

Abstract
Mitochondria have been proposed as targets for toxicity in amyotrophic lateral sclerosis (ALS), a progressive, fatal adult-onset neurodegenerative disorder characterized by the selective loss of motor neurons. A decrease in the capacity of spinal cord mitochondria to buffer calcium (Ca2+) has been observed in mice expressing ALS-linked mutants of SOD1 that develop motor neuron disease with many of the key pathological hallmarks seen in ALS patients. In mice expressing three different ALS-causing SOD1 mutants, we now test the contribution of the loss of mitochondrial Ca2+-buffering capacity to disease mechanism(s) by eliminating ubiquitous expression of cyclophilin D, a critical regulator of Ca2+-mediated opening of the mitochondrial permeability transition pore that determines mitochondrial Ca2+ content. A chronic increase in mitochondrial buffering of Ca2+ in the absence of cyclophilin D was maintained throughout disease course and was associated with improved mitochondrial ATP synthesis, reduced mitochondrial swelling, and retention of normal morphology. This was accompanied by an attenuation of glial activation, reduction in levels of misfolded SOD1 aggregates in the spinal cord, and a significant suppression of motor neuron death throughout disease. Despite this, muscle denervation, motor axon degeneration, and disease progression and survival were unaffected, thereby eliminating mutant SOD1-mediated loss of mitochondrial Ca2+ buffering capacity, altered mitochondrial morphology, motor neuron death, and misfolded SOD1 aggregates, as primary contributors to disease mechanism for fatal paralysis in these models of familial ALS.
Introduction
Amyotrophic lateral sclerosis (ALS) is an adult-onset neurodegenerative disease characterized by the selective loss of motor neurons (Boillée et al., 2006a). Twenty percent of inherited ALS is caused by mutations in Cu/Zn superoxide dismutase (SOD1) (Rosen et al., 1993). At least nine mechanisms for mutant SOD1 toxicity have been proposed, including dysregulation of intracellular calcium homeostasis [especially from glutamate-mediated excitotoxicity (Rothstein et al., 1990, 1992)]; aggregation of misfolded mutant SOD1; and alterations in mitochondrial morphology, function, and distribution (Ilieva et al., 2009).
Ca2+-mediated excitotoxicity, following entry of Ca2+ through ionotropic glutamate receptors permeable to Ca2+, has been proposed as a critical component of ALS (Rothstein et al., 1990, 1992). Indeed, increased intracellular Ca2+ levels within motor neurons have been reported in patients (Siklós et al., 1996, 1998) and mouse models (von Lewinski et al., 2008; Jaiswal and Keller, 2009), as has decreased capacity of mitochondria to buffer Ca2+ (Damiano et al., 2006; Kawamata and Manfredi, 2010). Reducing cytosolic Ca2+ levels in motor neurons expressing mutant SOD1 in culture (Roy et al., 1998) and in mice has been reported to attenuate motor neuron death (Beers et al., 2001; Van Damme et al., 2003; Tateno et al., 2004; Van Den Bosch et al., 2006).
Mitochondria play a pivotal role in regulating Ca2+ levels (Nicholls, 2009). Indeed, a significant decrease in the Ca2+ loading capacity of mitochondria from spinal cords of mutant SOD1 transgenic mice has been reported to appear presymptomatically (Damiano et al., 2006). Mutant SOD1 is preferentially deposited on the surface of, or imported into, spinal cord mitochondria in mice that express ALS-linked mutants in SOD1 (Mattiazzi et al., 2002; Liu et al., 2004; Vijayvergiya et al., 2005; Bergemalm et al., 2006; Deng et al., 2006; Vande Velde et al., 2008), where it has been reported to interact with multiple components of the mitochondrial outer membrane and alters their activities (Israelson et al., 2010; Li et al., 2010; Pedrini et al., 2010).
It is well accepted that irreversible opening of the mitochondrial permeability transition pore (mPTP), a nonselective high conductance channel located in the inner mitochondrial membrane (Azzolin et al., 2010), leads to mitochondrial depolarization, decreased ATP synthesis, matrix swelling, and mitochondrial degeneration (Hunter and Haworth, 1979; Bernardi, 1999; Petronilli et al., 2001; Bernardi et al., 2006). Genetic ablation of the gene encoding cyclophilin D (CypD) (named Ppif in mice) has demonstrated that CypD is a key regulator of Ca2+-induced opening of the mPTP. Mitochondria isolated from CypD-null animals store significantly increased quantities of Ca2+ before mPTP opening (Baines et al., 2005; Basso et al., 2005; Nakagawa et al., 2005; Schinzel et al., 2005; Barsukova et al., 2011).
By eliminating CypD expression in each of the three most prominently used mouse models of familial ALS from expression of ALS-causing mutants of SOD1 of divergent biochemical properties, we have now tested whether rescuing the loss of mitochondrial Ca2+ buffering capacity throughout disease can alter ALS-like pathogenesis.
Materials and Methods
Animals.
All mouse lines were on a pure C57BI/6 background: cyclophilin D-null mice with ubiquitous deletion of the Ppif gene, which encodes the cyclophilin D protein (Basso et al., 2005) and ALS mice [SOD1G93A, SOD1G85R, and SOD1G37R (Gurney et al., 1994; Bruijn et al., 1997; Boillée et al., 2006b)]. All the ALS mice are heterozygous for a 12 kb genomic DNA fragment encoding the human mutant SOD1 transgene, under its endogenous promoter.
Survival analysis.
CypD-null mice (CypD−/−) were mated to heterozygous SOD1G37R, SOD1G85R, and SOD1G93A ALS mice and the resulting CypD+/−/mutant SOD1 mice were mated to CypD+/− mice to obtain the experimental cohorts of CypD−/−/mutant SOD1 animals that were compared with CypD+/+/mutant SOD1 littermates. Mice were weighed weekly as an objective and unbiased measure of disease course (Lobsiger et al., 2009). Time of disease onset was retrospectively determined as the time when mice reached peak body weight, which is observed before any motor performance decline as measured by grip strength or rotarod (Liu et al., 2005; Schütz et al., 2005; Matsumoto et al., 2006). The time of early disease was defined as the age at which the animals had lost 10% of their maximal weight (which is accompanied by gait alterations, failure of hindlimb splaying reflex, and loss of grip strength without obvious signs of paralysis). This early disease stage is followed by the appearance of progressive paralysis. During the symptomatic phase, mice were observed daily and end stage was defined by paralysis so severe that the animal could not right itself within 30 s when placed on its side, an endpoint frequently used for SOD1 mutant-expressing mice and one that was consistent with the requirements of the Animal Care and Use Committee of the University of California.
Grip strength.
Grip strength was measured weekly using a Grip Strength Meter (Columbus Instruments) on cohorts (n ≥ 12) made up of approximately the same number of males and females. Mice were allowed to grip a triangular bar only with hindlimbs, followed by pulling the mice until they released; five force measurements were recorded in each separate trial.
Immunofluorescence.
Mice were perfused intracardially with 4% paraformaldehyde in 0.1 m Sorenson's phosphate buffer, pH 7.2. The entire spinal cord was dissected, postfixed for 2 h in fixative, and transferred in a 30% sucrose phosphate buffer for at least 2 d. The lumbar spinal cord was embedded in OCT compound (Sakura) and snap frozen in isopentane cooled at −40°C on dry ice. Floating lumbar spinal cord cryosections (30 μm) were incubated in a blocking solution containing PBS, 0.5% Tween 20, 1.5% bovine serum albumin (BSA) for 1 h 30 min at room temperature and then in PBS, 0.3% Triton X-100 overnight at room temperature with the following primary antibodies: polyclonal rabbit antibodies against ChAT (1:300; Millipore Bioscience Research Reagents), GFAP (1:1000; Dako), and Iba1 (1:500) or monoclonal mouse antibodies against misfolded SOD1: DSE2 3H1 (1:5000; kindly provided by Neil Cashman; Vande Velde et al., 2008), B8H10 (1:1000) and C4F6 (1:500, MediMabs), and D3H5 (1:500, kindly provided by Jean-Pierre Julien; Gros-Louis et al., 2010). Primary antibodies were washed with PBS and then detected using donkey anti-rabbit or anti-mouse FITC or Cy3 (1:500)-coupled secondary antibodies (Jackson ImmunoResearch). The sections were washed with PBS and mounted. Analysis was performed on a Nikon Eclipse laser scanning confocal microscope. Fluorescence intensity from unsaturated images captured with identical confocal settings (minimum of four spinal cord sections were imaged per animal) was quantified using NIS elements software (Nikon).
Motor neuron counting.
ChAT-positive ventral horn motor neurons were counted from 25–35 lumbar spinal cord cryosections (per animal) spaced 360 μm apart and expressed as the average of total motor neurons counted divided by the number of sections.
Neuromuscular junction denervation
Gastrocnemius muscle was dissected from perfused mice and prepared as described (see above, Immunofluorescence). Floating 40-μm-thick longitudinal sections of gastrocnemius were incubated in a blocking solution containing PBS, 0.5% Tween 20, 1.5% BSA for 4 h at room temperature and then in PBS, 0.3% Triton X-100 overnight at room temperature with the polyclonal rabbit anti-synaptophysin antibody at 1:50 (Invitrogen). The sections were washed with PBS and then incubated first with donkey anti-rabbit Cy3 (Jackson ImmunoResearch) and α-bungarotoxin–Alexa 488 (Invitrogen) at 1:500 for 1 h at room temperature and then with fluoromyelin red (Invitrogen) at 1:300 for 30 min. The sections were further washed with PBS and mounted. Analysis was performed on a Nikon Eclipse laser scanning confocal microscope. A total of ∼1000 neuromuscular junctions (NMJs) were counted from at least 10 sections of gastrocnemius. Individual NMJs were considered as innervated when synaptophysin staining covered at least 50% of the area of α-bungarotoxin staining.
Morphometric analysis of axons.
Mice were perfused intracardially with 4% paraformaldehyde in 0.1 m Sorenson's phosphate buffer, pH 7.2, and the L5 lumbar roots were dissected and conserved in fixative at 4°C. L5 roots were embedded in Epon-Araldite as described in the electron microscopy section, thick sections (0.75 μm) were prepared and stained for light microscopy with toluidine blue. Cross sections of L5 motor axons were analyzed at each age group. Axonal diameters were measured using the Bioquant Software and the number of large caliber axons with diameters >3.5 μm (α motor axons) was determined.
Electron microscopy.
Mice were perfused intracardially with 0.1 m cacodylate buffer, pH 7.2–7.4, containing 200 mm sucrose at room temperature for 5 min followed by fixative solution containing 4% formaldehyde, and 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.2–7.4, supplemented with 200 mm sucrose at 4°C. Following perfusion, the spinal cord and the lumbar L5 motor root were dissected and placed in fixative until embedding. A 2 mm thick section was obtained from the lumbar spinal cord. The tissue was rinsed in 0.1 m sodium cacodylate buffer, osmicated with 2% osmium tetroxide in cacodylate buffer, dehydrated through ascending ethyl alcohol concentrations, rinsed with acetone and incubated overnight in 1:1 acetone/Epon-Araldite (Araldite 502 Resin, dodecenyl succinic anhydride, Eponate 12 resin, components purchased from Ernest F. Fullam). The sections of spinal cord and the motor roots were rotated for 2 h in 2:1 acetone/Epon-Araldite, 2 h in pure Epon-Araldite and transferred to silicon molds to harden 65°C for 48 h. Ultrathin cross-sections (70 nm) were cut from the ventral horn of the spinal cord and the L5 motor root and stained with 1% uranyl acetate, prepared in 50% ethanol, for 10 min, and then with Reynold's lead citrate for 5 min. In ventral horn ultrathin sections, the soma of motor neurons were identified by size and nuclear morphology. Images were taken of the somal mitochondria (5–20 mitochondria per motor neuron soma) from all the motor neurons present in the section (5–10 motor neurons were found per section of ventral horn). In L5 motor roots, larger caliber axons lacking obvious features of degeneration, when observed at low magnification, were selected and high-power images were taken of all the mitochondria within each axon (1–10 mitochondria were found per axon). The area of all the somal and axonal mitochondria in each image was determined using ImageJ software. 50 to 170 spinal cord motor neuron mitochondria were analyzed per animal and two animals were analyzed per condition. 150 to 300 L5 motor axon mitochondria were analyzed per animal and three animals were analyzed per condition.
Mitochondrial isolation and purification.
The spinal cord was dissected from disease-matched CypD−/−/mutant SOD1 and CypD+/+/mutant SOD1 mice and mitochondria were isolated and purified as previously described (Vande Velde et al., 2008). Spinal cords were homogenized on ice in five volumes of ice-cold homogenization buffer (HB) composed of 210 mm mannitol, 70 mm sucrose, 1 mm EDTA-(Tris), and 10 mm Tris-HCl, pH 7.2, supplemented with protease inhibitors (Roche) and 0.45% of free fatty acid BSA. Homogenates were centrifuged at 1000 × g for 5 min. Supernatants were recovered and centrifuged again at 1000 × g for 5 min. Supernatants were centrifuged at 12,000 × g for 10 min to yield crude mitochondrial pellets (heavy membrane fraction). Mitochondria-enriched pellets were gently resuspended in HB and then adjusted to 12% Optiprep (iodixanol) and centrifuged at 17,000 × g for 10 min (SW-55; Beckman). The majority of the myelin (at the top of the sample) was removed and the mitochondria were washed once with HB (without BSA and EDTA) to remove the Optiprep. The mitochondria-enriched pellets were gently resuspended in a minimal amount of HB buffer and protein concentration was measured by Bradford assay. Mitochondria from nontransgenic animals isolated with this method have a respiratory capacity that is in the expected range (average 270 pmol/min/μg protein using glutamate/malate/pyruvate as respiratory substrates) and are tightly coupled (respiratory control ratio average of 8.9).
Measurement of mitochondrial calcium buffering capacity.
Mitochondrial Ca2+ capacity was estimated with the Ca2+-sensitive Calcium Green 5N fluorescent dye (Invitrogen). Incubation medium was composed of 125 mm KCl, 20 mm HEPES, pH 7.2, 2 mm KH2PO4, 2 mm MgCl2, 5 mm succinate, 1 μm rotenone, and 0.2 mm ADP, with 1 μg/ml oligomycin and 1 μm Calcium Green 5N. Calcium Green 5N does not enter into mitochondria, and its fluorescence reflects the extramitochondrial Ca2+ concentration. Bolus additions of CaCl2 were made to the 60 μg of mitochondria in suspension in 30 nmol increments and changes in Calcium Green 5N fluorescence were recorded at an emission of 532 nm with a QuantaMaster spectrofluorometer (Photon Technology International, Inc).
Measurement of ATP synthesis.
Spinal cord mitochondrial proteins were used to measure ATP synthesis with a luciferase/luciferin-based system as described previously (Johnston et al., 2000). Spinal cord mitochondria-enriched pellets (30 μg) were resuspended in 100 μl of buffer A (150 mm KCl, 25 mm Tris–HCl, 2 mm potassium phosphate, 0.1 mm MgCl2, pH 7.4) with 0.1% BSA, 1 mm malate, 1 mm glutamate, and buffer B (containing 0.8 mm luciferin and 20 mg/ml luciferase in 0.5 m Tris-acetate, pH 7.75). The reaction was initiated by addition of 0.1 mm ADP and monitored for 5 min using a microplate reader (Tecan). Luminescence of an ATP standard curve (containing 0, 0.05, 0.1, 0.25, 0.5, and 1.0 mm ATP solution in buffer A and buffer B) was also measured in parallel to determine the ATP synthesis rate (in nmol/min/mg mitochondrial protein).
Immunoprecipitation and immunoblotting.
Spinal cords from CypD−/− or CypD+/+ mutant SOD1 mice (either 12 or 13–14 months of age) were homogenized in HB buffer plus protease inhibitors. The lysates were centrifuged for 10 min at 1000 × g and either the resulting supernatants (clarified tissue extract) or the heavy membrane fractions resuspended in HB buffer as described above were supplemented with 150 mm KCl and 0.5% NP-40 and further incubated overnight at 4°C with misfolded SOD1 B8H10 antibody (MediMabs) previously cross-linked to Dynabeads protein G (Invitrogen) with dimethyl pimelimidate (Pierce) according to the manufacturer's instructions. The beads were magnetically isolated and washed three times with immunoprecipitation (IP) buffer. Samples were eluted with boiling in 2.0× sample buffer without dithiothreitol. Immunoprecipitated proteins were separated by SDS-PAGE, transferred to nitrocellulose membranes, and probed with the indicated antibodies followed by horseradish peroxidase-conjugated secondary antibodies (Jackson Immunochemicals). Pico ECL (Pierce) was used to detect immunoreactive bands. Primary antibodies against SOD1 (C-17; Santa Cruz Biotechnology), GAPDH (Abcam), prohibitin (Neomarkers), and cyclophilin D (Mitosciences) were used for immunoblotting.
Size exclusion chromatography
One mouse spinal cord was homogenized in HB buffer (10 mm Tri-HCl, pH 7.4, 150 mm KCl, 1 mm EDTA, 100 mm iodoacetamide, 0.5% NP-40) and spun at 5000 g for 5 min at 4°C. The supernatant (1 mg) was loaded on a Superose 6 column (GE Healthcare) equilibrated in 10 mm Tri-HCl, pH 7.4, 150 mm KCl, 1 mm EDTA, 0.5% NP-40 at a flow rate of 0.25 ml/min at 4°C. Fractions of 1 ml were collected. Then 75 and 40 μl of the collected fractions 4–11 and 13–19, respectively, were further analyzed by Slot blotting using a goat anti-SOD1 antibody (C-17; Santa Cruz Biotechnology). The calibration of the size exclusion column was performed with molecular weight markers ranging from 29 kDa to 2 MDa.
Results
Deleting cyclophilin D improves mitochondrial Ca2+ buffering capacity, ATP synthesis, and attenuates mitochondrial damage in mutant SOD1 mice
To determine whether increasing mitochondrial Ca2+ capacity alters SOD1 mutant-mediated ALS disease course and pathogenesis, we bred transgenic mice expressing mutant SOD1 mice that develop adult-onset fatal paralysis with Ppif −/− mice, which completely lack CypD expression (Basso et al., 2005). To ensure that conclusions drawn from deleting CypD would be broadly applicable to mutant SOD1-mediated ALS, all three most prominently used ALS mouse models were chosen so as to test mutants of markedly different biochemical characteristics, including fully dismutase active (SOD1G37R) (Boillée et al., 2006b), partially active SOD1G93A (Gurney et al., 1994), and inactive (SOD1G85R) mutant SOD1 (Bruijn et al., 1997). Additionally, the SOD1G85R- and SOD1G37R-expressing mouse lines used develop more slowly progressing disease from mutant SOD1 accumulation at levels equal to or modestly above, respectively, those of the endogenous mouse protein, thus mimicking more closely the levels of mutant SOD1 accumulation in ALS patients. Finally, SOD1G93A-expressing mice are the most widely used ALS models and develop very rapidly progressing disease from high level accumulation of mutant SOD1 (to levels 5- to 15-fold above endogenous SOD1).
Deleting CypD resulted in a doubling throughout disease of the Ca2+ storage capacity in mitochondria isolated from spinal cords of CypD−/−/SOD1G37R and CypD−/−/SOD1G85R animals (Fig. 1A,B). Importantly, the ∼50% reduction in Ca2+ storage capacity observed at disease end stage in CypD+/+/SOD1G37R and CypD+/+/SOD1G85R animals, compared with presymptomatic stages or age-matched nontransgenic mice, was completely reversed by deletion of CypD in mutant SOD1 animals compared with presymptomatic stages or age-matched CypD-deleted mice. Of note, in the SOD1G93A mice with normal CypD, the decrease in mitochondrial Ca2+ capacity preceded appearance of motor symptoms [occurring as early as 6 weeks of age (see presymptomatic CypD+/+/SOD1G93A animals in Fig. 1B)] and was completely reversed by deleting CypD.
Figure 1.

Deleting cyclophilin D improves mitochondrial calcium buffering capacity and mitochondrial ATP synthesis throughout disease in mutant SOD1-expressing mice. A, Representative traces of mitochondrial Ca2+ uptake capacity from spinal cords of asymptomatic nontransgenic animals (CypD+/+; green) (CypD−/−; orange) and from end stage CypD+/+/SOD1G93A (red) and CypD−/−/SOD1G93A (blue) animals. The peaks correspond to sequential bolus additions of 30 nmol of Ca2+. The downward deflections reflect mitochondrial Ca2+ uptake. B, Relative Ca2+ capacity of mitochondria isolated from spinal cords of CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at the presymptomatic stage and end stage of disease. The bar graph represents mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; ns: p > 0.05. The dotted line at 100% corresponds to Ca2+ capacity of mitochondria isolated from spinal cords of age-matched nontransgenic control animals (CypD+/+, green; CypD−/−, yellow). C, Levels of ATP synthesis in mitochondria isolated from spinal cords of CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at end stage disease and from age-matched nontransgenic controls at 6 and 14 months of age. The bar graph represents mean ± SEM. *p < 0.05.
CypD deletion has previously been reported to restore the activity of mitochondrial respiratory complexes and ATP synthesis in the cortices of aged mice overexpressing a mutant human form of amyloid precursor protein (Du et al., 2008). Compared with age-matched CypD knock-out mice, deletion of CypD in mutant SOD1 transgenic mice completely rescued the ∼30–50% decrease in mitochondrial ATP production observed in isolated spinal cord mitochondria from CypD+/+/SOD1G93A or CypD+/+/SOD1G85R animals at disease end stage when compared with nontransgenic controls (Fig. 1C).
Mitochondrial swelling resulting in damage to the organelle is a signature of mPTP opening (Bernardi et al., 2006). Electron microscopy was used to determine mitochondrial ultrastructure in spinal cord motor neurons (Fig. 2A), as well as lumbar motor axons (Fig. 3A) of CypD+/+/SOD1G93A and CypD−/−/SOD1G93A mice. Deleting CypD significantly reduced mitochondrial swelling (measured as the increase in average mitochondrial area and intracristal space) observed in CypD+/+/SOD1G93A mice at a symptomatic phase of disease (100 d) in both the soma of motor neurons (Fig. 2A–D) and in motor axons (Fig. 3A–E). Mitochondrial swelling was a general feature of the mitochondrial population in motor neurons (Fig. 2C) and their axons (Fig. 3D) as shown by the shift toward larger mitochondrial size in the mitochondrial area distribution of symptomatic CypD+/+/SOD1G93A mice versus CypD−/−/SOD1G93A mice and age-matched controls. Except for mitochondrial swelling, no other features of mitochondrial abnormalities were observed in the soma of motor neurons of CypD+/+/SOD1G93A mice at a symptomatic stage of disease. However, at this time point mitochondrial damage was very clear in the motor axons of CypD+/+/SOD1G93A mice, with a large number of mitochondria showing distension of the intermembrane space (sometimes containing membranous material), diluted matrix with disorganized cristae, and vacuolization (Fig. 3A,B). Mitochondrial damage in motor axons was significantly attenuated by deletion of CypD in symptomatic SOD1G93A mice (Fig. 3E).
Figure 2.

Deleting cyclophilin D reduces mitochondrial damage in spinal cord motor neurons throughout disease in mutant SOD1-expressing mice. A, Electron micrographs of motor neuron mitochondria from cross-sections of spinal cords from CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and from nontransgenic age-matched controls. Scale bar, 1 μm. B, Average area of mitochondria in spinal cord motor neurons of CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and in nontransgenic age-matched controls. The bar graph represents mean ± SEM. ***p < 0.001. C, Distribution of mitochondrial area as a percentage of total mitochondrial population analyzed in B. The percentage of mitochondria with area >1.1 μm2 for each genotype is represented in the bar graph. D, Average area of mitochondrial intracristae space in spinal cord motor neurons of CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and in nontransgenic age-matched controls. The bar graph represents mean ± SEM. ***p < 0.001.
Figure 3.

Deleting cyclophilin D reduces mitochondrial damage and degeneration in lumbar motor axons throughout disease in mutant SOD1-expressing mice. A, Electron micrographs of mitochondria from cross sections of lumbar L5 motor axon from CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and from nontransgenic age-matched controls. Scale bar, 1 μm. B, Electron micrographs of mitochondria from cross sections of lumbar L5 motor axon from CypD+/+/SOD1G93A animals at a symptomatic stage of disease. Scale bar, 250 nm. 1 illustrates an example of a healthy mitochondrion and 2–5 illustrate representative examples of damaged mitochondria: mitochondria with disorganized cristae (2), degenerating within vacuoles (3), with diluted (low electron density) matrix, (4) or abnormal distention between the inner and outer membranes with rupture of the outer membrane (5). C, Average area of mitochondria in lumbar motor axons of CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and in nontransgenic age-matched controls. The bar graph represents mean ± SEM. ***p < 0.001. D, Distribution of mitochondrial area as a percentage of the total mitochondrial population analyzed in C. The percentage of mitochondria with area >1.1 μm2 for each genotype is represented in the bar graph. E, Percentage of damaged mitochondria with the type of damage illustrated in B (2–5) in lumbar motor axons of CypD+/+/SOD1G93A and CypD−/−/SOD1G93A animals at a symptomatic stage of disease and in nontransgenic age-matched controls. The bar graph represents mean ± SEM. ***p < 0.001.
Thus, deleting CypD improved mitochondrial Ca2+ buffering and ATP synthesis capacity in the spinal cord and attenuated mitochondrial swelling within motor neurons and damage of mitochondria in motor axons of ALS mouse models expressing both dismutase active and inactive SOD1 mutants. Furthermore, the simplest interpretation for improved mitochondrial ATP synthesis in the CypD−/− nervous system is as a consequence of a reduction in Ca2+-dependent mitochondrial damage.
Improved mitochondrial function significantly reduces loss of motor neurons and glial activation, but does not delay muscle denervation or axonal degeneration
Sustained improvement in mitochondrial function resulting from deletion of CypD in mutant SOD1 animals did not correspond to a delay in the appearance of classical hallmarks of disease. The percentages of denervated NMJs in the gastrocnemius muscles were 73 and 67% (Fig. 4A,C), respectively, for CypD+/+/SOD1G37R and CypD−/−/SOD1G37R mice. Similarly, 86 and 85%, respectively, of junctions were denervated in CypD+/+/SOD1G85R and CypD−/−/SOD1G85R mice. Of note, deleting CypD did not prevent or delay the loss of NMJs at any stage of disease in mutant SOD1G37R mice (Fig. 4C). Similarly, there was no significant difference in the loss of α-motor axons throughout disease between CypD−/−/SOD1G37R or CypD−/−/SOD1G85R animals, compared with CypD+/+ littermates carrying the corresponding mutant SOD1 transgene (Fig. 4B,D; for SOD1G37R, 74 and 73%, lost by a symptomatic stage and 86% or 85% lost at disease end stage, while for SOD1G85R mice, 58% or 56%, lost at a symptomatic stage and 76% or 77% lost by disease end stage).
Figure 4.

Improving mitochondrial function by deleting CypD in mutant SOD1 animals does not prevent muscle denervation and axonal degeneration. A, Representative micrographs of gastrocnemius muscle from CypD+/+/SOD1G37R and CypD−/−/SOD1G37R animals at end stage disease and from age-matched nontransgenic littermates processed for immunofluorescence to reveal axons (red) using an antibody recognizing synaptophysin and fluoromyelin red and using α-bungarotoxin (green) to reveal muscle endplates. Scale bar, 20 μm. B, Representative micrographs of lumbar motor axons from CypD+/+/SOD1G37R and CypD−/−/SOD1G37R animals at end stage disease and from age-matched nontransgenic littermates. Scale bar, 25 μm. C, Quantification of innervation at the NMJ of the gastrocnemius muscle from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals throughout disease stages for mutant SOD1G37R and at end stage for CypD+/+/SOD1G85R, CypD−/−/SOD1G85R, and age-matched nontransgenic littermates. The bar graph represents mean ± SEM. ns: p > 0.05. D, Quantification of the total number of α-motor axons in the lumbar L5 motor root from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage and at end stage of disease.
In contrast, the loss of α-motor neurons in the spinal cords of all three mutant SOD1 mice at disease end stage was significantly reduced by deletion of CypD (Fig. 5A,B) (e.g., with 48% vs 27%, respectively, of the α-motor neurons lost at disease end stage in CypD+/+/SOD1G37R and CypD−/−/SOD1G37R mice; 48% vs 24%, respectively, of α-motor neurons lost at disease end stage in CypD+/+/SOD1G93A and CypD−/−/SOD1G93A mice). Indeed, α-motor neuron loss was almost completely suppressed when CypD was deleted in mutant SOD1G85R-expressing mice (Fig. 5B) (with 39% vs 4%, respectively, of α-motor neurons lost by disease end stage).
Figure 5.
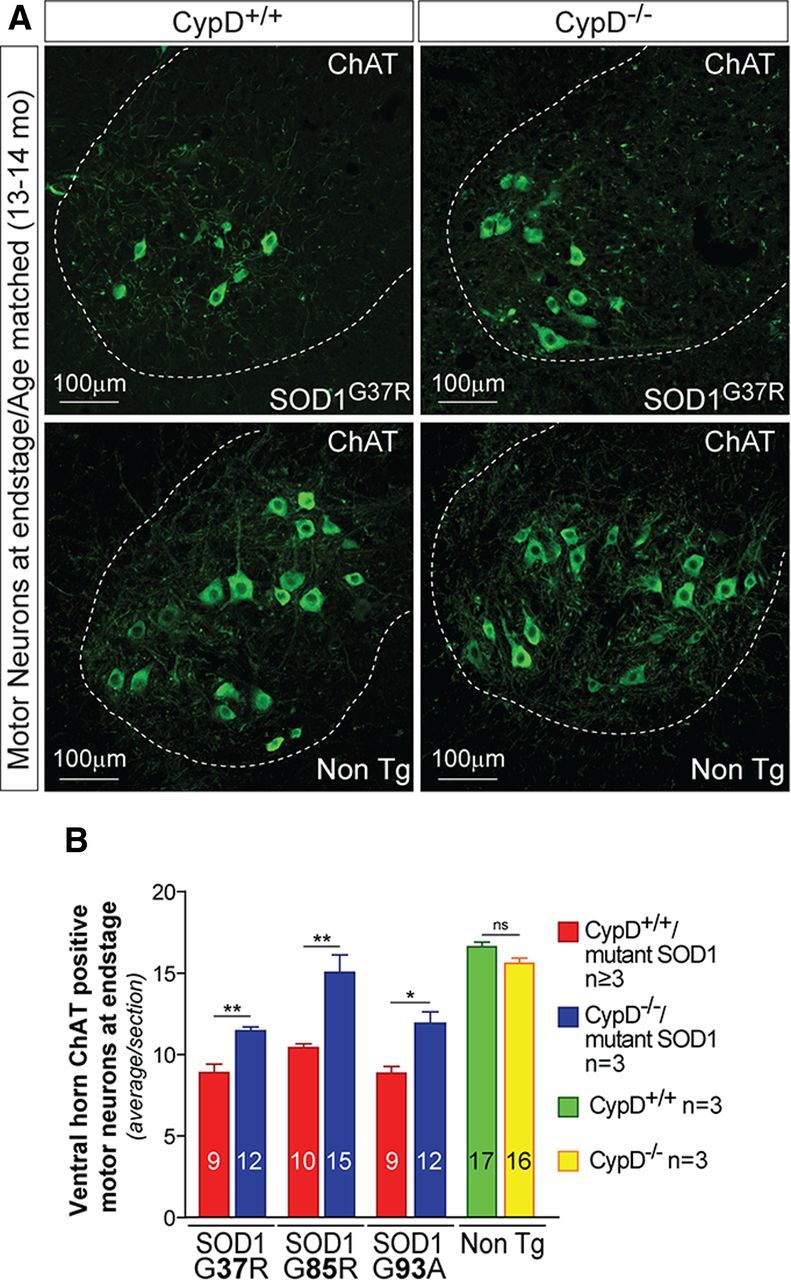
Improving mitochondrial function by deleting CypD in mutant SOD1 animals significantly reduces the loss of motor neurons. A, Representative micrographs of lumbar spinal cord sections from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at end stage of disease and from age-matched nontransgenic littermates processed for immunofluorescence using an antibody recognizing ChAT to reveal cholinergic motor neurons. Dashed outlines correspond to the boundary between gray and white matter. Scale bar, 100 μm. B, Quantification of the average number of large cholinergic ventral horn motor neurons per section of lumbar spinal cord from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at end stage disease and in age-matched nontransgenic littermates. The bar graph represents mean ± SEM. *p < 0.05, **p > 0.01; ns: p > 0.05. The number within each bar represents the average number of motor neurons per spinal cord section.
Additionally, improved mitochondrial function significantly reduced induction of microglial (Fig. 6A,B) and astroglial (Fig. 6C,D) activation (scored with Iba1 and GFAP immunoreactivity, respectively) in the spinal cord ventral horns of CypD−/−/SOD1G37R, SOD1G85R, and SOD1G93A mice at a symptomatic stage of disease (Fig. 6B,D; with reduction in Iba1 immunoreactivity of 95, 60, and 87%, respectively, in SOD1G37R, SOD1G85R, and SOD1G93A CypD-deleted mice and reduction in GFAP immunoreactivity of 71, 72, and 59%, respectively, in SOD1G37R, SOD1G85R and SOD1G93A CypD-deleted mice).
Figure 6.

Improving mitochondrial function by deleting CypD in mutant SOD1 animals significantly reduces glial activation. A, Representative micrographs of lumbar spinal cord sections from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage of disease processed for immunofluorescence using an antibody detecting activated microglia (IbaI). Dashed outlines correspond to the boundary between gray and white matter. Scale bar, 100 μm. B, Quantification of the relative fluorescence intensity of the IbaI staining of lumbar spinal cord from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage of disease. The bar graph represents mean ± SEM. **p < 0.01, ***p < 0.001. C, Representative micrographs of lumbar spinal cord sections from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at symptomatic stage of disease processed for immunofluorescence using an antibody-detecting activated astrocytes (GFAP). Dashed outlines correspond to the boundary between gray and white matter. Scale bar, 100 μm. D, Quantification of the relative fluorescence intensity of the GFAP staining of lumbar spinal cord from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage of disease. The bar graph represents mean ± SEM. **p < 0.01, ***p < 0.001.
Overall, deletion of CypD improved mitochondrial function in mice expressing SOD1 mutants of different biochemical properties and significantly reduced glial activation and the loss of spinal cord motor neurons, but did not alter motor axon degeneration and neuromuscular denervation.
Improved mitochondrial function reduces accumulation of misfolded SOD1 aggregates
SOD1-mediated toxicity to motor neurons has long been proposed to be linked to protein aggregates of SOD1 (Deng et al., 1993; Bruijn et al., 1998) that contain misfolded SOD1 as determined using antibodies that recognize epitopes unavailable in the natively folded protein and that bind preferentially or exclusively to misfolded conformers (Rakhit et al., 2007; Gros-Louis et al., 2010; Grad et al., 2011). To determine whether improving mitochondrial function attenuates SOD1-mediated toxicity to motor neurons by reducing the accumulation of misfolded SOD1, we used the monoclonal antibody B8H10, which recognizes epitopes within exon 3 that are exposed only upon misfolding or denaturation of SOD1 (Gros-Louis et al., 2010; Pickles and Vande Velde, 2012) to detect misfolded SOD1 forms by immunofluorescence on spinal cord sections.
Deleting CypD in all three models of mutant SOD1-mediated disease significantly reduced (by >80%) the accumulation of misfolded SOD1 detected by the B8H10 antibody at a symptomatic stage of disease (Fig. 7A,B) (with 84, 84, and 83%, respectively, of reduction in misfolded SOD1 accumulation in CypD-deleted SOD1G37R, SOD1G85R, and SOD1G93A mice). Similarly, a significant reduction in the accumulation of misfolded SOD1 was observed in CypD−/−/SOD1G37R at a symptomatic stage of disease when we used different monoclonal antibodies (A5C3 and D3H5), which recognize epitopes within exon 4 or 2, respectively, that are exposed upon misfolding of SOD1 (Gros-Louis et al., 2010; Pickles and Vande Velde, 2012) (Fig. 7C).
Figure 7.

Improving mitochondrial function by deleting CypD in mutant SOD1 animals reduces the accumulation of misfolded forms of mutant SOD1 in the spinal cord at a symptomatic stage of disease. A, C, D, Representative micrographs of lumbar spinal cord sections from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage of disease processed for immunofluorescence using an antibody-detecting misfolded SOD1: B8H10 (A), A5C3 and D3H5 (C), DSE2 (D). Dashed outlines correspond to the boundary between gray and white matter. Scale bar, 100 μm. B, Quantification of the relative fluorescence intensity of the B8H10 (A) and DSE2 (D) stainings of lumbar spinal cord from CypD+/+/mutant SOD1 and CypD−/−/mutant SOD1 animals at a symptomatic stage of disease. The bar graph represents mean ± SEM. **p < 0.01, ***p < 0.001.
To examine whether CypD deletion also reduced the accumulation of misfolded SOD1 exposing other disease-specific epitopes, we used a monoclonal antibody (DSE2) developed against the electrostatic loop of SOD1 (amino acids 125–142), a structural element accessible to antibody binding only upon misfolding or denaturation of SOD1 (Grad et al., 2011). Like that seen for the B8H10, A5C3, and D3H5 antibodies, the accumulation of misfolded SOD1 detected using the DSE2 antibody was also markedly reduced (by >70%) in CypD-deleted mutant SOD1 mice at a symptomatic stage of disease (Fig. 7B,D) (with 76, 88, and 73%, respectively, reductions in CypD-deleted SOD1G37R, SOD1G85R, and SOD1G93A mice).
To further confirm biochemically that CypD deletion reduces the accumulation of misfolded SOD1, we used immunoprecipitation to measure the levels of non-natively folded SOD1 conformers in detergent lysates of whole spinal cord (Fig. 8A). Significantly less misfolded SOD1 was detected in spinal cords from disease-matched symptomatic CypD−/−/SOD1G37R animals despite similar total levels of SOD1 accumulation (Fig. 8B). Moreover, because a proportion of misfolded SOD1 in the spinal cord of mice expressing SOD1 mutants of different biochemical characteristics is deposited on the surface of, or imported into, mitochondria (Mattiazzi et al., 2002; Liu et al., 2004; Vande Velde et al., 2008), spinal cord fractions enriched in mitochondria were examined for misfolded SOD1 (Fig. 8A). This accumulation, too, was very significantly reduced in SOD1G37R (Fig. 8C) and SOD1G85R (Fig. 8D) mice following CypD deletion.
Figure 8.

Improving mitochondrial function by deleting CypD in mutant SOD1 animals reduces the accumulation of misfolded SOD1 aggregates in whole spinal cord lysates and on the surface of spinal cord mitochondria. A, Schematic outlining the different purification steps used. Whole spinal cord lysate (B) from CypD+/+ or CypD−/− mutant SOD1 mice or mitochondria-enriched heavy membrane fraction (C, D) from spinal cords of CypD+/+ or CypD−/− mutant SOD1 mice were immunoprecipitated with an antibody specifically detecting misfolded forms of SOD1 (B8H10). Whole spinal cord lysate from CypD+/+ or CypD-/- mutant SOD1 mice was fractionated using size exclusion chromatography (E). B, Immunoblot of whole tissue extracts and the misfolded SOD1 immunoprecipitated from clarified spinal cord extract using the indicated antibodies. C, D, Immunoblot of the misfolded SOD1 immunoprecipitated from a heavy membrane fraction of spinal cords enriched in mitochondria. E, Immunoblot of SOD1 containing fractions separated by size exclusion chromatography and resolved by slot blot from spinal cord homogenates of CypD+/+ or CypD−/− mutant SOD1 mice. The molecular weight of each fraction was estimated from the separation of the indicated recombinant proteins.
In vitro evidence has led to the proposal that misfolded SOD1 protein is present as small soluble forms before coalescing into large aggregates (Rakhit et al., 2004). To determine whether the reduction in misfolded SOD1 levels in CypD-deleted mutant SOD1 mice reflects a decrease in the accumulation of the soluble and/or aggregated misfolded SOD1, size exclusion chromatography was used to separate SOD1 forms from CypD+/+/SOD1G93A or CypD−/−/SOD1G93A spinal cords (Fig. 8A). In CypD+/+/SOD1G93A spinal cords SOD1 eluted in low molecular weight complexes ∼29 kDa (corresponding to the soluble SOD1 dimers) and also in high molecular weight complexes from several hundred kDa to 2 MDa (Fig. 8E). While SOD1 levels in the low molecular weight complexes (<29 kDa) in CypD−/−/SOD1G93A spinal cords were similar to (or slightly higher) than that for CypD+/+/SOD1G93A, SOD1 levels in high molecular weight complexes were significantly diminished in CypD-deleted mutant SOD1 animals (Fig. 8E).
Since both high levels of cytosolic Ca2+ and increased oxidative damage may contribute to SOD1 aggregation (Beers et al., 2001; Kim et al., 2002; Rakhit et al., 2002; Tateno et al., 2004; Bosco et al., 2010), a plausible explanation is that the decrease in aggregated SOD1 in the CypD−/− nervous system arises from a combination of reduced oxygen radical production by Ca2+-mediated damage to mitochondria (Du et al., 2008) and a decrease in cytosolic levels of Ca2+ (Barsukova et al., 2011). In any event, deletion of CypD improves mitochondrial function and reduces both the overall level of misfolded SOD1 (including association with spinal cord mitochondria) and the accumulation of large aggregates within the spinal cord of misfolded SOD1 of different biochemical characteristics.
Improving mitochondrial function by deleting CypD in animals expressing SOD1 mutants with different biochemical properties does not alter disease course
Disease onset and progression in cohorts of SOD1G37R, SOD1G85R, and SOD1G93A with CypD+/+ or CypD−/− (n ≥ 22 for each genotype in each cohort) were followed (all matings were congenic on a C57Bl/6 background to eliminate confounding genetic influences). Despite (1) sustained improvement in mitochondrial function throughout disease, (2) attenuated motor neuron death, (3) reduced astrogliosis and microgliosis, and (4) suppression of aggregates of misfolded SOD1 in CypD−/− mice, there were no significant differences in age of disease onset, age of reaching a symptomatic stage, or survival compared with CypD+/+ mice (Fig. 9A–C). For example, for SOD1G37R (Fig. 9B; Table 1), disease onset was reached at 238 d for CypD+/+ versus 241 d for CypD−/−mice and survival was 397 versus 408 d, respectively. For each mutant SOD1 line, the increased mitochondrial Ca2+ buffering by deletion of CypD provided no benefit to males and females alike (as demonstrated, for example, by segregating the animals in the SOD1G93A cohort by sex; Table 1). Further, the gradual disease-related decrease in hindlimb muscle grip strength was also unaffected throughout disease despite sustained improvement of mitochondrial function upon CypD deletion in SOD1G37R, SOD1G85R, and SOD1G93A-expressing mice (Fig. 9D–F).
Figure 9.

Improving mitochondrial function by deleting CypD in mutant SOD1 animals does not alter disease course. A–C, Plot of ages (in days) at which disease onset (as determined by the weight peaks. At onset animals do not display any obvious motor phenotype), symptomatic stage (as determined by 10% weight loss from onset. This disease stage is characterized by clear gait abnormalities and tremor) and end stage (as determined by hindlimb paralysis and inability right itself) were reached for CypD+/+/mutant SOD1 (red) and CypD−/−/mutant SOD1 (blue) animals. D–F, Plot of averaged grip strength for CypD+/+/mutant SOD1 (red) and CypD−/−/mutant SOD1 (blue) animals at indicated age.
Table 1.
Ages of disease onset, reaching an early symptomatic stage and survival of mutant SOD1 mice in the presence or absence of CypD
| SOD1G93A |
SOD1G37R |
SOD1G85R Combined genders |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Combined genders |
Females |
Males |
Combined genders |
|||||||
| CypD+/+ | CypD−/− | CypD+/+ | CypD−/− | CypD+/+ | CypD−/− | CypD+/+ | CypD−/− | CypD+/+ | CypD−/− | |
| Onset | 92 ± 4 (n = 29) | 88 ± 4 (n = 31) | 92 ± 5 (n = 18) | 92 ± 6 (n = 16) | 93 5 (n = 11) | 80 ± 5 (n = 15) | 238 6 (n = 43) | 241 ± 7 (n = 33) | 270 ± 7 (n = 29) | 273 ± 8 (n = 24) |
| Early symptoms | 134 ± 1 (n = 24) | 133 ± 2 (n = 30) | 137 ± 2 (n = 14) | 136 ± 3 (n = 16) | 132 1 (n = 10) | 130 ± 2 (n = 14) | 337 6 (n = 40) | 350 ± 8 (n = 33) | 328 ± 6 (n = 29) | 340 ± 9 (n = 22) |
| End stage | 149 ± 1 (n = 37) | 150 ± 2 (n = 35) | 152 ± 2 (n = 20) | 154 ± 2 (n = 17) | 146 ± 2 (n = 17) | 147 ± 2 (n = 18) | 397 ± 6 (n = 35) | 408 ± 5 (n = 29) | 355 ± 6 (n = 33) | 369 ± 9 (n = 26) |
Values (days) are averages SEM.
Altogether, despite improved mitochondrial function throughout disease by CypD deletion, disease course was unaffected in all three models of familial ALS from expression of ALS-linked SOD1 mutants of divergent biochemical character.
Discussion
We have established that desensitizing Ca2+-mediated mPTP opening in mutant SOD1-expressing mice rescues the loss of mitochondrial Ca2+ buffering capacity and ATP synthesis, reduces mitochondrial swelling in spinal cord motor neurons and mitochondrial damage in motor axons even at late stages of disease, and protects motor neurons against cell death in the three most widely used models of ALS-like disease in mice. Despite this, attenuating mitochondrial damage, reducing or eliminating motor neuron cell death, attenuating gliosis, and inhibiting aggregation of misfolded SOD1 in the spinal cord leaves disease course unaffected. Thus, the mitochondrial dysfunction(s) following Ca2+-mediated opening of the mPTP, the formation of large misfolded SOD1 aggregates, and motor neuron death cannot be central to the mechanism of progressive, paralytic disease. That attenuated motor neuron death provides no benefit in affecting disease course complements earlier evidence (Gould et al., 2006; Rouaux et al., 2007) in an aggressive early onset model of ALS from high levels of mutant SOD1, extending those findings to more slowly progressing disease from dismutase active or inactive mutant SOD1 accumulation at levels modestly above (SOD1G37R) or equal to (SOD1G85R) those of the endogenous mouse protein, thus mimicking more closely the levels of mutant SOD1 accumulation in ALS patients.
Our findings also provide a mechanistic test of the proposal that Ca2+-induced opening of the mPTP is a key effector of axonal degeneration, a hypothesis that emerged in part from a report that reduced levels or pharmacological inhibition of CypD delayed the disintegration process (Wallerian degeneration) of the distal portion of an injured nerve in transected sciatic nerve explants (Barrientos et al., 2011). To this, not only have we and others shown that delaying Wallerian degeneration in SOD1 mutant models of ALS in mice (by introducing the “slow Wallerian degeneration” mutation Wlds) does not (Vande Velde et al., 2004), or only modestly (Fischer et al., 2005), prolongs survival, our current evidence demonstrates no slowing of degeneration from improved Ca2+ buffering and delayed mPTP opening. Therefore, although opening of the mPTP upon Ca2+ overload may contribute to distal axonal degeneration following injury, it is unlikely to be a primary contributor to degeneration of motor axons in mouse models of familial ALS that develop adult-onset fatal paralysis.
The absence of effect on any aspect of disease course in both males and females following removal of CypD contrasts with claims from two previous studies (Martin et al., 2009; Kim et al., 2012) reporting noncongenic matings of CypD−/− mice with mutant SOD1G93A animals and which reported conflicting outcomes on how CypD deletion affected survival. While Martin et al. (2009) claimed a delay in disease onset, which was then accompanied by an unexplained doubling of the rate of disease progression, with effect sizes differing between males and females, Kim et al. (2012) reported a lack of effect in males with slight acceleration of disease in females. The divergent outcomes of these two reports (Martin et al., 2009; Kim et al., 2012) are likely to be attributable to confounding genetic changes in the CypD gene disruption mouse lines used in these earlier studies. The CypD−/− strain used by Martin et al. (2009) develops multiple disorders (cardiomegaly and lymphoma, as well as liver disease and ovarian cancer [L.J. Martin, personal communication]) that are not seen in the CypD deletion mouse line used here. The CypD+/+/SOD1G93A mice of the experimental cohort in the Kim et al. (2012) study lived 50 d longer than their CypD+/+/SOD1G93A parents, demonstrating (as the authors acknowledged) the presence of a confounding genetic contributor(s) other than the mutant SOD1 transgene or CypD alleles. In contrast, the common outcome from our efforts using congenic matings and all three of the most frequently used SOD1 mutant mouse models provides what we believe is a comprehensive test demonstrating that loss of mitochondrial Ca2+ capacity is not a primary contributor to disease course in mutant SOD1 mice.
Astroglial and microglial activation is a well accepted feature of ALS in both patients and ALS mice models (Engelhardt and Appel, 1990; Kushner et al., 1991; Wong et al., 1995; Bruijn et al., 1997; Alexianu et al., 2001; Howland et al., 2002; Henkel et al., 2004). In addition, decreased mitochondrial respiration and ATP synthesis, as well as increased mitochondrial oxygen radical production, have been reported in primary astrocytes isolated from SOD1G93A mice (Cassina et al., 2008). This has suggested that glial activation may be linked to mitochondrial defects within those glia. Our results show that preventing mitochondrial damage in symptomatic mutant SOD1 mice decreases astroglial and microglial activation. Thus, these results suggest that mitochondrial damage mediated at least in part by Ca2+ induced mPTP opening may contribute to increased glial activation. Alternatively, the attenuation in glial activation that we observe in mutant SOD1 mice deleted in CypD may be due to the concomitant reduction in motor neuron cell death in these animals. This latter possibility is supported by evidence showing that glial activation is clearly increased only postsymptomatically in different mutant SOD1 mice mouse models (Wong et al., 1995; Bruijn et al., 1997; Hall et al., 1998; Levine et al., 1999). In addition, markers of glial activation were found to be significantly elevated in mice expressing high levels of mutant SOD1 selectively within motor neurons and which do not develop adult-onset fatal ALS-like paralysis but that sustain loss of motor neurons in late adulthood (Jaarsma et al., 2008). Importantly, our evidence of decreased GFAP and IbaI staining in CypD-deleted mutant SOD1 mice is fully consistent with the now well accepted proposal that mutant SOD1-mediated toxicity within astrocytes and microglia is a central component of the mechanism of disease progression (Beers et al., 2006; Boillée et al., 2006b; Di Giorgio et al., 2007; Nagai et al., 2007; Yamanaka et al., 2008). Our results show, however, that elevated astrocytic and microglial activation in symptomatic mutant SOD1 animals is not an indicator of the component of disease mechanism produced by mutant SOD1 synthesis within glia.
The formation of mutant SOD1 aggregates during disease in the spinal cords of both ALS patients carrying mutations in SOD1 and in transgenic mouse models has been proposed by many investigators to contribute to mutant SOD1-mediated toxicity (Durham et al., 1997; Bruijn et al., 1998; Johnston et al., 2000; Shinder et al., 2001; Wang et al., 2002). The generation of antibodies binding to epitopes exposed only in non-natively folded forms of SOD1 has revealed that misfolded SOD1 conformers form aggregates in ALS patients and mutant SOD1 mice (Rakhit et al., 2007; Gros-Louis et al., 2010; Kerman et al., 2010; Grad et al., 2011). Our use of a combination of IP, size exclusion chromatography, and immunofluorescence with multiple antibodies recognizing disease-specific misfolded SOD1 conformers has established that aggregated, but not soluble, misfolded SOD1 is significantly reduced in symptomatic animals expressing any of three ALS-linked mutations of SOD1 and deleted in CypD. Since this is not accompanied by an alteration in disease course, misfolded SOD1 aggregates must be a consequence rather than a central component of initiating or propagating disease pathogenesis. In agreement with this, formation of the large misfolded SOD1 aggregates in the spinal cord is largely undetectable before the onset of neuromuscular denervation (Rakhit et al., 2007; Karch et al., 2009; Gros-Louis et al., 2010). Furthermore, we have shown that aggregated misfolded SOD1, scored with different misfolded SOD1-specific antibodies, accumulates at different levels in disease-matched SOD1G37R, SOD1G85R, and SOD1G93A mice, adding further evidence against it as a component of disease mechanism.
Thus, the accumulated evidence supports the pool of soluble misfolded protein as a primary contributor to mutant SOD1 toxicity. This conclusion is consistent with passive and active immunization approaches aimed at neutralizing misfolded SOD1 species, which have reported that a reduction in the soluble pool of misfolded SOD1 [some of which is extracellular (Urushitani et al., 2006)] correlates with an increase in survival in mutant SOD1G37R and SOD1G93A mice (Urushitani et al., 2006; Gros-Louis et al., 2010; Liu et al., 2012). This insight brings SOD1-mediated ALS in line with the situation in several other neurodegenerative diseases where large inclusions correlate poorly with either onset or severity of neurodegeneration (Gutekunst et al., 1999; Kirkitadze et al., 2002; Arrasate et al., 2004; Van Raamsdonk et al., 2005; Treusch et al., 2009). The misfolded molecules so produced may mediate toxicity by binding to and/or interfering with specific cellular pathways [including hyperactivation of AMPK metabolism (Lim et al., 2012), chronic activation of NADPH oxidase by binding to Rac 1 (Harraz et al., 2008), induction of endoplasmic reticulum stress (Kikuchi et al., 2006; Nishitoh et al., 2008; Saxena et al., 2009), and mitochondrial dysfunction including inhibition of the voltage-dependent anion channel (Israelson et al., 2010) and errors in mitochondrial trafficking (Vande Velde et al., 2011; Magrane et al., 2012)] and also through secretion into the extracellular space (Urushitani et al., 2006).
Footnotes
This work has been supported by grants from the Muscular Dystrophy Association and the National Institutes of Health to D.W.C. S.D.C. was supported by a postdoctoral fellowship from the Amyotrophic Lateral Sclerosis Association. Salary support for D.W.C. is provided by the Ludwig Institute for Cancer Research. We are very grateful to Michael Forte and Paolo Bernardi for generously providing us with the CypD−/− mice. We thank Neil Cashman and Jean-Pierre Julien for generously providing the misfolded SOD1 antibodies. We thank Timo Merloo and Ying Jones for their help with the electron microscopy study.
References
- Alexianu ME, Kozovska M, Appel SH. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology. 2001;57:1282–1289. doi: 10.1212/wnl.57.7.1282. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, Bernardi P. The mitochondrial permeability transition from yeast to mammals. FEBS Lett. 2010;584:2504–2509. doi: 10.1016/j.febslet.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- Barrientos SA, Martinez NW, Yoo S, Jara JS, Zamorano S, Hetz C, Twiss JL, Alvarez J, Court FA. Axonal degeneration is mediated by the mitochondrial permeability transition pore. J Neurosci. 2011;31:966–978. doi: 10.1523/JNEUROSCI.4065-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsukova A, Komarov A, Hajnóczky G, Bernardi P, Bourdette D, Forte M. Activation of the mitochondrial permeability transition pore modulates Ca2+ responses to physiological stimuli in adult neurons. Eur J Neurosci. 2011;33:831–842. doi: 10.1111/j.1460-9568.2010.07576.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Beers DR, Ho BK, Siklós L, Alexianu ME, Mosier DR, Mohamed AH, Otsuka Y, Kozovska ME, McAlhany RE, Smith RG, Appel SH. Parvalbumin overexpression alters immune-mediated increases in intracellular calcium, and delays disease onset in a transgenic model of familial amyotrophic lateral sclerosis. J Neurochem. 2001;79:499–509. doi: 10.1046/j.1471-4159.2001.00582.x. [DOI] [PubMed] [Google Scholar]
- Beers DR, Henkel JS, Xiao Q, Zhao W, Wang J, Yen AA, Siklos L, McKercher SR, Appel SH. Wild-type microglia extend survival in PU. 1 knock-out mice with familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2006;103:16021–16026. doi: 10.1073/pnas.0607423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergemalm D, Jonsson PA, Graffmo KS, Andersen PM, Brännström T, Rehnmark A, Marklund SL. Overloading of stable and exclusion of unstable human superoxide dismutase-1 variants in mitochondria of murine amyotrophic lateral sclerosis models. J Neurosci. 2006;26:4147–4154. doi: 10.1523/JNEUROSCI.5461-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Bernardi P, Krauskopf A, Basso E, Petronilli V, Blachly-Dyson E, Di Lisa F, Forte MA. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006;273:2077–2099. doi: 10.1111/j.1742-4658.2006.05213.x. [DOI] [PubMed] [Google Scholar]
- Boillée S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their non-neuronal neighbors. Neuron. 2006a;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Boillée S, Yamanaka K, Lobsiger CS, Copeland NG, Jenkins NA, Kassiotis G, Kollias G, Cleveland DW. Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 2006b;312:1389–1392. doi: 10.1126/science.1123511. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak NM, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien JP, Brady ST, Brown RH., Jr Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijn LI, Becher MW, Lee MK, Anderson KL, Jenkins NA, Copeland NG, Sisodia SS, Rothstein JD, Borchelt DR, Price DL, Cleveland DW. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron. 1997;18:327–338. doi: 10.1016/s0896-6273(00)80272-x. [DOI] [PubMed] [Google Scholar]
- Bruijn LI, Houseweart MK, Kato S, Anderson KL, Anderson SD, Ohama E, Reaume AG, Scott RW, Cleveland DW. Aggregation and motor neuron toxicity of an ALS-linked SOD1 mutant independent from wild-type SOD1. Science. 1998;281:1851–1854. doi: 10.1126/science.281.5384.1851. [DOI] [PubMed] [Google Scholar]
- Cassina P, Cassina A, Pehar M, Castellanos R, Gandelman M, de León A, Robinson KM, Mason RP, Beckman JS, Barbeito L, Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiano M, Starkov AA, Petri S, Kipiani K, Kiaei M, Mattiazzi M, Flint Beal M, Manfredi G. Neural mitochondrial Ca2+ capacity impairment precedes the onset of motor symptoms in G93A Cu/Zn-superoxide dismutase mutant mice. J Neurochem. 2006;96:1349–1361. doi: 10.1111/j.1471-4159.2006.03619.x. [DOI] [PubMed] [Google Scholar]
- Deng HX, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung WY, Getzoff ED, Hu P, Herzfeldt B, Roos RP. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science. 1993;261:1047–1051. doi: 10.1126/science.8351519. [DOI] [PubMed] [Google Scholar]
- Deng HX, Shi Y, Furukawa Y, Zhai H, Fu R, Liu E, Gorrie GH, Khan MS, Hung WY, Bigio EH, Lukas T, Dal Canto MC, O'Halloran TV, Siddique T. Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria. Proc Natl Acad Sci U S A. 2006;103:7142–7147. doi: 10.1073/pnas.0602046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giorgio FP, Carrasco MA, Siao MC, Maniatis T, Eggan K. Noncell autonomous effect of glia on motor neurons in an embryonic stem cell-based ALS model. Nat Neurosci. 2007;10:608–614. doi: 10.1038/nn1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durham HD, Roy J, Dong L, Figlewicz DA. Aggregation of mutant Cu/Zn superoxide dismutase proteins in a culture model of ALS. J Neuropathol Exp Neurol. 1997;56:523–530. doi: 10.1097/00005072-199705000-00008. [DOI] [PubMed] [Google Scholar]
- Engelhardt JI, Appel SH. IgG reactivity in the spinal cord and motor cortex in amyotrophic lateral sclerosis. Arch Neurol. 1990;47:1210–1216. doi: 10.1001/archneur.1990.00530110068019. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Culver DG, Davis AA, Tennant P, Wang M, Coleman M, Asress S, Adalbert R, Alexander GM, Glass JD. The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol Dis. 2005;19:293–300. doi: 10.1016/j.nbd.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Guest WC, Yanai A, Pokrishevsky E, O'Neill MA, Gibbs E, Semenchenko V, Yousefi M, Wishart DS, Plotkin SS, Cashman NR. Intermolecular transmission of superoxide dismutase 1 misfolding in living cells. Proc Natl Acad Sci U S A. 2011;108:16398–16403. doi: 10.1073/pnas.1102645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros-Louis F, Soucy G, Larivière R, Julien JP. Intracerebroventricular infusion of monoclonal antibody or its derived Fab fragment against misfolded forms of SOD1 mutant delays mortality in a mouse model of ALS. J Neurochem. 2010;113:1188–1199. doi: 10.1111/j.1471-4159.2010.06683.x. [DOI] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Oostveen JA, Gurney ME. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia. 1998;23:249–256. doi: 10.1002/(sici)1098-1136(199807)23:3<249::aid-glia7>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schöneich C, Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkel JS, Engelhardt JI, Siklós L, Simpson EP, Kim SH, Pan T, Goodman JC, Siddique T, Beers DR, Appel SH. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol. 2004;55:221–235. doi: 10.1002/ana.10805. [DOI] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD. Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS) Proc Natl Acad Sci U S A. 2002;99:1604–1609. doi: 10.1073/pnas.032539299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter DR, Haworth RA. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys. 1979;195:453–459. doi: 10.1016/0003-9861(79)90371-0. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarsma D, Teuling E, Haasdijk ED, De Zeeuw CI, Hoogenraad CC. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28:2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal MK, Keller BU. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol Pharmacol. 2009;75:478–489. doi: 10.1124/mol.108.050831. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Dalton MJ, Gurney ME, Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karch CM, Prudencio M, Winkler DD, Hart PJ, Borchelt DR. Role of mutant SOD1 disulfide oxidation and aggregation in the pathogenesis of familial ALS. Proc Natl Acad Sci U S A. 2009;106:7774–7779. doi: 10.1073/pnas.0902505106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Mitochondrial dysfunction and intracellular calcium dysregulation in ALS. Mech Ageing Dev. 2010;131:517–526. doi: 10.1016/j.mad.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerman A, Liu HN, Croul S, Bilbao J, Rogaeva E, Zinman L, Robertson J, Chakrabartty A. Amyotrophic lateral sclerosis is a nonamyloid disease in which extensive misfolding of SOD1 is unique to the familial form. Acta Neuropathol. 2010;119:335–344. doi: 10.1007/s00401-010-0646-5. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Almer G, Yamashita S, Guégan C, Nagai M, Xu Z, Sosunov AA, McKhann GM, 2nd, Przedborski S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A. 2006;103:6025–6030. doi: 10.1073/pnas.0509227103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Kim M, Kim SH, Sung JJ, Lee KW. Alteration in intracellular calcium homeostasis reduces motor neuronal viability expressing mutated Cu/Zn superoxide dismutase through a nitric oxide/guanylyl cyclase cGMP cascade. Neuroreport. 2002;13:1131–1135. doi: 10.1097/00001756-200207020-00012. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Magran è J, Starkov AA, Manfredi G. The mitochondrial calcium regulator cyclophilin D is an essential component of oestrogen-mediated neuroprotection in amyotrophic lateral sclerosis. Brain. 2012;135:2865–2874. doi: 10.1093/brain/aws208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Kushner PD, Stephenson DT, Wright S. Reactive astrogliosis is widespread in the subcortical white matter of amyotrophic lateral sclerosis brain. J Neuropathol Exp Neurol. 1991;50:263–277. doi: 10.1097/00005072-199105000-00008. [DOI] [PubMed] [Google Scholar]
- Levine JB, Kong J, Nadler M, Xu Z. Astrocytes interact intimately with degenerating motor neurons in mouse amyotrophic lateral sclerosis (ALS) Glia. 1999;28:215–224. [PubMed] [Google Scholar]
- Li Q, Vande Velde C, Israelson A, Xie J, Bailey AO, Dong MQ, Chun SJ, Roy T, Winer L, Yates JR, Capaldi RA, Cleveland DW, Miller TM. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151. doi: 10.1073/pnas.1014862107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim MA, Selak MA, Xiang Z, Krainc D, Neve RL, Kraemer BC, Watts JL, Kalb RG. Reduced activity of AMP-activated protein kinase protects against genetic models of motor neuron disease. J Neurosci. 2012;32:1123–1141. doi: 10.1523/JNEUROSCI.6554-10.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HN, Tjostheim S, Dasilva K, Taylor D, Zhao B, Rakhit R, Brown M, Chakrabartty A, McLaurin J, Robertson J. Targeting of monomer/misfolded SOD1 as a therapeutic strategy for amyotrophic lateral sclerosis. J Neurosci. 2012;32:8791–8799. doi: 10.1523/JNEUROSCI.5053-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lillo C, Jonsson PA, Vande Velde C, Ward CM, Miller TM, Subramaniam JR, Rothstein JD, Marklund S, Andersen PM, Brännström T, Gredal O, Wong PC, Williams DS, Cleveland DW. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron. 2004;43:5–17. doi: 10.1016/j.neuron.2004.06.016. [DOI] [PubMed] [Google Scholar]
- Liu J, Shinobu LA, Ward CM, Young D, Cleveland DW. Elevation of the Hsp70 chaperone does not effect toxicity in mouse models of familial amyotrophic lateral sclerosis. J Neurochem. 2005;93:875–882. doi: 10.1111/j.1471-4159.2005.03054.x. [DOI] [PubMed] [Google Scholar]
- Lobsiger CS, Boillee S, McAlonis-Downes M, Khan AM, Feltri ML, Yamanaka K, Cleveland DW. Schwann cells expressing dismutase active mutant SOD1 unexpectedly slow disease progression in ALS mice. Proc Natl Acad Sci U S A. 2009;106:4465–4470. doi: 10.1073/pnas.0813339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magran é J, Sahawneh MA, Przedborski S, Estévez ÁG, Manfredi G. Mitochondrial dynamics and bioenergetic dysfunction is associated with synaptic alterations in mutant SOD1 motor neurons. J Neurosci. 2012;32:229–242. doi: 10.1523/JNEUROSCI.1233-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol. 2009;218:333–346. doi: 10.1016/j.expneurol.2009.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Okada Y, Nakamichi M, Nakamura M, Toyama Y, Sobue G, Nagai M, Aoki M, Itoyama Y, Okano H. Disease progression of human SOD1 (G93A) transgenic ALS model rats. J Neurosci Res. 2006;83:119–133. doi: 10.1002/jnr.20708. [DOI] [PubMed] [Google Scholar]
- Mattiazzi M, D'Aurelio M, Gajewski CD, Martushova K, Kiaei M, Beal MF, Manfredi G. Mutated human SOD1 causes dysfunction of oxidative phosphorylation in mitochondria of transgenic mice. J Biol Chem. 2002;277:29626–29633. doi: 10.1074/jbc.M203065200. [DOI] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial calcium function and dysfunction in the central nervous system. Biochim Biophys Acta. 2009;1787:1416–1424. doi: 10.1016/j.bbabio.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22:1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrini S, Sau D, Guareschi S, Bogush M, Brown RH, Jr, Naniche N, Kia A, Trotti D, Pasinelli P. ALS-linked mutant SOD1 damages mitochondria by promoting conformational changes in Bcl-2. Hum Mol Genet. 2010;19:2974–2986. doi: 10.1093/hmg/ddq202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- Pickles S, Vande Velde C. Misfolded SOD1 and ALS: zeroing in on mitochondria. Amyotroph Lateral Scler. 2012;13:333–340. doi: 10.3109/17482968.2012.648645. [DOI] [PubMed] [Google Scholar]
- Rakhit R, Cunningham P, Furtos-Matei A, Dahan S, Qi XF, Crow JP, Cashman NR, Kondejewski LH, Chakrabartty A. Oxidation-induced misfolding and aggregation of superoxide dismutase and its implications for amyotrophic lateral sclerosis. J Biol Chem. 2002;277:47551–47556. doi: 10.1074/jbc.M207356200. [DOI] [PubMed] [Google Scholar]
- Rakhit R, Crow JP, Lepock JR, Kondejewski LH, Cashman NR, Chakrabartty A. Monomeric Cu,Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic lateral sclerosis. J Biol Chem. 2004;279:15499–15504. doi: 10.1074/jbc.M313295200. [DOI] [PubMed] [Google Scholar]
- Rakhit R, Robertson J, Vande Velde C, Horne P, Ruth DM, Griffin J, Cleveland DW, Cashman NR, Chakrabartty A. An immunological epitope selective for pathological monomer-misfolded SOD1 in ALS. Nat Med. 2007;13:754–759. doi: 10.1038/nm1559. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O'Regan JP, Deng HX. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Tsai G, Kuncl RW, Clawson L, Cornblath DR, Drachman DB, Pestronk A, Stauch BL, Coyle JT. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann Neurol. 1990;28:18–25. doi: 10.1002/ana.410280106. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin LJ, Kuncl RW. Decreased glutamate transport by the brain and spinal cord in amyotrophic lateral sclerosis. N Engl J Med. 1992;326:1464–1468. doi: 10.1056/NEJM199205283262204. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Panteleeva I, Ren é F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding protein-dependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy J, Minotti S, Dong L, Figlewicz DA, Durham HD. Glutamate potentiates the toxicity of mutant Cu/Zn-superoxide dismutase in motor neurons by postsynaptic calcium-dependent mechanisms. J Neurosci. 1998;18:9673–9684. doi: 10.1523/JNEUROSCI.18-23-09673.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12:627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A. 2005;102:12005–12010. doi: 10.1073/pnas.0505294102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schütz B, Reimann J, Dumitrescu-Ozimek L, Kappes-Horn K, Landreth GE, Schürmann B, Zimmer A, Heneka MT. The oral antidiabetic pioglitazone protects from neurodegeneration and amyotrophic lateral sclerosis-like symptoms in superoxide dismutase-G93A transgenic mice. J Neurosci. 2005;25:7805–7812. doi: 10.1523/JNEUROSCI.2038-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinder GA, Lacourse MC, Minotti S, Durham HD. Mutant Cu/Zn-superoxide dismutase proteins have altered solubility and interact with heat shock/stress proteins in models of amyotrophic lateral sclerosis. J Biol Chem. 2001;276:12791–12796. doi: 10.1074/jbc.M010759200. [DOI] [PubMed] [Google Scholar]
- Siklós L, Engelhardt J, Harati Y, Smith RG, Jo ó F, Appel SH. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann Neurol. 1996;39:203–216. doi: 10.1002/ana.410390210. [DOI] [PubMed] [Google Scholar]
- Siklós L, Engelhardt JI, Alexianu ME, Gurney ME, Siddique T, Appel SH. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J Neuropathol Exp Neurol. 1998;57:571–587. doi: 10.1097/00005072-199806000-00005. [DOI] [PubMed] [Google Scholar]
- Tateno M, Sadakata H, Tanaka M, Itohara S, Shin RM, Miura M, Masuda M, Aosaki T, Urushitani M, Misawa H, Takahashi R. Calcium-permeable AMPA receptors promote misfolding of mutant SOD1 protein and development of amyotrophic lateral sclerosis in a transgenic mouse model. Hum Mol Genet. 2004;13:2183–2196. doi: 10.1093/hmg/ddh246. [DOI] [PubMed] [Google Scholar]
- Treusch S, Cyr DM, Lindquist S. Amyloid deposits: protection against toxic protein species? Cell Cycle. 2009;8:1668–1674. doi: 10.4161/cc.8.11.8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urushitani M, Sik A, Sakurai T, Nukina N, Takahashi R, Julien JP. Chromogranin-mediated secretion of mutant superoxide dismutase proteins linked to amyotrophic lateral sclerosis. Nat Neurosci. 2006;9:108–118. doi: 10.1038/nn1603. [DOI] [PubMed] [Google Scholar]
- Van Damme P, Leyssen M, Callewaert G, Robberecht W, Van Den Bosch L. The AMPA receptor antagonist NBQX prolongs survival in a transgenic mouse model of amyotrophic lateral sclerosis. Neurosci Lett. 2003;343:81–84. doi: 10.1016/s0304-3940(03)00314-8. [DOI] [PubMed] [Google Scholar]
- Van Den Bosch L, Van Damme P, Bogaert E, Robberecht W. The role of excitotoxicity in the pathogenesis of amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:1068–1082. doi: 10.1016/j.bbadis.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk JM, Murphy Z, Slow EJ, Leavitt BR, Hayden MR. Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum Mol Genet. 2005;14:3823–3835. doi: 10.1093/hmg/ddi407. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Garcia ML, Yin X, Trapp BD, Cleveland DW. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromolecular Med. 2004;5:193–203. doi: 10.1385/NMM:5:3:193. [DOI] [PubMed] [Google Scholar]
- Vande Velde C, Miller TM, Cashman NR, Cleveland DW. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc Natl Acad Sci U S A. 2008;105:4022–4027. doi: 10.1073/pnas.0712209105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Velde C, McDonald KK, Boukhedimi Y, McAlonis-Downes M, Lobsiger CS, Bel Hadj S, Zandona A, Julien JP, Shah SB, Cleveland DW. Misfolded SOD1 associated with motor neuron mitochondria alters mitochondrial shape and distribution prior to clinical onset. PloS One. 2011;6:e22031. doi: 10.1371/journal.pone.0022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayvergiya C, Beal MF, Buck J, Manfredi G. Mutant superoxide dismutase 1 forms aggregates in the brain mitochondrial matrix of amyotrophic lateral sclerosis mice. J Neurosci. 2005;25:2463–2470. doi: 10.1523/JNEUROSCI.4385-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lewinski F, Fuchs J, Vanselow BK, Keller BU. Low Ca2+ buffering in hypoglossal motoneurons of mutant SOD1 (G93A) mice. Neurosci Lett. 2008;445:224–228. doi: 10.1016/j.neulet.2008.08.084. [DOI] [PubMed] [Google Scholar]
- Wang J, Xu G, Gonzales V, Coonfield M, Fromholt D, Copeland NG, Jenkins NA, Borchelt DR. Fibrillar inclusions and motor neuron degeneration in transgenic mice expressing superoxide dismutase 1 with a disrupted copper-binding site. Neurobiol Dis. 2002;10:128–138. doi: 10.1006/nbdi.2002.0498. [DOI] [PubMed] [Google Scholar]
- Wong PC, Pardo CA, Borchelt DR, Lee MK, Copeland NG, Jenkins NA, Sisodia SS, Cleveland DW, Price DL. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron. 1995;14:1105–1116. doi: 10.1016/0896-6273(95)90259-7. [DOI] [PubMed] [Google Scholar]
- Yamanaka K, Chun SJ, Boillee S, Fujimori-Tonou N, Yamashita H, Gutmann DH, Takahashi R, Misawa H, Cleveland DW. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat Neurosci. 2008;11:251–253. doi: 10.1038/nn2047. [DOI] [PMC free article] [PubMed] [Google Scholar]