functional BLYP method (33, 34). The TZVP triple-ζ valence basis set augmented with polarization functions p on hydrogenand d on first and second row atoms, as implemented in the TURBOMOLE version 5-8-0 software package, was used (32). For the iron and copper metals, an effective core potential (ECP) was used. It is named “ecp-10-mdf” for iron and “ecp-18 arep” for copper. We implemented the computational advantage of the RI-J (Resolution of Identity) approximation as defined in the TURBOMOLE software package with default parameterization.
RESULTS AND DISCUSSION
Mixing of oxygen with the CO-bound MV oxidase yields long-lived oxygenated intermediate(s) that occur after the decay of the oxy intermediate (26). The visible region optical absorption spectra of the wild-type oxygenated species formed this way, and their time evolution is shown in Fig. 2A. The optical data demonstrate that incubation of the oxidized enzyme with CO in the presence of O2 generated the P intermediate (607 nm), but not the F intermediate (580 nm). It also demonstrates that the 607 nm species was generated first and that the heme a3 returned to the oxidized form without forming the 580 nm (F) species. It should be noted that in the reaction of oxidized CcO with H2O2, the decay of the 607 nm species to the oxidized form occurs through the 580 nm species (23, 28).
FIGURE 2.

A, optical absorption difference spectra of the reaction products by direct mixing of O2 with CO-MV (mixed valence) aa3 oxidase from P. denitrificans minus the oxidized form of the enzyme at the indicated times subsequent to mixing, pH 7.5. The enzyme concentration was 5 μm, and the path length of the cell was 0.5 cm. B, resonance Raman spectra of CO-MV at 0–5 min subsequent to mixing with 16O2 (spectrum a) and 18O2 (spectrum b). The inset shows the difference a-b spectrum. Several 0–5-min spectra were collected and added for the 16O2 and 18O2 experiments. The enzyme concentration was 50 μm, pH 7.5. The excitation wavelength was 428.7 nm, and the incident power was 1 milliwatt.
We have used continuous wave 428.7 nm RR excitation to detect the long-lived oxygenated intermediates. In Fig. 2B, we present low frequency RR spectra of the MV-CcO/16O2 and MV-CcO/18O2 reactions at 0–5 min at pH 7.5. The inset shows the 16O2/18O2 RR difference spectrum in which the oxygen-associated vibrations at 804/764 and 790/750 cm−1 are the only isotope-sensitive mode present. The frequency, bandwidth, and isotope shift of the 804/764 cm−1 mode match closely those observed in the reaction of fully reduced bovine aa3 with O2, the reaction of MV CcO with O2, and that of the oxidized CcO with H2O2, and they have been assigned to a heme a3 Fe(IV)=O species of the P intermediate (23, 25, 28). The other O2-sensitive mode at 790 cm−1 and its isotope shift was also consistent with those previously reported in the reaction of fully reduced CcO with O2 and in the reaction of oxidized CcO with H2O2 (12, 14, 16, 23, 28). The two ferryl-oxo intermediates we have detected are in an equilibrium state. The 790 cm−1 mode, however, has been previously assigned to a heme a3 Fe(IV)=O species of the F intermediate (580 nm) that is one oxidation level lower than the P intermediate (607 nm) in the reactions of both the bovine and the P. denitrificans aa3 enzymes with H2O2 (23, 28). Fig. 3B shows the RR spectra of the MV-Y167F/16O2 and MV-Y167F/18O2 reactions at 0–5 min subsequent to mixing the enzyme with oxygen at pH 7.5. The difference spectrum (trace a minus trace b) is shown in the inset. In contrast to the wild-type MV CcO/O2 reaction, only the 804/764 cm−1 pair was present, as well as decays at times similar to those observed in the wild-type MV-CcO/O2 reaction.
FIGURE 3.

A, optical absorption difference spectra of the reaction products by direct mixing of O2 with CO-MV (mixed valence) Y167F aa3 oxidase from P. denitrificans minus the oxidized form of the enzyme at the indicated times subsequent to mixing, pH 7.5. The enzyme concentration was 5 μm, and the path length of the cell was 0.5 cm. B, resonance Raman spectra of CO-MV Y167F at 0–5 min subsequent to mixing with 16O2 (spectrum a) and 18O2 (spectrum b). The inset shows the difference a-b spectrum. Several 0–5-min spectra were collected and added for the 16O2 and 18O2 experiments. The enzyme concentration was 50 μm, pH 7.5. The excitation wavelength was 428.7 nm, and the incident power was 1 milliwatt.
It has been established that the properties of the trans-ligand of ferryl-oxo complexes can affect the bonding between the iron and the oxygen, and thus, its vibrational frequency (28); the vibrational frequency decreases as the ligand becomes more electron-donating to Fe(IV)=O. The major contributor to this effect is the proximal to the heme Fe His-411 ligand in which a high Fe-His mode arises from a population in which the H-bond is intact, whereas a lower frequency of the Fe-His vibration reflects a form of the enzyme lacking the H-bond. Based on the crystal structure of aa3 from P. denitrificans (7) and from mammals (8), the proximal His is capable of forming an H-bond with Gly and Thr (Fig. 1). We attribute the 14 cm−1 difference of the 790 and 804 cm−1 modes to the H-/non-H-bonding interaction, respectively, of the proximal His-411 with Gly-386 and/or Thr-389 (Fig. 1). In the case of the H-bonded His-411, electron density is pushed into the antibonding π* orbitals of Fe(IV)=O, weakening the bond, and thus, shifting the Fe(IV)=O stretching frequency from 804 to 790 cm−1. The data presented here also demonstrate that although there are similarities in the O2-associated vibrations in the peroxide and O2 reactions at the P and F level, suggesting that the two reactions proceed through common intermediates, the conformational changes induced to the proximal site by the cleavage of the O–O bond in the distal site in these two reactions are different.
In the DFT calculations, the binuclear center was composed of CuB(II) and His heme Fe(IV)=O (Fig. 4). Due to the nature of these large sites, theoretical calculations were performed on simplified models with a restricted number of atoms. In these models, the CuB metal site was represented by imidazole ligands instead of histidines, and the cross-linked His-Tyr residue was represented by a cross-linked imidazole phenol unit. In the heme iron, the proximal histidine was represented by an imidazole ligand, and the heme was represented by a simplified porphyrin substituted with a CH3CH2(OH)- group to account for the hydroxyethylfarnesyl interactions in the active site of CcO. We investigated the effect of H-bonding to the ferryl-oxo species that is provided by the distal environment of CuB and also by the H-bonding interaction of the proximal histidine with the protein environment. Binuclear models with zero (A1), one (A2), or two hydrogen bonds (A3) to the proximal histidine were treated along with a CuB(II) site coordinated to hydroxyl (–OH) group. One or two ethylic acids are modeled near the proximal histidine to reproduce the effect of H-bonding. No restrictions were applied in the models. Charge population analysis based on Mulliken approach was performed for the binuclear models, and the results are shown on selected atoms in Fig. 4. Population analysis allowed us to check the validity of the optimized structures for the intermediates under study. The calculated spins for iron and copper for all models (A1–A3) demonstrated that the heme iron was indeed in the +4 oxidation state and that copper was in the +2 oxidation state. These results were consistent with spin population analysis for ferryl-oxo species (35).
FIGURE 4.

Geometry-optimized structures of ferryl conformations in the heme a3-CuB binuclear center with different hydrogen bonding to proximal histidine. Spin (with charge in parentheses) population analysis based on the Mulliken approach is also shown for selected atoms. Numbering of amino acids is the same as in Fig. 1.
Structures A1, A2, and A3 differed in the distance of CuB from Fea3 (∼4 Å) at a maximum of ± 0.098 Å as CuB approached Fe(IV)=O and as the H-bonding interaction of proximal His changed from null to two. The TURBOMOLE software was applied for the calculation of Δν, and as shown in Table 1, these values represented real shifts that do not suffer from accuracy restrictions of the method used because the peaks involved have large intensities. For example, the modes at 787 (A1) and 763 (A3) cm−1 have intensities of 89.28 and 106.98 km/mol, respectively (significantly higher than adjacent normal modes). Calculated frequencies are shown with and without scaling in Table 1. The Fe-CuB distance calculated from the unconstrained group of models did not differ significantly from that observed in the crystal structures of CcO (7–9). Based on the crystal structures, the hydrogen bonds are asymmetric, and O–N(H) distances vary from 2.78 to 3.52 Å. In our models, the O–H(N) distances varied from 1.94 to 2.51 Å, representing the hydrogen-bonding network in vivo. Cleavage of the O–O bond was linked to an Fe(V)=O moiety. However, iron in +5 oxidation state was not stabilized and was converted to a more stable Fe(IV) π-cation radical iso-electronic configuration. Thus, in addition to oxo-ferryl intermediates (doublet species) with different proximal hydrogen-bonding strength presented above, we have investigated the possibility that either a porphyrin π-cation radical or an Fe(V) species appeared in the CcO catalytic cycle. Although π-cation radical models (triplet and quintet with total charge of +2) containing Fe(IV) and Cu(II) metal sites were optimized in geometry (riBLYP/TZVP), the DFT calculations gave no Fe(V) species in a scan of different multiplicities and geometries. These results are in agreement with those reported by Dey and Ghosh (36). They have reported that Fe(V) porphyrin complexes are rare and less stable by 3.7–5.8 kcal/mol as compared with π-cation radicals (A2u, A1u). A triplet iron-oxo π-cation radical gave a spin population 1.27 on iron and 0.42 on CuB, suggesting, based on Mulliken population analysis, Fe(IV) and CuB(II) oxidation states. A spin population of 0.77 was found on the porphyrin ring. For the quintet iron-oxo π-cation radical, spin populations were calculated to be 1.24, 0.43, and 0.80 on iron, copper, and the porphyrin ring, respectively.
TABLE 1.
Calculated frequencies (in cm−1) in TURBOMOLE riBLYP/TZVP level are shown without and with (×1.022) scaling factor
Scaling factor was chosen to derive the experimental 804 cm−1 ν(Fe-O) vibrational frequency.
| Fe(IV) models | ν(Fe-16O)a | ν(Fe-18O)a | ν(Fe-16O) ×1,022 | ν(Fe-18O) ×1,022 |
|---|---|---|---|---|
| Ferryl + 0× H-bond | 787 | 755 (Δν = 32) | 804 | 772 (Δν = 32) |
| Ferryl + 1× H-bond | 779 | 750 (Δν = 29) | 796 | 766 (Δν = 30) |
| Ferryl + 2× H-bond | 763 | 728 (Δν = 35) | 780 | 744 (Δν = 36) |
| Ferryl π-cation radical (triplet) | 774 | — | 791 | — |
| Ferryl π-cation radical (quintet) | 774 | — | 791 | — |
a No scaling.
A close inspection of the data presented in Table 1 shows that the νFe(IV)=O is mainly affected by the interaction of His-411 with the protein environment and by the formation of a π-cation radical. In the latter case, significant differences were calculated for the ν3 and ν4 porphyrin modes as shown in Table 2. Moreover, the ν(Fe(IV)=O) = 774 cm−1 remained unaffected in the case of the two calculated π-cation radicals. In the case of a ferryl species, the calculated ν10, ν2, ν3, and ν4 modes shown in Table 2 are in agreement with those reported in the RR experiments (12, 19, 25). The His proximal to heme a3 exerted similar distances to residues such as Gly, Thr, or His found in the proximal area on the crystal structures of different cytochrome oxidases (7–10). Although these H-bonding interactions appear rigid, we have observed their dependence on the protonation state of the heme a3 propionate A/Asp-399 pair (37). In detail, based on these earlier simulations, we probed the His-411-Gly-386 and His-411-Thr-389 distances for the aa3 from the P. denitrificans model system. Molecular dynamics simulation trajectories exerted variable distances of H-bonding interactions for the proximal area for the same enzyme. Fig. 5 shows the correlation diagrams for selected interactions: combined mean value of His-411-Gly-386/Thr-389 distance and mean value for His-411-Gly-386 distance. There was an increase in the proximal H-bonding strength to His-411 when the propionate-A/Asp-399 pair and Glu-278 were deprotonated. In contrast to the static picture of a crystal structure, dynamical methodologies accurately probe a variable proximal interaction altering resonances in the His-411-Fe=O system (38). The dynamic of the proximal to the heme Fe environment controls the properties of the Fe(IV)=O intermediate found in CcOs.
TABLE 2.
Calculated high frequency Raman frequencies in TURBOMOLE riBLYP/TZVP level
| Fe(IV) (16O) models | ν10a | ν2a | ν3a | ν4a |
|---|---|---|---|---|
| cm−1 | cm−1 | cm−1 | cm−1 | |
| Ferryl + 0× H-bond | 1558 | 1508 | 1422 | 1373 (1173) |
| Ferryl π-cation radical (triplet) | 1561 | 1512 | 1411 | 1360 (1179) |
| Ferryl π-cation radical (quintet) | 1549 | 1509 | 1413 | 1329 (1176) |
a No scaling.
FIGURE 5.
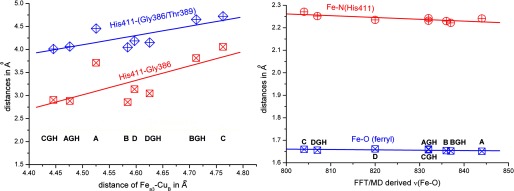
Correlation diagrams for selected interactions. Left panel: the combined mean value of His-411-Gly-386/Thr-389 distance and mean value for His-411-Gly-386 distance. Right panel: the effect of the variation in Fe(IV)=O (ferryl) and Fe-N (His-411) bond lengths on the ν(Fe=O) mode. The abbreviations used are: C, fully deprotonated propionate-A/Asp-399 pair; A, fully protonated propionate-A/Asp-399 pair; D, only the propionate-A is protonated; B, only Asp-399 is protonated; GH, Glu-278 is protonated.
In summary, the following points emerge from the present study. First, the detection of two ferryl-oxo intermediates at the same oxidation level (P) establishes crucial links between their structures. Second, it clearly demonstrates that the frequencies detected for the ferryl-oxo intermediates are independent of the formation of the P or F intermediate but rather depend on the equilibrium between the H- and the non-H-bonded O=Fe-His-411, yielding either the 790 cm−1 or the 804 cm−1 species. This idea finds support in the experiments on oxygenated MV mammalian aa3, where only the 804 cm−1 ferryl-oxo species has been detected at the P level (24, 26). Obviously, in the mammalian enzyme, the equilibrium of the proximal site is shifted toward the non-H-bonded O=Fe-His-411 conformer, and thus, it was possible to conclude that the 804 cm−1 species represents the P intermediate in this enzyme. The present data and those previously reported in the peroxide reaction demonstrate that the H-/non-H-bonded proximal His-411 equilibrium determines the strength of the distal Fe(IV)=O bond (28).
Finally, the determination of the structures of the ferryl-oxo intermediates in the reduction of O2 to H2O by CcO is of profound importance in elucidating the mechanism of proton translocation in the enzyme. In particular, it has been suggested that the chemical energy released in the O2 reduction is stored at the end of the oxidative phase prior to the re-reduction of the protein and is utilized for proton translocation. We postulate that the proximal to the heme a3 environment plays a crucial role in the strength of the Fe(IV)=O bond, and thus, in the chemical energy produced by the cleavage of the O–O bond. Comparison of the ferryl-oxo intermediates formed in solution conditions with those formed in membranous preparations has revealed very similar structures (39). This observation may indicate that the chemical energy produced by the cleavage of the O–O bond is stored in the chemical bonds of the oxygen reduction products in the heme a3-CuB center.
This work was supported by funds from the Cyprus Research Promotion Foundation (to C. V.) (TEXNOLOGIA/THEPIS/0609(BE)/05), the Minister of Science, Sports and Culture, Japan (Grant 14001004) (to T. K.), and Deutsche Forschungsgemeinschaft (DFG) (Grant SFB472) (to B. L.).
- CcO
- cytochrome c oxidase
- RR
- resonance Raman
- MV
- mixed valence
- DFT
- density functional theory
- ECP
- effective core potential
- P
- peroxy
- F
- ferryl
- RI
- resolution of identity
- TZVP
- triple zeta valence plus polarization basis set
- BLYP
- Becke (for the exchange part), Lee, Yang, and Parr (for the correlation part) functional
- riBLYP
- resolution of identity approximation to the BLYP functional calculations.
REFERENCES
- 1. Wikström M. (1989) Identification of the electron transfers in cytochrome oxidase that are coupled to proton-pumping. Nature 338, 776–778 [DOI] [PubMed] [Google Scholar]
- 2. Babcock G. T., Wikström M. (1992) Oxygen activation and the conservation of energy in cell respiration. Nature 356, 301–309 [DOI] [PubMed] [Google Scholar]
- 3. Michel H. (1998) The mechanism of proton pumping by cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 95, 12819–12824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Michel H., Behr J., Harrenga A., Kannt A. (1998) Cytochrome c oxidase: Structure and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 27, 329–356 [DOI] [PubMed] [Google Scholar]
- 5. Ferguson-Miller S., Babcock G. T. (1996) Heme/Copper terminal oxidases. Chem. Rev. 96, 2889–2908 [DOI] [PubMed] [Google Scholar]
- 6. Gennis R. B. (1998) Multiple proton-conducting pathways in cytochrome oxidase and a proposed role for the active-site tyrosine. Biochim. Biophys. Acta 1365, 241–248 [Google Scholar]
- 7. Iwata S., Ostermeier C., Ludwig B., Michel H. (1995) Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Nature 376, 660–669 [DOI] [PubMed] [Google Scholar]
- 8. Tsukihara T., Aoyama H., Yamashita E., Tomizaki T., Yamaguchi H., Shinzawa-Itoh K., Nakashima R., Yaono R., Yoshikawa S. (1995) Structures of metal sites of oxidized bovine heart cytochrome c oxidase at 2.8 Å. Science 269, 1069–1074 [DOI] [PubMed] [Google Scholar]
- 9. Yoshikawa S., Shinzawa-Itoh K., Nakashima R., Yaono R., Yamashita E., Inoue N., Yao M., Fei M. J., Libeu C. P., Mizushima T., Yamaguchi H., Tomizaki T., Tsukihara T. (1998) Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science 280, 1723–1729 [DOI] [PubMed] [Google Scholar]
- 10. Lyons J. A., Aragão D., Slattery O., Pisliakov A. V., Soulimane T., Caffrey M. (2012) Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature 487, 514–518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varotsis C., Woodruff W. H., Babcock G. T. (1989) Time-resolved Raman detection of ν(Fe-O) in an early intermediate in the reduction of oxygen by cytochrome oxidase. J. Am. Chem. Soc. 111, 6439–6440 [Google Scholar]
- 12. Varotsis C., Babcock G. T. (1990) Appearance of the ν(FeIV=O) vibration from a ferryl-oxo intermediate in the cytochrome oxidase/dioxygen reaction. Biochemistry 29, 7357–7362 [DOI] [PubMed] [Google Scholar]
- 13. Varotsis C., Woodruff W. H., Babcock G. T. (1990) Direct detection of a dioxygen adduct of cytochrome a3 in the mixed valence cytochrome oxidase/dioxygen reaction. J. Biol. Chem. 265, 11131–11136 [PubMed] [Google Scholar]
- 14. Ogura T., Takahashi S., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (1990) Observation of the Fe4+=O stretching Raman band for cytochrome oxidase compound B at ambient temperature. J. Biol. Chem. 265, 14721–14723 [PubMed] [Google Scholar]
- 15. Ogura T., Takahashi S., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (1990) Observation of the iron(II)-oxygen stretching Raman band for cytochrome oxidase compound A at ambient temperature. J. Am. Chem. Soc. 112, 5630–5631 [PubMed] [Google Scholar]
- 16. Han S., Ching Y.-C., Rousseau D. L. (1990) Ferryl and hydroxy intermediates in the reaction of oxygen with reduced cytochrome c oxidase. Nature 348, 89–90 [DOI] [PubMed] [Google Scholar]
- 17. Han S. H., Ching Y.-C., Rousseau D. L. (1990) Cytochrome c oxidase: decay of the primary oxygen intermediate involves direct electron transfer from cytochrome a. Proc. Natl. Acad. Sci. U.S.A. 87, 8408–8412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Varotsis C., Zhang Y., Appelman E. H., Babcock G. T. (1993) Resolution of the reaction sequence during the reduction of O2 by cytochrome oxidase. Proc. Natl. Acad. Sci. U.S.A. 90, 237–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ogura T., Takahashi S., Hirota S., Shinzawa-Itoh K., Yoshikawa S., Appleman E. H., Kitagawa T. (1993) Time-resolved resonance Raman elucidation of the pathway for dioxygen reduction by cytochrome c oxidase. J. Am. Chem. Soc. 115, 8527–8536 [Google Scholar]
- 20. Proshlyakov D. A., Ogura T., Shinzawa-Itoh K., Yoshikawa S., Appelman E. H., Kitagawa T. (1994) Selective resonance Raman observation of the “607 nm” form generated in the reaction of oxidized cytochrome c oxidase with hydrogen peroxide. J. Biol. Chem. 269, 29385–29388 [PubMed] [Google Scholar]
- 21. Varotsis C., Babcock G. T. (1995) Photolytic activity of early intermediates in dioxygen activation and reduction by cytochrome oxidase. J. Am. Chem. Soc. 117, 11260–11269 [Google Scholar]
- 22. Ogura T., Hirota S., Proshlyakov D. A., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (1996) Time-resolved resonance raman evidence for tight coupling between electron transfer and proton pumping of cytochrome c oxidase upon the change from the FeV oxidation level to the FeIV oxidation level. J. Am. Chem. Soc. 118, 5443–5449 [Google Scholar]
- 23. Proshlyakov D. A., Ogura T., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (1996) Microcirculating system for simultaneous determination of Raman and absorption spectra of enzymatic reaction intermediates and its application to the reaction of cytochrome c oxidase with hydrogen peroxide. Biochemistry 35, 76–82 [DOI] [PubMed] [Google Scholar]
- 24. Proshlyakov D. A., Pressler M. A., Babcock G. T. (1998) Dioxygen activation and bond cleavage by mixed-valence cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 95, 8020–8025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Han S., Takahashi S., Rousseau D. L. (2000) Time dependence of the catalytic intermediates in cytochrome c oxidase. J. Biol. Chem. 275, 1910–1919 [DOI] [PubMed] [Google Scholar]
- 26. Kim Y., Shinzawa-Itoh K., Yoshikawa S., Kitagawa T. (2001) Presence of the heme-oxo intermediate in oxygenation of carbon monoxide by cytochrome c oxidase revealed by resonance Raman spectroscopy. J. Am. Chem. Soc. 123, 757–758 [DOI] [PubMed] [Google Scholar]
- 27. Pinakoulaki E., Pfitzner U., Ludwig B., Varotsis C. (2002) The role of the cross-link His-Tyr in the functional properties of the binuclear center in cytochrome c Oxidase. J. Biol. Chem. 277, 13563–13568 [DOI] [PubMed] [Google Scholar]
- 28. Pinakoulaki E., Pfitzner U., Ludwig B., Varotsis C. (2003) Direct detection of Fe(IV)=O intermediates in the cytochrome aa3 oxidase from Paracoccus denitrificans/H2O2 reaction. J. Biol. Chem. 278, 18761–18766 [DOI] [PubMed] [Google Scholar]
- 29. Proshlyakov D. A., Pressler M. A., DeMaso C., Leykam J. F., DeWitt D. L., Babcock G. T. (2000) Oxygen activation and reduction in respiration: Involvement of redox-active tyrosine 244. Science 290, 1588–1591 [DOI] [PubMed] [Google Scholar]
- 30. Wiertz F. G. M., Richter O.-M. H., Cherepanov A. V., MacMillan F., Ludwig B., de Vries S. (2004) An oxo-ferryl tryptophan radical catalytic intermediate in cytochrome c and quinol oxidases trapped by microsecond freeze-hyperquenching (MHQ). FEBS Lett. 575, 127–130 [DOI] [PubMed] [Google Scholar]
- 31. Budiman K., Kannt A., Lyubenova S., Richter O.-M. H., Ludwig B., Michel H., MacMillan F. (2004) Tyrosine 167: The origin of the radical species observed in the reaction of cytochrome c oxidase with hydrogen peroxide in Paracoccus denitrificans. Biochemistry 43, 11709–11716 [DOI] [PubMed] [Google Scholar]
- 32. TURBOMOLE GMbH (2005) TURBOMOLE, Version 5-8-0, TURBOMOLE GMbH, Karlsruhe, Germany [Google Scholar]
- 33. Becke A. D. (1988) A multicenter numerical integration scheme for polyatomic molecules. J. Chem. Phys. 88, 2547–2553 [Google Scholar]
- 34. Lee C., Yang W., Parr R. G. (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 37, 785–789 [DOI] [PubMed] [Google Scholar]
- 35. Blomberg M. R. A., Siegbahn P. E. M., Wikström M. (2003) Metal-bridging mechanism for O–O bond cleavage in cytochrome c oxidase. Inorg. Chem. 42, 5231–5243 [DOI] [PubMed] [Google Scholar]
- 36. Dey A., Ghosh A. (2002) “True” iron(V) and iron(VI) porphyrins: A first theoretical exploration. J. Am. Chem. Soc. 124, 3206–3207 [DOI] [PubMed] [Google Scholar]
- 37. Daskalakis V., Farantos S. C., Varotsis C. (2008) Assigning vibrational spectra of ferryl-oxo intermediates of cytochrome c oxidase by periodic orbits and Molecular Dynamics. J. Am. Chem. Soc 130, 12385–12393 [DOI] [PubMed] [Google Scholar]
- 38. Daskalakis V., Farantos S. C., Guallar V., Varotsis C. (2010) Vibrational resonances and CuB displacement controlled by proton motion in cytochrome c oxidase. J. Phys. Chem. B 114, 1136–1143 [DOI] [PubMed] [Google Scholar]
- 39. Takahashi T., Kuroiwa S., Ogura T., Yoshikawa S. (2005) Probing the oxygen activation reaction in intact whole mitochondria through analysis of molecular vibrations. J. Am. Chem. Soc. 127, 9970–9971 [DOI] [PubMed] [Google Scholar]