


Abstract
RAD51 is the central strand exchange recombinase in somatic homologous recombination, providing genomic stability and promoting resistance to DNA damage. An important tool for mechanistic studies of RAD51 is the D-loop or strand assimilation assay, which measures the ability of RAD51-coated single-stranded DNA (ssDNA) to search for, invade and exchange ssDNA strands with a homologous duplex DNA target. As cancer cells generally overexpress RAD51, the D-loop assay has also emerged as an important tool in oncologic drug design programs for targeting RAD51. Previous studies have adapted the traditional gel-based D-loop assay by using fluorescence-based substrates, which in principle allow for use in high-throughput screening platforms. However, these existing D-loop methods depend on linear oligonucleotide DNA duplex targets, and these substrates enable recombinase-independent ssDNA annealing that can obscure the recombinase-dependent strand assimilation signal. This compelled us to fundamentally re-design this assay, using a fluorescent target substrate that consists of a covalently closed linear double-hairpin dsDNA. This new microplate-based method represents a fast, inexpensive and non-radioactive alternative to existing D-loop assays. It provides accurate kinetic analysis of strand assimilation in high-throughput and performs well with human RAD51 and Escherichia coli RecA protein. This advance will aid in both mechanistic studies of homologous recombination and drug screening programs.
INTRODUCTION
Homologous recombination (HR) is an essential process in eukaryotic cells that provides repair for chromosomal damage, including DNA double-strand breaks, replication-blocking DNA lesions and collapsed replication forks. HR is generally restricted to the S and G2 phases of the cell cycle, during which the undamaged sister chromatid provides a template for faithful repair of the damaged chromosome (1). The proteins that carry out HR are evolutionarily conserved, with RAD51 being the central strand exchange protein during mitotic HR in eukaryotic cells (2). In mitotic double-strand break repair by HR, DNA ends are resected to produce 3′ single-stranded DNA (ssDNA) tails, onto which RAD51 assembles to form nucleoprotein filaments (3). These nucleoprotein filaments conduct a search for regions of homology within duplex DNA and subsequently invade and exchange ssDNA strands with homologous sequences in the target duplex. This process generates a base-paired heteroduplex, consisting of the 3′ ssDNA tail and the complementary strand of the duplex, together with a displaced loop of ssDNA originating from the target dsDNA. This configuration is referred to as a D-loop.
This RAD51-mediated strand invasion represents a critical step in HR, and mechanistic studies of HR have depended heavily on biochemical assays that measure strand assimilation and strand exchange (4,5). In the modern version of the D-loop assay, a recombinase protein is allowed to assemble on a radiolabeled ssDNA oligonucleotide, and the resulting nucleoprotein filament is subsequently incubated with target dsDNA consisting of a supercoiled circular plasmid (Figure 1A). The reaction product is then deproteinated and separated by gel electrophoresis. D-loop-containing molecules migrate more slowly than the ssDNA substrate and can, therefore, be detected and quantified (6).
Figure 1.
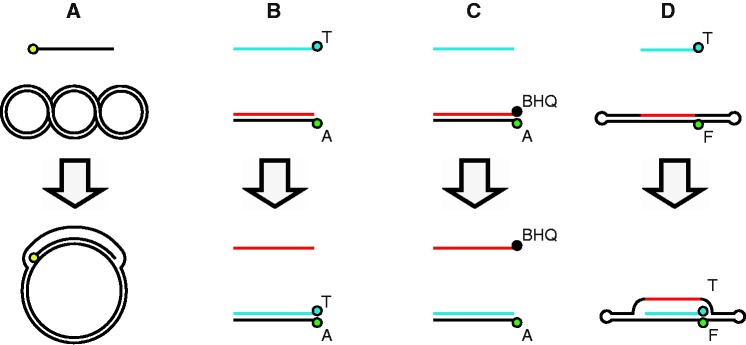
Methods of detecting homologous strand assimilation. The following abbreviations are used throughout: A = Alexa Fluor 488, F = fluorescein, BHQ = black hole quencher 1 and T = terbium. (A) The standard gel-based D-loop assay detects assimilation of a 5′-radiolabeled ssDNA into a supercoiled plasmid duplex. The radiolabel is depicted as a yellow circle. (B) T-labeled ssDNA is incubated with an A-labeled linear duplex. Pairing results in elevated TR-FRET. (C) An unlabeled ssDNA is incubated with a duplex, which consists of an A-labeled strand annealed to BHQ-labeled strand. The fluorescence intensity is quenched while the duplex remains paired, and fluorescence increases on dissociation of the starting duplex. (D) T-labeled ssDNA is incubated with a double-hairpin duplex, which contains an internal F label. Pairing results in elevated TR-FRET.
HR has emerged as an important target in oncology drug development, and many RAD51-modulating compounds have been developed to modulate RAD51 activities (7–14). Although the gel-based D-loop assay remains an important method in such studies, it allows for only low-throughput experiments, making it impractical for screening large collections of compounds. Higher-throughput assays of strand assimilation have been developed using fluorescently labeled DNA substrates (15,16), and one of these has been adapted for high-throughput drug screening (13). These fluorescent substrates generally make use of linear duplexed oligos serving as the target, which are incubated with a RAD51-coated ssDNA oligo (Figure 1B and C). A central challenge with this strategy is the tendency for linear duplex oligonucleotides to ‘breathe’, which in turn allows RAD51-independent strand annealing to occur with the ssDNA oligo. Thus, these assays have an inherent background that mimics the result of strand exchange in control reactions in which no protein is added. This technical challenge led us to re-design the assay. We constructed a linear covalently closed double-hairpin dsDNA to reduce the ability of the duplex to spontaneously unwind and allow annealing (Figure 1D). This method offers a non-radioactive alternative to D-loop assays that does not generate background signal in the absence of strand exchange protein. It enables accurate measurements of strand assimilation on a high-throughput scale, and it functions well with two key strand exchange proteins human RAD51 and bacterial RecA.
MATERIALS AND METHODS
Proteins
RAD51 protein was purified as previously described (7). RecA protein was purchased from New England Biolabs.
DNA oligonucleotides
Single-stranded DNA oligonucleotides (listed in Supplementary Table S1) were purchased from IDT or Eurofins MWG Operon and purified on denaturing urea–polyacrylamide gels. The linear duplexes RG1(±)A and RG1(±)AQ were generated by thermal annealing of RG1(+)A and RG1(−) or RG1(+)A and RG1(−)Q, respectively, in annealing buffer (10 mM Tris–HCl, pH 7.5, and 40 mM NaCl) by heating to 95°C for 2 min and cooling to 25°C at −1°C per min.
Construction of closed double-hairpin duplex DNA substrate
The 162-bp linear double-hairpin duplex, called DHD162, was constructed from four component ssDNA oligonucleotides (Figure 2A): DHD162-HP-L, DHD162-HP-R, DHD162-CD-O and DHD162-CD-CF. The two hairpin-forming oligos (DHD162-HP-L and DHD162-HP-R) and the oligos forming the central duplex (DHD162-CD-O and DHD162-CD-CF) were annealed separately in annealing buffer as described earlier in the text, and the 5′-ends were phosphorylated for 30 min at 37°C by T4 polynucleotide kinase (New England Biolabs) using 10 U of kinase per 300 pmol 5′-ends. The annealed and phosphorylated duplexes were then combined into a single tube and ligated with T4 DNA ligase (New England Biolabs) overnight at 16°C using 25 U of ligase per 30 pmol ligatable ends. The final product was purified on a denaturing urea–polyacrylamide gel and re-annealed. The molar yield of DHD162 relative to the component oligonucleotides was typically 30–40%.
Figure 2.
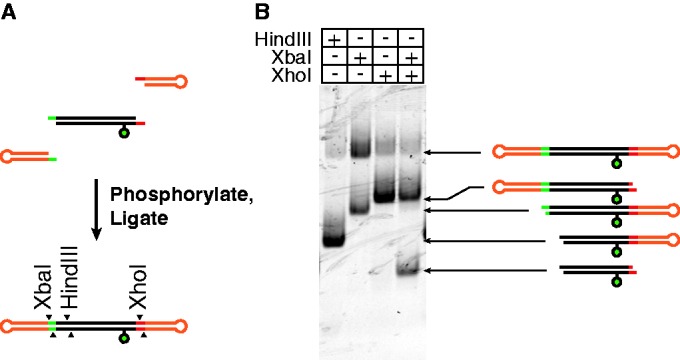
Construction and verification of the double-hairpin duplex. (A) The two single-hairpin forming duplexes and a single internal duplex were separately annealed and phosphorylated, then combined and ligated to generate the double-hairpin duplex as described in detail in the ‘Materials and Methods’ section. The fluorescein label is depicted as a green circle. (B) The double-hairpin duplex was verified by restriction digestion, separation on a native polyacrylamide gel and visualization of fluorescein-labeled fragments on a phosphorimager.
Gel-based D-loop assay
D-loop assays for strand assimilation were performed essentially as described previously (7). Briefly, 0.4 µM RAD51 and 13.3 nM (1.2 µM nucleotide concentration) 32P-end-labeled ssDNA was combined in 9 µl of reaction buffer containing 25 mM HEPES–NaOH (pH 7.5), 3 mM adenosine triphosphate (ATP), 5 mM CaCl2 and 1 mM TCEP [tris(2-carboxyethyl)phosphine] and incubated at 37°C for 5 min. Then 1 µl of supercoiled pRS306 was added to a final concentration of 12.1 nM and incubation at 37°C was continued for 40 min. The reaction was stopped by addition of sodium dodecyl sulfate to 1% and proteinase K to 1 mg/ml. Deproteinated reaction products were run on a 0.9% agarose 1× TAE gel and visualized by phosphorimaging.
Fluorescence-based D-loop assays
To detect homologous strand assimilation by human RAD51 using a fluorophore and quenching dye combination, 11.1 nM (oligonucleotide concentration) of ssDNA was combined with sufficient protein for one protomer per 3-nt ssDNA in 40 µl of reaction buffer and incubated at 37°C for 5 min. This corresponds to 0.3 µM RAD51 in methods B and C (Figure 1), and 0.2 µM RAD51 in method D (Figure 1). Then dsDNA was added in 5 µl at an equimolar ratio to the ssDNA, and incubation was continued at 37°C; the final reaction buffer component concentrations were 25 mM HEPES–NaOH (pH 7.5), 3 mM ATP, 5 mM CaCl2, 1 mM TCEP and 1.5 µM bovine serum albumin. Fluorescence measurements were taken at 37°C on a Tecan Infinite F200 Pro using a 480 (±20)-nm excitation filter and a 535 (±30)-nm emission filter; the gain was set manually to 77. Time-resolved fluorescence resonance energy transfer (TR-FRET)-based assays were set-up in the same way, except 3′-biotinylated ssDNA was used to form pre-synaptic filaments, and after the final incubation at 37°C, an amount terbium–streptavidin chelate (Invitrogen) equimolar to the biotinylated ssDNA was added to the reaction in 5 µl and incubated at room temperature for 5 min. TR-FRET was measured at 30°C using 340 (±30)-nm excitation filters and 495 (±10)- or 520 (±25)-nm emission filters to measure emission from terbium and fluorescein/Alexa Fluor 488, respectively, with a 100-µs delay between the excitation flash and emission reading and the gain set manually to 200 for 495- and 520-nm readings. Homologous strand assimilation by Escherichia coli RecA was assayed using optimal conditions for generating stable joint molecules (17), by combining 0.4 µM RecA, 1.2 µM (nucleotide concentration) DHD-HQ or DHD-NQ ssDNA and 23.5 nM (equimolar to the ssDNA) DHD162 in 50 µl of reaction buffer containing 70 mM Tris–HCl, pH 7.6, 1.1 mM adenosine diphosphate (ADP), 0.3 mM adenosine 5′-[γ-thio]triphosphate (ATPγS), 10 mM MgCl2, 5 mM dithiothreitol (DTT) and 1.5 µM bovine serum albumin. Fluorescence readings were taken at 37°C using the 480 (±20)-nm excitation filter and a 535 (±30)-nm emission filter as described earlier in the text. For comparing different assays, measurements were taken at 40 min, a time at which optimal reactions reached ∼80% of final levels observed in more extensive time course experiments.
RESULTS
We sought to develop a high-throughput method for measuring RAD51-mediated strand invasion that would not be susceptible to RAD51-independent strand annealing, as this artifact can obscure the biologically relevant HR signal. This recombinase-independent annealing can occur in assays that depend on a linear oligonucleotide duplex substrate. Therefore, we developed a novel method, based on the prediction that adding a DNA hairpin to each end of the dsDNA target would overcome this problem. In parallel, we also re-constructed some previously published D-loop methods for comparison. Figure 1 displays schematic representations of different assays we evaluated, with method A depicting the standard gel-based method. In the fluorescence-based methods, D-loop formation is quantified by one of two general detection strategies. In methods B and D, the pairing of a fluorescently labeled DNA strand and a terbium-labeled DNA strand generates an increase in TR-FRET. Alternatively, in method C, an unlabeled ssDNA is incubated with a duplex, consisting of a fluorescently labeled DNA strand annealed to a black hole quencher 1 (BHQ1)-labeled DNA strand. The BHQ1 quenches the fluorescence intensity when the duplex remains paired, and the fluorescence increases on dissociation of the quencher and the fluorophore. These methods also differ based on the category of product that is measured in each. Methods A, B and D all directly measure the pairing of two DNA strands that results from homology recognition and strand exchange. Method C quantifies the dissociation of the two strands of the target duplex that occurs as a consequence of strand exchange and thus represents a less direct measure of strand assimilation.
A fluorescein-labeled double-hairpin dsDNA substrate was constructed as shown in Figure 2A. The proper assembly of this target duplex was verified by resistance to digestion by λ exonuclease (data not shown) and by restriction endonuclease digestion that revealed the expected cleavage fragments Figure 2B. Of note, certain restriction digestion reactions (particularly those with XbaI) were incomplete; the reason for this is unclear, but it may result from a conformational problem created by the hairpin structure. This double-hairpin dsDNA substrate was used as outlined in Figure 1 (method D), and it successfully detected homology-dependent D-loop signal exclusively when RAD51 was present (Figure 3D). The specificity of this signal was excellent, with 0.2 µM RAD51 generating a maximum signal-to-noise ratio of 28:1 relative to the control lacking RAD51. Similarly, RAD51 was required for homology-dependent D-loop signal in the standard gel-based D-loop assay (Figure 3A), which uses a supercoiled closed-circular duplex.
Figure 3.

The double-hairpin assay is not subject to RAD51-independent strand annealing. (A–D) These correspond to assay designs depicted in Figure 1. H and N denote homologous and non-homologous (scrambled) versions of ssDNA strands. In all cases, RAD51 nucleoprotein filaments were assembled on ssDNA, introduced to target duplex DNA and allowed to form D-loops at 37°C for 40 min. For B–D, TR-FRET and fluorescence intensities are in arbitrary units, and error bars represent the standard error of four replicates. A = Alexa Fluor 488, F = fluorescein, BHQ = black hole quencher 1 and T = terbium. (A) In the standard gel-based D-loop assay, 32P-labeled ssDNA consisted of 306.90, and the duplex target consisted of the supercoiled plasmid pRS306. Reaction mixture was deproteinated and separated by electrophoresis. (B) ssDNA consisted of T-labeled RG1(−)B and duplex target RG1(±)A consisted of an unlabeled RG1(−) strand annealed to a A-labeled RG1(+)A. Pairing was detected by TR-FRET. (C) ssDNA consisted of an unlabeled RG1(−), and the duplex target RG1(±)AQ consisted of A-labeled RG1(+)A strand annealed to BHQ-labeled RG1(−)Q strand. Dissociation of the duplex was measured based on an increase in fluorescence intensity. (D) ssDNA consisted of a T-labeled DHD-HB, and the duplex target consisted of the double-hairpin construct DHD162, which contains an internal F label. Pairing was detected by TR-FRET.
The performance of this novel double-hairpin substrate was directly compared with the two fluorescent D-loop methods (methods B and C) that use linear DNA duplex targets. Consistent with previous reports (13,15,16), methods B and C could successfully detect homology-dependent signal when RAD51 was included in the reaction. However, methods B and C also generated abundant RAD51-independent signal in the no-recombinase control, and this false-positive signal met or exceeded the true-positive signal observed in RAD51-containing conditions.
The double-hairpin–based D-loop method was modified slightly, such that the terbium-labeled ssDNA was replaced with BHQ1-labeled ssDNA (Figure 4). In this version of the assay, a positive D-loop signal is detected based on quenching of fluorescence intensity. As with the TR-FRET–based set-up, this method successfully detected homology-dependent D-loops exclusively when RAD51 was present. This quenching version of the assay has several advantages over the TR-FRET–based version. First, it allows for dynamic measurements of reaction kinetics. This is not possible with the TR-FRET version because terbium labeling is performed at the end of the reaction, by adding terbium–streptavidin to biotin-labeled DNA immediately before readings are collected by the plate reader. A second advantage is that the quenching version directly quantifies the fraction of substrate that has been converted to D-loop. For example, ∼25% of incoming ssDNA was paired with the target duplex after 60 min (Figure 4B), relative to the signal from the RAD51-free control. This activity is comparable with that observed in the gel-based assay with similar protein and buffer conditions (Figure 3A).
Figure 4.
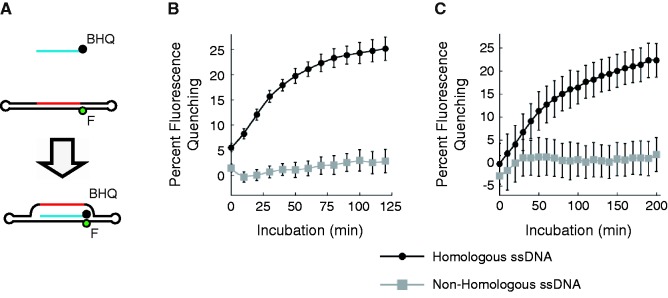
A fluorescence quenching-based double-hairpin D-loop assay. (A) D-loops are detected as a decrease in fluorescence intensity on pairing of the BHQ-labeled DHD-HQ ssDNA with the complementary strand of the double-hairpin duplex DHD162, which contains an internal fluorescein (F) label. DHD-NQ is used as a non-homologous (scrambled) ssDNA control. (B and C) Kinetics of D-loop formation by human RAD51 or E. coli RecA are displayed as the per cent decrease in fluorescence, relative to the protein-free control. Error bars represent the standard error of four replicates.
The quenching version of this double-hairpin–based D-loop assay allowed us to demonstrate that human RAD51 protein continues forming D-loops well beyond the typical 30-min incubations in standard assays, reaching 25% depletion of substrate fluorescence after 2 h at 37°C (Figure 4B). The assay also performs well with the E. coli RecA protein as demonstrated in Figure 4C. The accumulation of stable joint molecules in the RecA-mediated reaction is in agreement with a previous report, showing that a cofactor set of ADP and ATPγS prevents the dissociation of these joint molecules (17).
The shape of these kinetic curves (Figures 4B and C) suggests a slowing of reactions after ∼1 h, which is similar to published reports using other fluorescent D-loop methods (13,15,16). We examined whether this represented either (i) depletion of active substrate or (ii) equilibrium between D-loop product formation and dissolution. D-loop dissolution has been previously observed at late time points by Shibata et al. (18) using E. coli RecA and ATP in filter-based D-loop assays. In assays with our double-hairpin target substrate, however, we found that RecA in ATP generated the expected time-dependent D-loop formation but no measurable D-loop dissolution (Supplementary Figure S1). To address this question further using human RAD51, an equimolar amount of photobleached DHD162 was added to an ongoing D-loop reaction at the 2 h time point. This addition of photobleached target substrate prevented any further accumulation of D-loop signal (Supplementary Figure S2), consistent with the photobleached DHD162 competing with the fluorescent DHD162 target. However, the addition of photobleached target did not reduce D-loop signal over time, as would have been expected if the reaction was nearing equilibrium of D-loop formation and dissolution. This suggests that recombinase-mediated D-loops formed with DHD162 are stable over the time assayed. This stability of D-loop product may depend on the presence of protein; there is no deproteination step in our method, in contrast to the standard gel method that requires deproteination.
DISCUSSION
We developed a microplate-based assay to measure the strand assimilation intermediate (D-loop) of HR. This method represents a fast, inexpensive and non-radioactive alternative to standard gel-based D-loop assays. It also enables kinetic analysis of D-loop formation, and it is ideal for high-throughput platforms. Most importantly, this assay solves the problem of recombinase-independent strand annealing, which is a problem when using previously described fluorescence-based assays that depend on linear duplex targets. This method seems suitable for use with other strand exchange proteins, which will enable mechanistic studies of HR in many model systems.
Despite the limitation of linear duplex DNA substrates, the older fluorescent D-loop methods have functioned well enough to enable some mechanistic studies of HR and a productive chemical screen (12,13,15,16). This is because when strand exchange proteins bind to ssDNA, they block its ability to participate in spontaneous annealing. However, the challenges posed by recombinase-independent annealing prompted us to re-design the assay. As the melting temperatures of these linear duplexes were well above our reaction temperature, the spontaneous annealing reaction likely proceeds as follows: (i) partial melting of the initial duplex, (ii) formation of a branched nucleation complex and (iii) isothermal branch migration leading to complete displacement (19). Alternatively, the recombinase-free annealing we observed might reflect pairing of DNA ends only. This possibility is based on the terminal location of the fluorescent dyes on linear DNA substrates, combined with the ‘breathing’ that is known to occur preferentially at DNA ends (20,21). Additionally, we considered possibility that this recombinase-free annealing was an artifact caused by dsDNA being partially denatured during preparation of this substrate. However, this possibility seems unlikely, as (i) polyacrylamide gel electrophoresis analysis of the dsDNA substrates showed no remaining single-stranded oligos, and as (ii) this recombinase-free annealing signal occurred to the same extent when experiments were repeated using polyacrylamide gel electrophoresis-purified dsDNA substrate (data not shown).
It is important to reiterate that fluorescence-based D-loop assays differ from the standard gel-based D-loop method in that recombinase proteins are not removed from DNA products before quantification. In fact, the D-loops formed in our assay are not stable enough to tolerate deproteination and electrophoresis (data not shown). This instability of reaction products is reminiscent of earlier findings with gel-based D-loops showing that negative supercoiling of the target duplex is essential for D-loops to tolerate deproteination and electrophoresis (22).
This double-hairpin–based D-loop assay represents an important advance for mechanistic studies of HR intermediates. In particular, it provides an important tool for research programs that are conducting HR-related drug screening. Several groups are working to develop compounds that block RAD51’s D-loop activity, as a means to generate anti-cancer drugs. As discussed earlier, fluorescent substrates that use linear duplex substrates are susceptible to abundant RAD51-independent annealing. Based on this observation, a drug screen that uses linear duplex-based targets might mistakenly miss the most active RAD51 inhibitors, as such compounds are expected to generate false-negative signals. Our new double-hairpin–based D-loop method is expected to entirely overcome this technical hurdle.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1 and Supplementary Figures 1 and 2.
FUNDING
National Institutes of Health [CA142642-02 2010-2015 to P.P.C., 2T32CA009594 to B.B. and GM50936 to D.K.B.]. Funding for open access charge: [CA142642-02 2010-2015].
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Symington LS, Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 2.Thompson LH, Schild D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001;477:131–153. doi: 10.1016/s0027-5107(01)00115-4. [DOI] [PubMed] [Google Scholar]
- 3.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 4.Cox MM, Lehman IR. recA protein of Escherichia coli promotes branch migration, a kinetically distinct phase of DNA strand exchange. Proc. Natl Acad. Sci. USA. 1981;78:3433–3437. doi: 10.1073/pnas.78.6.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shibata T, Cunningham RP, DasGupta C, Radding CM. Homologous pairing in genetic recombination: complexes of recA protein and DNA. Proc. Natl Acad. Sci. USA. 1979;76:5100–5104. doi: 10.1073/pnas.76.10.5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konforti BB, Davis RW. 3′ homologous free ends are required for stable joint molecule formation by the RecA and single-stranded binding proteins of Escherichia coli. Proc. Natl Acad. Sci. USA. 1987;84:690–694. doi: 10.1073/pnas.84.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jayathilaka K, Sheridan SD, Bold TD, Bochenska K, Logan HL, Weichselbaum RR, Bishop DK, Connell PP. A chemical compound that stimulates the human homologous recombination protein RAD51. Proc. Natl Acad. Sci. USA. 2008;105:15848–15853. doi: 10.1073/pnas.0808046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Budke B, Logan HL, Kalin JH, Zelivianskaia AS, Cameron McGuire W, Miller LL, Stark JM, Kozikowski AP, Bishop DK, Connell PP. RI-1: a chemical inhibitor of RAD51 that disrupts homologous recombination in human cells. Nucleic Acids Res. 2012;40:7347–7357. doi: 10.1093/nar/gks353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Budke B, Kalin JH, Pawlowski M, Zelivianskaia AS, Wu M, Kozikowski AP, Connell PP. An optimized RAD51 inhibitor that disrupts homologous recombination without requiring Michael acceptor reactivity. J. Med. Chem. 2013;56:254–263. doi: 10.1021/jm301565b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ishida T, Takizawa Y, Kainuma T, Inoue J, Mikawa T, Shibata T, Suzuki H, Tashiro S, Kurumizaka H. DIDS, a chemical compound that inhibits RAD51-mediated homologous pairing and strand exchange. Nucleic Acids Res. 2009;37:3367–3376. doi: 10.1093/nar/gkp200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takaku M, Kainuma T, Ishida-Takaku T, Ishigami S, Suzuki H, Tashiro S, van Soest RW, Nakao Y, Kurumizaka H. Halenaquinone, a chemical compound that specifically inhibits the secondary DNA binding of RAD51. Genes Cells. 2011;16:427–436. doi: 10.1111/j.1365-2443.2011.01494.x. [DOI] [PubMed] [Google Scholar]
- 12.Huang F, Mazina OM, Zentner IJ, Cocklin S, Mazin AV. Inhibition of homologous recombination in human cells by targeting RAD51 recombinase. J. Med. Chem. 2012;55:3011–3020. doi: 10.1021/jm201173g. [DOI] [PubMed] [Google Scholar]
- 13.Huang F, Motlekar NA, Burgwin CM, Napper AD, Diamond SL, Mazin AV. Identification of specific inhibitors of human RAD51 recombinase using high-throughput screening. ACS Chem. Biol. 2011;6:628–635. doi: 10.1021/cb100428c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu J, Zhou L, Wu G, Konig H, Lin X, Li G, Qiu XL, Chen CF, Hu CM, Goldblatt E, et al. A novel small molecule RAD51 inactivator overcomes imatinib-resistance in chronic myeloid leukaemia. EMBO Mol. Med. 2013;5:353–365. doi: 10.1002/emmm.201201760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta RC, Golub EI, Wold MS, Radding CM. Polarity of DNA strand exchange promoted by recombination proteins of the RecA family. Proc. Natl Acad. Sci. USA. 1998;95:9843–9848. doi: 10.1073/pnas.95.17.9843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gupta RC, Bazemore LR, Golub EI, Radding CM. Activities of human recombination protein Rad51. Proc. Natl Acad. Sci. USA. 1997;94:463–468. doi: 10.1073/pnas.94.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsieh P, Camerini-Otero CS, Camerini-Otero RD. The synapsis event in the homologous pairing of DNAs: RecA recognizes and pairs less than one helical repeat of DNA. Proc. Natl Acad. Sci. USA. 1992;89:6492–6496. doi: 10.1073/pnas.89.14.6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shibata T, Ohtani T, Iwabuchi M, Ando T. D-loop cycle. A circular reaction sequence which comprises formation and dissociation of D-loops and inactivation and reactivation of superhelical closed circular DNA promoted by recA protein of Escherichia coli. J. Biol. Chem. 1982;257:13981–13986. [PubMed] [Google Scholar]
- 19.Reynaldo LP, Vologodskii AV, Neri BP, Lyamichev VI. The kinetics of oligonucleotide replacements. J. Mol. Biol. 2000;297:511–520. doi: 10.1006/jmbi.2000.3573. [DOI] [PubMed] [Google Scholar]
- 20.Andreatta D, Sen S, Perez Lustres JL, Kovalenko SA, Ernsting NP, Murphy CJ, Coleman RS, Berg MA. Ultrafast dynamics in DNA: "fraying" at the end of the helix. J. Am. Chem. Soc. 2006;128:6885–6892. doi: 10.1021/ja0582105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jose D, Datta K, Johnson NP, von Hippel PH. Spectroscopic studies of position-specific DNA “breathing” fluctuations at replication forks and primer-template junctions. Proc. Natl Acad. Sci. USA. 2009;106:4231–4236. doi: 10.1073/pnas.0900803106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konforti BB, Davis RW. DNA substrate requirements for stable joint molecule formation by the RecA and single-stranded DNA-binding proteins of Escherichia coli. J. Biol. Chem. 1991;266:10112–10121. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.