

Abstract
TNF is an important mediator of glomerulonephritis. The two TNF-receptors TNFR1 and TNFR2 contribute differently to glomerular inflammation in vivo, but specific mechanisms of TNFR-mediated inflammatory responses in glomeruli are unknown. We investigated their expression and function in murine kidneys, isolated glomeruli ex vivo, and glomerular cells in vitro. In normal kidney TNFR1 and TNFR2 were preferentially expressed in glomeruli. Expression of both TNFRs and TNF-induced upregulation of TNFR2 mRNA was confirmed in murine glomerular endothelial and mesangial cell lines. In vivo, TNF exposure rapidly induced glomerular accumulation of leukocytes. To examine TNFR-specific inflammatory responses in intrinsic glomerular cells but not infiltrating leukocytes we performed microarray gene expression profiling on intact glomeruli isolated from wildtype and Tnfr-deficient mice following exposure to soluble TNF ex vivo. Most TNF-induced effects were exclusively mediated by TNFR1, including induced glomerular expression of adhesion molecules, chemokines, complement factors and pro-apoptotic molecules. However, TNFR2 contributed to TNFR1-dependent mRNA expression of inflammatory mediators in glomeruli when exposed to low TNF concentrations. Chemokine secretion was absent in TNF-stimulated Tnfr1-deficient glomeruli, but also significantly decreased in glomeruli lacking TNFR2. In vivo, TNF-induced glomerular leukocyte infiltration was abrogated in Tnfr1-deficient mice, whereas Tnfr2-deficiency decreased mononuclear phagocytes infiltrates, but not neutrophils. These data demonstrate that activation of intrinsic glomerular cells by soluble TNF requires TNFR1, whereas TNFR2 is not essential, but augments TNFR1-dependent effects. Previously described TNFR2-dependent glomerular inflammation may therefore require TNFR2 activation by membrane-bound, but not soluble TNF.
Introduction
Cytokines produced by intrinsic renal cells and infiltrating leukocytes are central mediators of inflammatory kidney diseases. Tumor necrosis factor-α (TNF) is involved in glomerular inflammation and scarring [1], [2]. Renal expression of TNF is up-regulated in experimental animals and patients with glomerulonephritis (GN) [3], [4], [5], [6]. TNF is produced by macrophages, T cells, neutrophils and endothelium. Furthermore, intrinsic renal cells, including mesangial cells, podocytes, and tubular epithelial cells produce TNF upon stimulation [4], [7], [8], [9], [10], [11].
The functional role of TNF in GN has been demonstrated in animal models. Systemic administration of TNF induced glomerular damage in rabbits [12] and exacerbated glomerular injury in rats with nephrotoxic serum nephritis (NTN), a model of immune complex-mediated GN [13]. TNF-deficient mice subjected to NTN demonstrated reduced proteinuria, glomerular crescent formation, infiltration of leukocytes, and expression of vascular adhesion molecules [14]. In bone marrow chimeric mice it was shown that intrinsic renal cells are the major source of TNF contributing to renal injury in the NTN model [15], [16]. TNF blockade also attenuated glomerular lesions and crescent formation in rats developing anti-glomerular basement membrane antibody-induced crescentic GN [17], [18], [19].
Biological activities of TNF are mediated through two functionally distinct TNF receptors (TNFR), TNFR1 (CD120a) and TNFR2 (CD120b). An increased glomerular expression of TNFR1 and TNFR2 has been suggested by animal and human biopsy studies of GN [6], [20]. We have previously identified distinct proinflammatory roles of the two TNFRs in the development of glomerular injury in murine NTN. Tnfr1-deficiency delayed the onset of GN, whereas deficiency of intrinsic renal cell-expressed TNFR2 abrogated glomerular injury following immune complex deposition [20]. Underlying mechanisms of these differential TNFR-mediated inflammatory responses in the glomerulus are not known.
With TNF receptor-targeted interventions becoming a potential therapeutic strategy to reduce glomerular inflammation, mechanisms of TNFR-specific inflammatory responses in glomeruli remain to be elucidated. Here, we characterize TNFR1- and TNFR2-dependent glomerular inflammatory pathways in murine kidneys in vivo, in isolated glomeruli ex vivo, and in murine mesangial cells in vitro. Microarray gene expression profiling on intact glomeruli isolated from wildtype and Tnfr-deficient mice demonstrated that soluble TNF mediates most inflammatory effects in glomeruli via TNFR1-dependent mRNA induction of proinflammatory mediators, including adhesion molecules and chemokines. TNFR2 contributed to TNFR1-dependent effects after stimulation with low TNF concentrations. In addition, we identified a posttranscriptional role of TNFR2 in enhancing chemokine secretion. Consistent with these in vitro results, we demonstrated a predominant role of TNFR1 in mediating soluble TNF-induced glomerular leukocyte accumulation in vivo, with a selective contribution of TNFR2 to the accumulation of glomerular mononuclear phagocytes. Together, these data suggest that activation of intrinsic glomerular cells by soluble TNF requires TNFR1, whereas TNFR2 augments TNFR1-dependent effects. In contrast, no exclusively TNFR2-dependent inflammatory pathways were identified in soluble TNF-stimulated glomeruli. These data suggest that mice deficient for renal cell-expressed TNFR2 are protected from GN in vivo [20] as a result from blocked TNFR2 activation by membrane-bound TNF, but not soluble TNF in intrinsic glomerular cells.
Materials and Methods
Animals
Mouse strains with targeted deletion of TNFR1 (Tnfr1−/−, B6.129-Tnfrsf1atm1Mak) and 2 (Tnfr2−/−, B6.129-Tnfrsf1btm1Mwm) were originally purchased from the Jackson Laboratory (Bar Harbor, ME, USA) and bred in our Central Animal Facility (University Hospital Innenstadt, Ludwig-Maximilians-University Munich) under specific pathogen-free conditions. Tnfr1 and 2 double-deficient mice (Tnfr1,2−/−) and C57BL/6J wildtype controls were generated by cross-breeding of single Tnfr-deficient mice. Renal leukocyte infiltration was induced in 6 to 8 week old male mice by intraperitoneal injection of 5 µg recombinant murine TNF (Invitrogen, Karlsruhe, Germany). Perfused kidneys were harvested eight hours after TNF injection. Untreated male mice were used for paramagnetic isolation of glomeruli subsequently analyzed in ex vivo experiments. All experimental procedures involving mice were approved by the Ethical Committee of the Regierung von Oberbayern (AZ 55.2-1-54-2532.3-8-12).
Paramagnetic Isolation of Glomeruli
Isolation of glomeruli from mouse kidneys was essentially performed as described by Takemoto and coworkers [21]. Briefly, M-450 tosylactivated, 4.5 µm diameter Dynabeads (Invitrogen, Karlsruhe, Germany) were inactivated according to the manufacturer`s instructions. Mice were perfused with 8×107 paramagnetic Dynabeads diluted in 40 ml of phosphate-buffered saline (PBS; Pan Biotech, Aidenbach, Germany) through the heart with a constant pressure of 40 mmHg using a custom-made, pressure-controlled syringe. Kidneys were digested with 1 mg/ml collagenase A (Roche Diagnostics, Mannheim, Germany) in 1 ml Hanks` balanced salt solution (HBSS; Pan Biotech) at 37°C for 30 minutes. The digested tissue was passed through a 100 µm cell strainer (BD Biosciences, Heidelberg, Germany). The cell suspension containing glomeruli with trapped paramagnetic Dynabeads was placed into a magnetic particle concentrator (Invitrogen) for 7 minutes. The first supernatant was pipetted into a separate tube and stored on ice. The remaining tissue was resuspended in 10 ml PBS, and placed again into the magnetic particle concentrator for 5 minutes. The supernatant was removed and remaining paramagnetically labelled glomeruli were resuspended in 4 ml PBS and passed through a 100 µm cell strainer. This suspension was placed again in the magnetic particle separator for 5 minutes, the supernatant discarded, and the remaining tissue was washed three times with 10 ml PBS. Finally, purified glomeruli were resuspended in 1.5 ml of PBS. All procedures were performed on ice except the collagenase digestion at 37°C. Samples of all obtained fractions were viewed microscopically. The final paramagnetically labelled fraction contained highly purified glomeruli with a yield of approximately 10,000 glomeruli per mouse kidney and a purity >97% (intact glomeruli versus tubular fragments or single cells). As previously described, isolated glomeruli were lacking the Bowman`s capsule [21]. The first supernatant was free of glomeruli, but contained tubular fragments, single tubular cells and polymorphic interstitial cells.
Cell Culture
A murine glomerular endothelial cell line was originally derived from tsA58 immorto mice [22] and generously provided by Dr. Nese Akis (Division of Microbiology, Uludag University, Bursa, Turkey). Cells were grown in DMEM medium with GlutaMax (Invitrogen) supplemented with 10% fetal calf serum (FCS; Biochrom, Berlin, Germany), 100 U/ml penicillin and 100 µg/ml streptomycin (PAA Laboratories, Pasching, Austria). The endothelial cells were characterized by positive immunostaining for CD31, von Willebrand factor, and cytokeratin 18. A murine mesangial cell line was maintained in DMEM supplemented with 2.5% FCS and penicillin-streptomycin as described [23]. Reaching 80% confluency cells were serum starved for 24 hours and subsequently stimulated with murine TNF (Invitrogen) or IFN-γ (PeproTech, Rocky Hill, NJ, USA) in DMEM for 5 hours (endothelial cells) or 24 hours (mesangial cells).
Primary murine mesangial cells (pMC) were prepared from C57BL/6J wildtype and Tnfr-deficient mice following paramagnetic isolation of glomeruli as previously described [24]. After five passages, pMC were characterized by positive immunostaining for α-smooth muscle actin and desmin, and by negative staining for CD31, von Willebrand factor and cytokeratin 18. Being 80% confluent pMC from passages 7–8 were serum starved for 24 hours and subsequently stimulated with indicated concentrations of murine TNF in RPMI 1640 for 12 hours.
Glomerular Culture and Stimulation
After isolation 5,000 intact glomeruli were resuspended in 3 ml RPMI 1640 supplemented with 15% FCS, 15 mM HEPES buffer (Invitrogen), 0.66 U/ml insulin and penicillin-streptomycin, and were incubated in 6 well plates at 37°C for 24 hours. After serum starving for further 24 hours glomeruli were stimulated with indicated concentrations of murine TNF in RPMI 1640 for 12 or 24 hours.
Microarray Studies
For microarray experiments 5,000 glomeruli isolated from male C57BL/6J wildtype and Tnfr-deficient mice were serum starved and subsequently stimulated with 50 ng/ml TNF. After 12 hours total RNA was prepared using RNeasy Mini Kit (Qiagen, Hilden, Germany). Integrity of each RNA sample was tested by denaturing agarose gel electrophoresis. Three independent preparations of 2 µg total RNA per genotype was used for biotin-labeled complementary RNA probe synthesis and hybridization of Affymetrix Mouse Genome 430 2.0 arrays according to the Affymetrix Expression Analysis Technical Manual. Triplicate arrays per genotype were scanned and analyzed using the Affymetrix GeneChip Operating Software (GCOS1.0). The complete data set was deposited into the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/; submission no. GSE43928).
For microarray analysis robust multichip average (RMA) [25] was performed. All twelve Affymetrix Microarray CEL files were normalized together using RMA Express, version 1.0 beta 2 (http://rmaexpress.bmbolstad.com/). For probe set annotation the default Mouse 430_2.cdf was used. Following normalized RMA, significance analysis of microarrays (SAM) was applied using the Microsoft Excel plugin of SAM, version 1.21 [26] to analyze differentially expressed transcripts in Tnfr1,2−/−, Tnfr1−/− and Tnfr2−/− glomeruli compared to wildtype. A q-value <5% was used to identify probe sets that detected transcipts with different expression levels in the analyzed groups. If the same gene was identified by multiple probe sets, fold-changes to wildtype were calculated from the averaged signal intensities of the respective probe sets. Genes with an at least 1.5-fold difference in mRNA abundance were considered as differentially expressed between groups.
Ranking of Differentially Expressed Genes by Gene Ontology
The Database for Annotation, Visualization and Integrated Discovery (DAVID; accessible at http://david.abcc.ncifcrf.gov) [27], [28] was used to group functionally related genes. DAVID gene functional classification was applied to rank the overall importance of functional gene groups that reached an enrichment score of ≥1.3. To identify over-represented biological categories (gene ontology classes) DAVID functional annotation chart was applied using the functional annotation categories of the Gene Ontology Consortium for cellular components, biological processes, and molecular function [27], [29]. Based on the Expression Analysis Systematic Explorer (EASE) [30], annotation terms with EASE scores of p ≤0.05, fold enrichment ≥1.5, and at least 4 genes per group were considered significant for over-representation in the given gene list [27]. For all analysis, redundant probe sets for a given gene were removed before analysis to prevent statistical biasing.
RNA Isolation and qRT-PCR
Total RNA from cells and isolated tissue was prepared using the RNeasy Mini Kit (Quiagen). Complementary DNA was generated from 500 ng of total RNA by reverse transcription (RT) using random priming and MMLV reverse transcriptase (Invitrogen).
TaqMan real-time PCR was performed on an AB Prism 7000 analyser (Applied Biosystems, Weiterstadt, Germany) using heat-activated TaqDNA polymerase (Amplitaq Gold, Applied Biosystems). For murine nephrin (NM_019459), VCAM-1 (NM_011693), P-selectin (NM_011347), and GAPDH (NM_008084) the following oligonucleotide primers (300 nmol/L) and hydrolysis probes (100 nmol/L) were used (all from Applied Biosystems): murine nephrin, sense primer 5′-ACCCTCCAGTTAACTTGTCTTTGG-3′, antisense primer 5′-ATGCAGCGG AGCCTTTGA-3′, fluorescence labelled probe (FAM) 5′-TCCAGCCTCTCTCC-3′; murine VCAM-1, sense primer 5′-AACCCAAACAGAGGCAGAGTGTAC-3′, antisense primer 5′-GACCCAGATGGTGGTTTCCTT-3′, fluorescence labelled probe (FAM) 5′-TGTCAACGTTGCCCC-3′, murine P-selectin, sense primer 5′-ATGAA CCCTGTTTTAAACGAAAGC-3′, antisense primer 5′-CTTGGTTGCTGCAGGAC ATG-3′, fluorescence labelled probe (FAM) 5′-ACACAGCCTCCTGCC-3′, and murine Gapdh, sense primer 5′-CATGGCCTTCCGTGTTCCTA-3′, antisense primer 5′-ATGCCTGCTTCACCACCTTCT-3′, fluorescence labelled probe (VIC) 5′-CCCAA TGTGTCCGTCGTGGATCTGA-3′: For murine FXYD2 domain-containing ion transport regulator 2 (Fxyd2; NM_007503), TNFR1 (NM_011609), TNFR2 (NM_011610), CCL2/MCP-1 (NM_011333), CCL3/MIP-1α (NM_011337), CCL5/RANTES (NM_013653), ICAM-1 (NM_010493), E-selectin (NM_011693), and 18S rRNA commercially available pre-developed TaqMan reagents (Applied Biosystems) were used. For some target genes amplification and PCR product detection was performed using SYBR Green PCR Master Mix reagents. The SYBR Green forward (f) and reverse (r) oligonucleotide primer sequences were as follows: murine Rab6B, 5′- GGTTGCCTGGTAGGTGTTGT-3′ (f), 5′- GCTGCGAAAATTC AAGTTGG-3′ (r); and 18S rRNA, 5′-GCAATTATTCCCCATGAACG-3′ (f), 5′-AGG GCCTCACTAAACCATCC-3′ (r). The expression of candidate genes was normalized to reference genes GAPDH or 18S rRNA. The mRNA expression was analyzed by standard curve quantification. All measurements were performed in duplicates. Controls consisting of bidistilled H2O and RT− controls were negative in all runs.
Immunohistochemistry
Protein expression of renal TNFR1 and TNFR2 was detected on 4 µm cryostat sections of OCT-embedded tissue stained with polyclonal goat anti-mouse TNFR1 and polyclonal goat anti-mouse TNFR2 antibodies (both from R&D Systems, Wiesbaden, Germany). Specificity of the antisera was shown by negative staining of tissue sections from Tnfr1- and Tnfr2-deficient mice, respectively.
Detection of TNF Receptor Surface Expression by Flow Cytometry
To detect TNFR1 surface expression glomerular endothelial or mesangial cells were incubated in fluorescence-activated cell sorting (FACS) buffer (PBS, 0.2% bovine serum albumin, 0.1% NaN2) with a monoclonal hamster anti-mouse TNFR1 antibody (clone 55R-286; BD Biosciences), followed by a biotin-labeled mouse anti-hamster IgG cocktail (BD Biosciences). Cells were then stained with phycoerythrin (PE)-conjugated streptavidin (BD Biosciences). Hamster IgG1 was used as the appropriate isotype control. TNFR2 expression was analyzed with a PE-conjugated monoclonal hamster anti-mouse TNFR2 antibody (clone TR75-89; BD Bioscience). PE-conjugated hamster IgG1 was used as isotype control.
Chemokine ELISA
Chemokine levels in glomerular culture supernatants were determined using commercially available ELISA kits for CCL2/MCP-1, CCL5/RANTES and CXCL10/IP-10 (R&D Systems) following the manufacturer’s protocols.
Quantification of Glomerular Leukocyte Subsets by Four-color Flow Cytometry
Single-cell suspensions from glomerular tissue preparations of individual kidneys were prepared as previously described [31]. Cells were incubated in FACS buffer on ice with combinations of fluorochrome-labeled antibodies to murine leukocyte surface markers including PE-conjugated anti-CD45 (clone 30-F11), FITC-conjugated Ly-6G (clone 1A8), FITC-conjugated CD3ε (clone 145-2C11), allophycocyanin (APC)-conjugated CD4 (clone RM4-5), PE-Cy5-conjugated CD8α (clone 53-6.7) (all from BD Biosciences), and APC-conjugated F4/80 (clone CL:A3-1; from AbD serotec, Oxford, UK). Stained cells were analysed with a FACS Calibur™ flow cytometer and Cellquest™ software (BD Biosciences). The number of positively stained cells was expressed as percentage of total glomerular cells.
Statistical Analysis
Numerical results of each experimental group were expressed as mean±SD or SE as indicated, and were compared using two-sided non-parametric Mann-Whitney U test. When more than 2 experimental groups were compared, Kruskal-Wallis test was carried out, followed by pairwise Mann-Whitney U tests. Significance was assigned to P<0.05.
Results
Expression of TNFR1 and TNFR2 in Mouse Kidney and Intrinsic Glomerular Cells
Glomeruli were isolated from 6 to 8 week old wildtype mice after perfusion with paramagnetic Dynabeads (Figure S1A). The first supernatant obtained during the washing procedure consisted of a tubulointerstitial tissue fraction that was free of glomeruli (Figure S1B). To assure the glomerular and tubulointerstitial origin of the two tissue fractions mRNA expression of glomerular and tubular marker genes was determined examining nephrin and FXYD2, the γ-subunit of the tubular Na,K-ATPase, respectively (Figure S1C, S1D).
Compartment-specific quantitative PCR revealed a constitutive expression of TNFR1 and TNFR2 in normal mouse glomeruli, with substantially lower transcript levels in tubulointerstitial tissue (Figure 1A). Consistently, immunohistochemistry demonstrated a prominent protein expression of TNFR1 and TNFR2 in glomeruli, but only weak expression in the tubulointerstitium of normal mice (Figure 1B).
Figure 1. Expression of TNFR1 and TNFR2 in mouse kidney.

A: Compartment-specific expression of TNFR1 and TNFR2 mRNA in glomeruli and tubulointerstitial tissue isolated from normal mouse kidney as analyzed by quantitative real-time PCR. Values were normalized to rRNA expression used as reference gene. ∗ p<0.05. B: Immunohistochemistry on frozen sections for TNFR1 and TNFR2 protein demonstrates a prominent glomerular expression of both receptors (black color product) in normal mouse kidney. Original magnification x200.
In vitro, glomerular endothelial (Figure 2A, 2B) and mesangial cells (Figure 2C, 2D) constitutively expressed mRNA of both TNFRs, with a substantially lower abundance of TNFR2 mRNA. After stimulation of both cell types with TNF and IFN-γ, TNFR1 mRNA was only upregulated after challenge with IFN-γ or TNF in combination with IFN-γ (Figure 2A, 2C). In contrast, TNFR2 expression was readibly inducable after incubation with TNF, IFN-γ or combined stimulation (Figure 2B, 2D). As TNF receptors are shed from the cell surface following ligand binding and activation we next analyzed surface expression of TNFR1 and TNFR2. In glomerular endothelial cells cytokine stimulation decreased surface expression of both TNFRs (Figure 2E, 2F). In contrast, TNFR1 and TNFR2 were not shed from the surface of mesangial cells upon stimulation, and surface expression of TNFR2 increased after combined stimulation with TNF and IFN-γ (Figure 2G, 2H), suggesting a robust mesangial TNFR signaling capacity in inflammatory conditions.
Figure 2. Expression of TNFR1 and TNFR2 in glomerular endothelial and mesangial cells in vitro.

A: Expression of TNFR1 mRNA in unstimulated, TNF-stimulated and IFN-γ-stimulated glomerular endothelial cells. Cells were stimulated with indicated concentrations of TNF and/or IFN-γ for 5 hours. B: Expression of TNFR2 mRNA in unstimulated, TNF-stimulated and IFN-γ-stimulated glomerular endothelial cells. Cells were stimulated as in A. C: Expression of TNFR1 mRNA in unstimulated, TNF-stimulated and IFN-γ-stimulated mesangial cells. Cells were stimulated with indicated concentrations of TNF and/or IFN-γ for 24 hours. D: Expression of TNFR2 mRNA in unstimulated, TNF-stimulated and IFN-γ-stimulated glomerular endothelial cells. Cells were stimulated as in C. mRNA expression levels were analyzed by quantitative real-time PCR. Values were normalized to GAPDH expression used as reference gene. ∗ p<0.05, ∗∗ <0.01 (A-D). E: TNFR1 surface expression in unstimulated, TNF-stimulated and IFN-γ-stimulated glomerular endothelial cells in vitro analyzed by flow cytometry. Cells were stimulated with indicated concentrations of TNF and/or IFN-γ for 5 hours. F: TNFR2 surface expression in unstimulated, TNF-stimulated and IFN-γ-stimulated glomerular endothelial cells. Cells were stimulated as in E. G: Flow cytometric analysis of TNFR1 surface expression in unstimulated, TNF-stimulated and IFN-γ-stimulated mesangial cells in vitro. Cells were treated with indicated concentrations of TNF and IFN-γ for 24 hours. H: TNFR2 surface expression in unstimulated, TNF-stimulated and IFN-γ-stimulated mesangial cells. Cells were stimulated as in G. The percentage of TNFR-positive cells was determined in comparison to the respective isotype controls. ∗ p<0.05 versus medium control (E-H). Data are mean and SD of 3 to 6 independent experiments per group.
Together, these data demonstrate a constitutive glomerular expression of both TNFRs in normal mouse kidneys, which can be induced by proinflammatory stimuli, with TNFR2 being more readily upregulated than TNFR1.
TNF-induced Expression of Glomerular Adhesion Molecules and Chemokines Correlates with Glomerular Leukocyte Infiltration in vivo
8 hours after intraperitoneal injection of 5 µg TNF, glomerular mRNA expression of adhesion molecules and chemokines substantially increased, with the highest induction (42.5-fold) seen for the proinflammatory chemokine CCL2/MCP-1 (Figure 3A). This was associated with glomerular influx of CD45+ leukocytes, mainly Ly6C+ neutrophils and F4/80+ mononuclear phagocytes, as revealed by compartment-specific flow cytometry (Figure 3B). These data demonstrate that TNF-induced expression of glomerular adhesion molecules and chemokines results in a rapid glomerular influx of leukocytes, which may substantially contribute to the glomerular production of inflammatory mediators. Thus, analysis of TNF-exposed glomeruli in vivo would not allow the characterization of local TNFR-mediated inflammatory responses in intrinsic glomerular cells as opposed to infiltrating leukocytes.
Figure 3. TNF-induced expression of glomerular adhesion molecules and chemokines correlates with leukocyte infiltration in vivo.

A: Glomerular expression of adhesion molecules and proinflammatory chemokines in glomeruli isolated from control mice (white bars) and from mice 8 hours after intraperitoneal injection of 5 µg TNF (grey bars). mRNA levels were determined by quantitative real-time PCR, and values were normalized to 18S rRNA as reference gene. Results are expressed as fold change compared to control glomeruli. Data shown are means and SE of n = 3 to 6 per group; ∗ p<0.05 versus control. B: Glomerular leukocyte infiltration 8 hours after intraperitoneal injection of TNF (5 µg) was analysed by compartment-specific flow cytometry. Leukocyte subpopulations were identified as follows: leukocytes: CD45+, neutrophils: CD45+ Ly6G+ F4/80−, F4/80 positive mononuclear phagocytes: CD45+ F4/80+ Ly6G−, and T cells: CD45+ CD3ε+. Leukocyte numbers are expressed as percentage of all glomerular cells. ∗ p<0.05 versus control.
Analysis of TNFR1- and TNFR2-specific Inflammatory Responses in TNF-stimulated Glomeruli ex vivo by Microarray Expression Profiling
To examine TNFR-specific inflammatory responses in intrinsic glomerular cells but not infiltrating leukocytes we stimulated intact glomeruli isolated from wildtype and Tnfr-deficient mice with soluble TNF ex vivo. Global gene expression levels were determined on Affymetrix Mouse Genome 430 2.0 gene chips. Representative genes differentially expressed in TNF-stimulated Tnfr1,2−/−, Tnfr1−/− and Tnfr2−/− glomeruli compared to wildtype are listed in Table S1. A comprehensive summary of all differentially expressed gene sets and genes in Tnfr1,2−/−, Tnfr1−/−, and Tnfr2−/− glomeruli compared to wildtype is shown in Table S2, S3, S4).
In Tnfr1,2−/− glomeruli, i.e. glomeruli without TNF-signaling capacity, we detected 290 differentially expressed genes compared to wildtype glomeruli following TNF stimulaton, with 219 genes being down-regulated, and 71 genes with up-reglulated expression (Figure 4A, 5, Table S2). Among these, 168 genes were regulated similarly (i.e. up- or down-regulated) as in Tnfr1−/− glomeruli (Figure 4B, 6A, Table S3), without altered expression in Tnfr2−/− glomeruli. Expression of 5 genes was decreased in Tnfr1,2−/− as well as Tnfr2−/− glomeruli (Figure 4C, 6B, Table S4), but not in Tnfr1−/−. The remaining 117 genes were differentially expressed in Tnfr1,2−/− glomeruli only. However, as shown in Figure 5 all of these genes had a trend towards similar regulation in Tnfr1−/− glomeruli, with mean fold changes of expression levels mostly >1.5, although not reaching statistical significance. In contrast, these genes were not differentially expressed in Tnfr2−/− glomeruli compared to wildtype (Figure 5). Functional groups of genes expressed in a TNFR1,2-dependent way included cell adhesion proteins, proinflammatory chemokines, cytokines and cytokine receptors, innate immune effectors, antigen presentation proteins, components of the NF-κB signaling cascade, matrix metalloproteinases (MMPs), transport proteins, and signalling molecules (see Table S1), indicating a robust proinflammatory stimulation of wildtype glomeruli by TNF.
Figure 4. Differentially expressed genes in Tnfr-deficient glomeruli compared to wildtype after TNF stimulation.
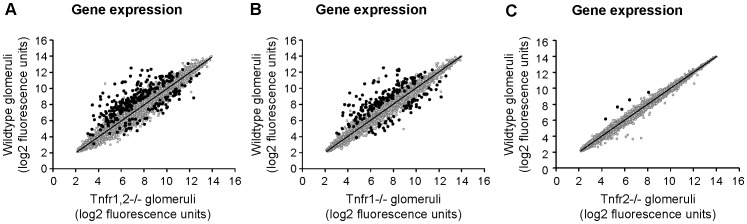
Expression profiling by microarrays was performed on isolated glomeruli from wildtype, Tnfr1,2−/−, Tnfr1−/− and Tnfr2−/− mice after stimulation with 50 ng/ml TNF for 12 hours ex vivo. A: Significance analysis using SAM detected differential regulation of 290 unique genes in Tnfr1,2−/− glomeruli with at least 1.5-fold difference compared to wildtype (black data points). 219 genes were down-regulated (upper left), and 71 genes were up-regulated (lower right). B: In Tnfr1−/− mice 219 differentially regulated genes were identified, with lower expression of 159 genes and increased expression of 60 genes. C: In Tnfr2-deficient glomeruli only 5 genes were differentially expressed, all of them with decreased expression compared to wildtype. In all three genotypes, expression of the majority of genes with fluorescence signals above background level did not significantly differ from wildtype (grey data points). Data represent the mean log2 value of fluorescence signals from three independent experiments per genotype.
Figure 5. Gene expression profiles in wildtype and Tnfr1,2-deficient glomeruli after TNF stimulation ex vivo.

Expression profiling by microarrays was performed on isolated glomeruli from wildtype and Tnfr1,2−/− mice 12 after stimulation with 50 ng/ml TNF for 12 hours ex vivo. A negativ “fold change” (black to green) indicates decreased expression and a postive “fold change” (black to red) indicates increased expression in a glomerular sample compared with the average of all analyzed samples. The 290 differentially regulated genes identified in Tnfr1,2−/− glomeruli are listed according to fold-change values versus wildtype, starting with the most down-regulated gene (CCL2; -50.0-fold). Note that almost all genes found significantly regulated in Tnfr1,2−/− glomeruli had a similar trend of expression change in Tnfr1−/− glomeruli, although not reaching statistical significance in Tnfr1−/− glomeruli for several genes. Table S2 lists all differentially expressed genes with respective probe set IDs, gene bank accession numbers, gene names and fold-changes according to fold-change values compared to wildtype.
Figure 6. Gene expression profiles in wildtype, Tnfr1- and Tnfr2-deficient glomeruli after TNF stimulation ex vivo.

Expression profiling by microarrays was performed on isolated glomeruli from wildtype, Tnfr1−/− and Tnfr2−/− mice 12 after stimulation with 50 ng/ml TNF for 12 hours ex vivo. A negativ “fold change” (black to green) indicates decreased expression and a postive “fold change” (black to red) indicates increased expression in a glomerular sample compared with the average of all analyzed samples. A: Compared to wildtype 219 significantly regulated genes were found in Tnfr1−/− glomeruli. The number of genes identified in Tnfr1−/− glomeruli was lower than in Tnfr1,2−/− glomeruli, but all genes differentially expressed in Tnfr1−/− glomeruli were regulated similarly in Tnfr1,2−/− glomeruli, although for some genes pair-wise comparison between Tnfr1,2−/− to wildtype did not reach significance or was below the pre-defined 1.5-fold cut-off (see also Table S3). B: 5 differentially expressed genes in Tnfr2−/− glomeruli are listed according to fold-change values compared to wildtype. These genes are not differentially expressed in Tnfr1−/− glomeruli, but 4 of the 5 genes are similarly regulated in Tnfr1,2−/− mice. Tables S3 (Tnfr1−/−) and S4 (Tnfr2−/−) list all differentially expressed genes with respective probe set IDs, gene bank accession numbers, gene names and fold-changes according to fold-change values compared to wildtype.
Comparing TNF-stimulated wildtype and Tnfr1−/− glomeruli, we identified 219 unique genes being differentially expressed between the two genotypes (Figure 4B, 6A, Table S3). Of these, 159 genes were down-regulated in Tnfr1−/− glomeruli, and 60 were up-regulated. None of these genes was differentially expressed in Tnfr2−/− glomeruli following TNF-stimulation. Representative genes differentially expressed in Tnfr1−/− glomeruli are shown in Table S1. The proinflammatory chemokine CCL2/MCP-1 was the most prominently down-regulated gene (-42.9-fold) in Tnfr1−/− glomeruli. Of note, the magnitude of this differential expression was similar to our in vivo result showing a 42.5-fold induction of glomerular CCL2/MCP-1 mRNA after intraperitoneal TNF injection in wildtype mice (Figure 3A). TNFR1-dependently expressed genes included several cell adhesion molecules, proinflammatory chemokines of the CXC and CC subfamilies, and prototypic proinflammatory cytokines like pro-interleukin (IL)-1β and IL-6, all of which were down-regulated in Tnfr1−/− glomeruli. These data suggest a prominent role for TNFR1 in mediating glomerular leukocyte accumulation in TNF-stimulated glomeruli. Additionally, effectors and receptors of the innate immune system, including complement components, the lipopolysaccharide binding protein, toll-like receptor (TLR) 2, and proteolytic enzymes like MMP 3, 9, and 13 were down-regulated in Tnfr1-deficient glomeruli. Many TNFR1-induced proinflammatory responses are mediated through NF-κB signaling pathways, and expression of various components and regulators of the NF-κB signaling cascade was reduced in Tnfr1−/− glomeruli. Besides proinflammatory genes, several mediators of apoptosis (including Fas and caspase 12) were expressed TNFR1-dependently.
In contrast to Tnfr1−/−, only 5 genes were found to be differentially expressed in Tnfr2−/− glomeruli, all of which were down-regulated compared to wildtype (Figure 4C, Figure 6B, see also Table S1, Table S4). These 5 genes were not differentially expressed in Tnfr1−/− glomeruli, but expression of 4 of the 5 genes was also reduced in Tnfr1,2−/− glomeruli (Table S1). Among the 4 TNFR2-dependently expressed genes (excluding TNFR2) we identified a 2.6-fold reduced expression of the small GTPase Rab6B in Tnfr2−/− glomeruli (Table S1, Table S4).
In composite, the array data suggest a prominent role of TNFR1, in contrast to TNFR2, in mediating proinflammatory effects in glomeruli exposed to soluble TNF.
Enriched Gene Groups and Gene Ontology Classes within TNFR-dependently Expressed Genes
To further characterize functional roles of TNFR-dependently expressed genes we analysed Tnfr1,2−/− and Tnfr1−/− regulated gene sets using DAVID and the EASE bioinformatic algorithms. Due to the few genes identified in Tnfr2−/− glomeruli, this analysis was not performed on the Tnfr2−/− specific gene set. The enriched functional gene groups differentially expressed in Tnfr1,2−/− glomeruli illustrate a robust TNF-mediated proinflammatory response in intrinsic glomerular cells (Table S5). Chemokines and cytokines constituted the most enriched group (enrichment score 8.86), with all members being downregulated in Tnfr1,2−/− glomeruli compared to wildtype. Applying the functional annotation chart feature of DAVID we identified 136 significantly enriched (i.e. overrepresented) gene ontology categories associated with the differentially regulated genes in Tnfr1,2−/− glomeruli. Many of the 10 most enriched gene ontology categories in Tnfr1,2−/− glomeruli were immune-related, including chemokine activity and chemokine receptor binding (Table 1).
Table 1. Overrepresented gene ontology terms within differentially expressed genes in TNF-stimulated Tnfr1,2−/− gomeruli 1.
| GO term ID | GO term name | Fold enrichment | P-value | Gene count | % of regulated genes |
| GO:0008009 | chemokine activity | 19.7 | 1.29E-10 | 11.0 | 3.57 |
| GO:0042379 | chemokine receptor binding | 19.2 | 1.70E-10 | 11.0 | 3.57 |
| GO:0001664 | G-protein-coupled receptor binding | 12.0 | 2.26E-08 | 11.0 | 3.57 |
| GO:0002474 | antigen processing and presentation of peptideantigen via MHC class I | 11.0 | 5.36E-03 | 4.0 | 1.30 |
| GO:0000270 | peptidoglycan metabolic process | 9.5 | 8.31E-03 | 4.0 | 1.30 |
| GO:0006958 | complement activation, classical pathway | 9.1 | 9.17E-03 | 4.0 | 1.30 |
| GO:0042108 | positive regulation of cytokine biosynthetic process | 8.8 | 5.48E-04 | 6.0 | 1.95 |
| GO:0008217 | regulation of blood pressure | 8.6 | 6.08E-04 | 6.0 | 1.95 |
| GO:0050727 | regulation of inflammatory response | 8.6 | 1.10E-02 | 4.0 | 1.30 |
| GO:0031347 | regulation of defense response | 8.6 | 1.10E-02 | 4.0 | 1.30 |
The 10 most overrepresented gene ontology (GO) terms are listed according to their enrichment scores. Terms with an EASE score of p ≤0.05, fold enrichment ≥1.5, and at least 4 genes per group were considered significant for overrepresentation.
Enriched gene groups in Tnfr1−/− glomeruli were similar to Tnfr1,2−/− (Table S6). Again, chemokines and cytokines were the group with the most significant differential expression (enrichment score 7.61). Compared to Tnfr1,2−/− glomeruli DAVID analysis identified fewer, but substantially overlapping enriched biological terms in Tnfr1−/− glomeruli, with 77 overrepresented categories being associated with the differentially regulated genes. Among the 10 most enriched gene ontology categories in Tnfr1−/− glomeruli (Table 2) chemokine activity, chemokine receptor binding, G-protein-coupled receptor binding, antigen processing and presentation, and regulation of blood pressure were identical to those found in Tnfr1,2−/− glomeruli. Additional highly enriched gene onotology categories were again immune-related, such as acute phase response, positive regulation of the I-κB kinase/NF-κB cascade, acute inflammatory response, and chemotaxis (Table 2).
Table 2. Overrepresented gene ontology terms within differentially expressed genes in TNF-stimulated Tnfr1−/− glomeruli 1.
| GO term ID | GO term name | Fold enrichment | P-value | Gene count | % of regulated genes |
| GO:0008009 | chemokine activity | 22.8 | 3.54E-10 | 10 | 4.26 |
| GO:0042379 | chemokine receptor binding | 22.3 | 4.53E-10 | 10 | 4.26 |
| GO:0002474 | antigen processing and presentation of peptideantigen via MHC class I | 14.1 | 2.69E-03 | 4 | 1.70 |
| GO:0001664 | G-protein-coupled receptor binding | 13.9 | 3.62E-08 | 10 | 4.26 |
| GO:0006953 | acute-phase response | 12.5 | 3.79E-03 | 4 | 1.70 |
| GO:0008217 | regulation of blood pressure | 9.2 | 2.05E-03 | 5 | 2.13 |
| GO:0043123 | positive regulation of I-κB kinase/NF-κB cascade | 8.5 | 1.15E-02 | 4 | 1.70 |
| GO:0002526 | acute inflammatory response | 8.3 | 4.97E-05 | 8 | 3.40 |
| GO:0006935 | chemotaxis | 8.2 | 8.50E-07 | 11 | 4.68 |
| GO:0042330 | taxis | 8.2 | 8.50E-07 | 11 | 4.68 |
The 10 most overrepresented gene ontology (GO) terms are listed according to their enrichment scores. Terms with an EASE score of p ≤0.05, fold enrichment ≥1.5, and at least 4 genes per group were considered significant for overrepresentation.
Taken together, these results suggest a predominant role of TNFR1 in mediating many aspects of soluble TNF-induced inflammation in the glomerulus. In the presence of TNFR1, TNFR2 may additionally contribute to TNF signaling in cooperation with TNFR1, as DAVID analysis identified more enriched gene ontology terms in Tnfr1,2−/− glomeruli compared to Tnfr1−/− glomeruli.
Quantitative RT-PCR Analysis of Differentially Regulated Genes in TNF-stimulated Glomeruli Isolated from Wildtype and Tnfr-deficient Mice
Microarray expression data were validated by qRT-PCR for chemokines, adhesion molecules and Rab6B. CCL2/MCP-1 expression was induced in a dose-dependent fashion in TNF-stimulated wildtype glomeruli, and this induction was completely abrogated in Tnfr1−/− and Tnfr1,2−/− glomeruli at 12 and 24 hours (Figure 7A). In contrast, CCL2 expression was not significantly altered in Tnfr2−/− glomeruli at 12 hours compared to wildtype. However, at 24 hours CCL2 mRNA levels were significantly reduced after exposure of Tnfr2−/− glomeruli to lower TNF concentrations, i.e 10 ng/ml, with a similar trend for 25 ng/ml. TNF stimulation with 50 ng/ml resulted in a comparable induction of CCL2 mRNAs in Tnfr2−/− and wildtype glomeruli (Figure 7A). Similar results were found for CCL5/RANTES (Figure 7B) and the adhesion molecules ICAM-1 (Figure 7C) and VCAM-1 (Figure 7D). We also analyzed the expression pattern of E-selectin (Figure 7E) and P-selectin (Figure 7F) in TNF-stimulated glomeruli of all four genotypes. As shown before in vivo (Figure 3A), we detected an induced expression of both mRNA species in wildtype glomeruli in a dose dependent fashion at 12 hours, but not 24 hours. No induction occurred in Tnfr1−/− and Tnfr1,2−/− glomeruli. This differential expression may have been missed in the less sensitive microarray studies due to relative low expression levels. In summary, qRT-PCR confirmed the microarray data demonstrating a TNFR1-dependent expression of chemokines and adhesion molecules. Importantly, in the presence of TNFR1, TNFR2 additionally contributed to a prolonged expression of inflammatory mediators when glomeruli were exposed to low TNF concentrations.
Figure 7. Validation of the array expression pattern by quantitative RT-PCR.
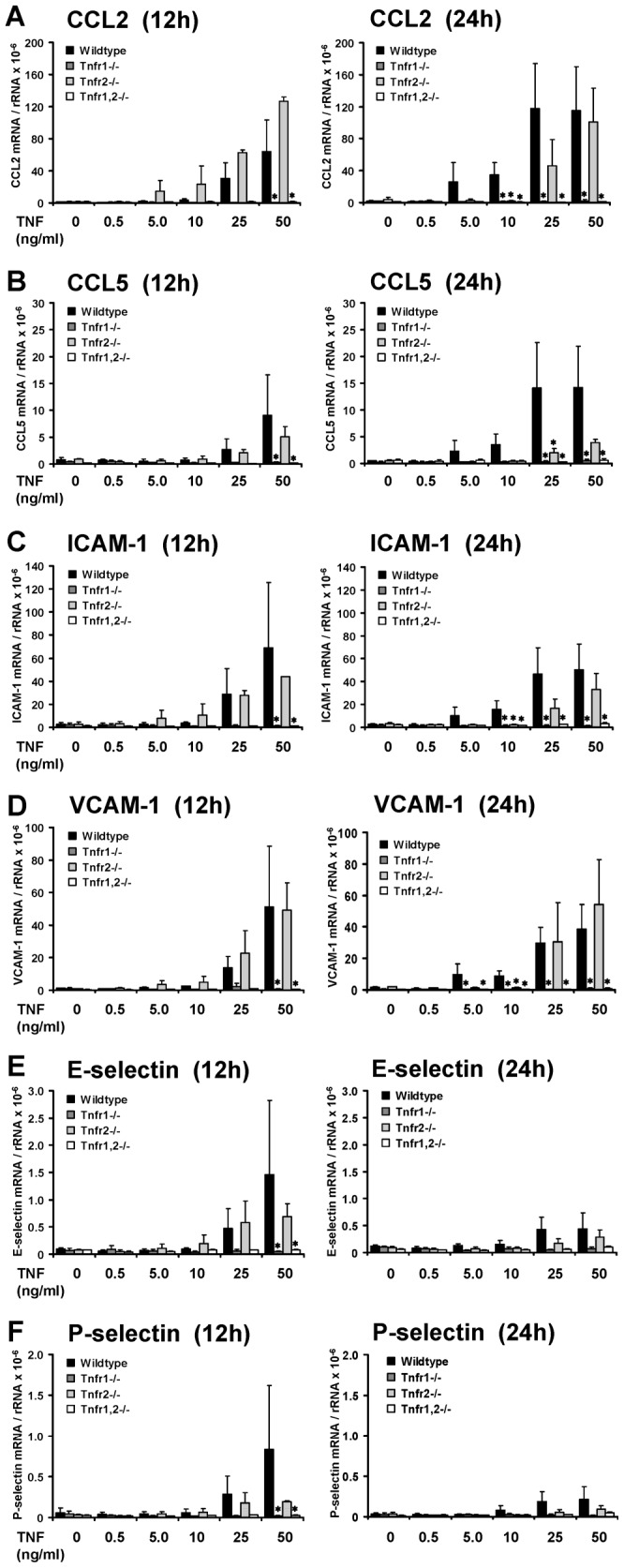
Expression of proinflammatory chemokines and adhesion molecules in glomeruli isolated from control mice (black bars), Tnfr1−/− (dark grey bars), Tnfr2−/− (bright grey bars), and Tnfr1,2−/− mice (white bars) was anlayzed after challenge with indicated concentrations of TNF in vitro for 12 or 24 hours. The mRNA levels of CCL2/MCP-1 (A), CCL5/RANTES (B), ICAM-1 (C), VCAM-1 (D), E-selectin (E), and P-selectin (F) were determined by quantitative real-time PCR, and values were normalized to 18S rRNA as reference gene. Data shown are means and SE of n = 3 to 4 per group; ∗ p<0.05 versus wildtype.
In contrast to the TNFR1-dependent induction of chemokines and adhesion molecules in TNF-stimulated glomeruli, expression of Rab6B was not substantially altered by TNF exposure in all four genotypes (Figure 8). However, in unstimulated Tnfr2−/− and Tnfr1,2−/−, but not Tnfr1−/− glomeruli we detected constitutively low expression levels of Rab6B. Compared to wildtype, glomerular expression of Rab6B remained significantly reduced after TNF stimulation of glomeruli lacking TNFR2 (Figure 8).
Figure 8. Glomerular expression of the small GTPase Rab6B.
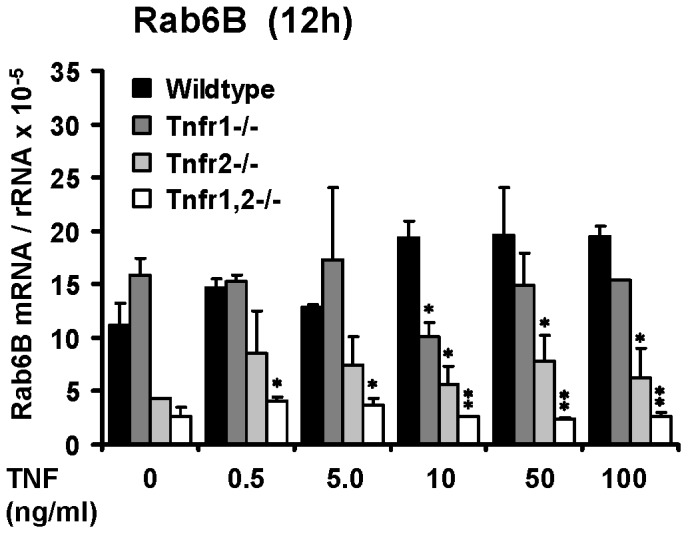
Rab6B expression was analyzed in glomeruli isolated from control mice (black bars), Tnfr1−/− (dark grey bars), Tnfr2−/− (bright grey bars), and Tnfr1,2−/− mice (white bars) after challenge with indicated concentrations of TNF in vitro for 12 or 24 hours. The mRNA levels of Rab6B were determined by quantitative real-time PCR, and values were normalized to 18S rRNA as reference gene. Data shown are means and SE of n = 3 per group; ∗ p<0.05, ∗∗ p<0.01 versus wildtype.
TNF-induced Expression of Chemokines and Adhesion Molecules in Wildtype and Tnfr-deficient Primary Mesangial Cells
Resident mesangial cells may substantially contribute to TNF-induced inflammatory responses in glomeruli. We therefore analyzed TNF-stimulated primary mesangial cells (pMCs) isolated from wildtype and Tnfr-deficient glomeruli. Responses of pMCs to TNF challenge were similar to isolated intact glomeruli. Tnfr1- or Tnfr1,2-deficiency, but not Tnfr2-deficiency prevented TNF-induced expression of chemokines and adhesion molecules (Figure 9A-D). We could not detect a significant expression of E- and P-selectin in unstimulated or TNF-stimulated pMCs (Figure 9E, 9F), although expression of both selectins was induced in TNF-treated intact glomeruli in vivo and in vitro (Figure 3A, Figure 7E, 7F). These data indicate that mesangial cells significantly contribute to TNF-induced expression of inflammatory mediators in the glomerulus, although some TNF effects like up-regulation of selectins are apparently not mediated by mesangial cells but other glomerular cell types.
Figure 9. Expression of proinflammatory chemokines and adhesion molecules in TNF-stimulated primary mesangial cells (pMCs).

pMCs were isolated from control mice (black bars), Tnfr1−/− (dark grey bars), Tnfr2−/− (bright grey bars), and Tnfr1,2−/− mice (white bars) after stimulation with indicated concentrations of TNF in vitro for 12 hours. The mRNA levels of CCL2/MCP-1 (A), CCL5/RANTES (B), ICAM-1 (C), VCAM-1 (D), E-selectin (E), and P-selectin (F) were determined by quantitative real-time PCR, and values were normalized to 18S rRNA as reference gene. Data shown are means and SE of two independently performed experiments.
Reduced Secretion of Chemokines in TNF-stimulated Glomeruli from Tnfr1−/−, Tnfr2−/− and Tnfr1,2−/− Mice
Consistent with the microarray and qRT-PCR data, TNF-induced secretion of CCL2/MCP-1, CCL5/RANTES, and CXCL10/IP-10 in wildtype glomeruli was completely abrogated in Tnfr1−/− and Tnfr1,2−/− glomeruli (Figure 10). Interestingly, TNF-induced chemokine secretion also significantly decreased in Tnfr2−/− glomeruli compared to wildtype (Figure 10), although mRNA expression of chemokines was not significantly different after stimulation with TNF under the same conditions (Figure 7, Table S1). Apparently, TNF-induced secretion of these chemokines is additionally dependent on TNFR2-mediated posttranscriptional modifications.
Figure 10. Secretion of proinflammatory chemokines by TNF-stimulated glomeruli ex vivo.
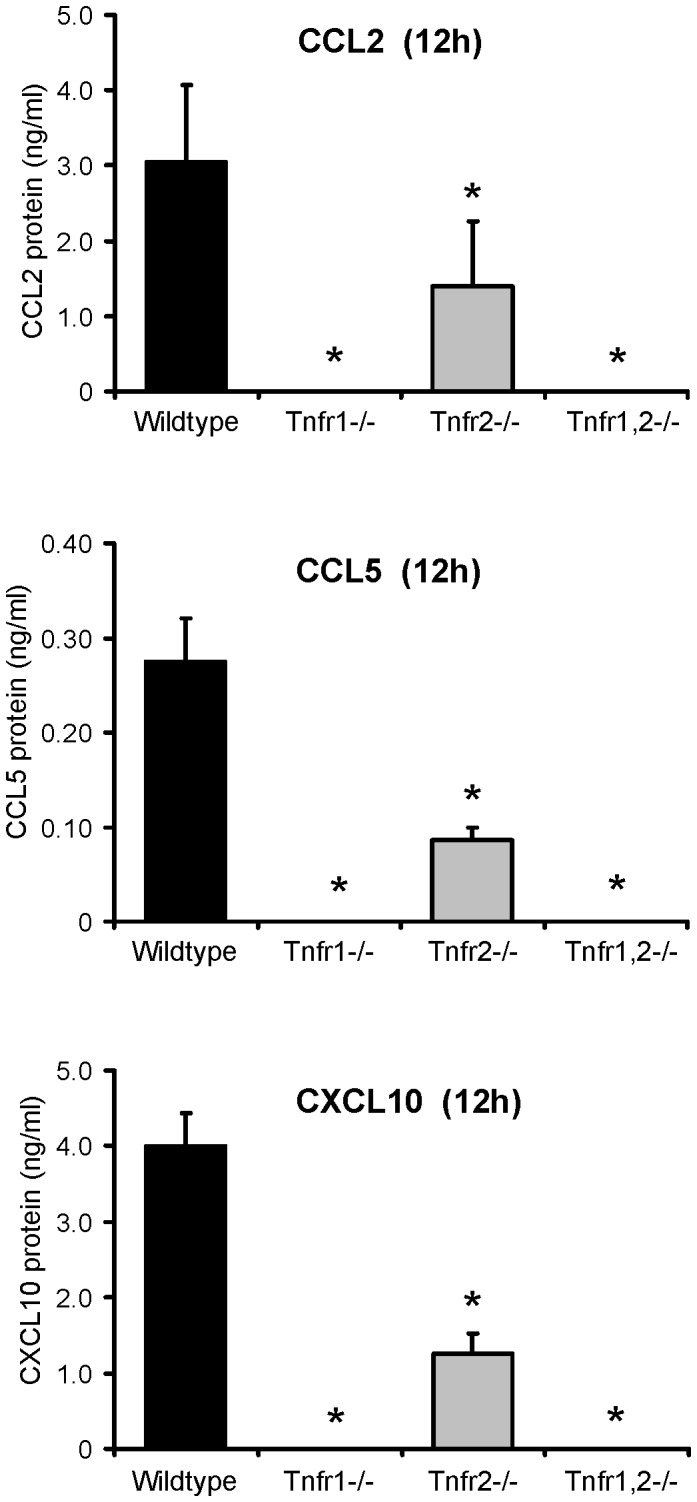
Concentrations of CCL2/MCP-1, CCL5/RANTES, and CXCL10/IP-10 were measured by ELISA in culture supernatants of wildtype, Tnfr1−/−, Tnfr2−/−, and Tnfr1,2−/− glomeruli 12 hours after stimulation with 50 ng/ml of TNF. Data shown are means and SD of n = 3 per group; ∗ p<0.05 versus wildtype.
Functional Roles of TNFR1 and TNFR2 in TNF-mediated Glomerular Inflammation in vivo
Does the predominantly TNFR1-mediated inflammatory response in TNF-stimulated glomeruli in vitro translate into a functional role of TNFR1 in glomerular inflammation in vivo? To address this question we analyzed glomerular leukocyte infiltration in wildtype and Tnfr-deficient mice 8 hours after intraperitoneal TNF injection by compartment-specific flow cytometry. TNF-induced infiltrates of CD45+ leukocytes and Ly6G+ granulocytes were significantly reduced in Tnfr1−/− and Tnfr1,2−/− mice, but not in Tnfr2−/− mice (Figure 11A, 11B). Glomerular accumulation of F4/80+ mononuclear phagocytes decreased in Tnfr1−/−, Tnfr1,2−/− as well as Tnfr2−/− mice, indicating a non-redundant functional role of both TNFRs for glomerular phagocyte infiltration in TNF-challenged glomeruli (Figure 11C). Numbers of glomerular CD3+ T cells were not significantly different in all four genotypes (Figure 11D), consistent with absent T cell infiltrates (Figure 3B) within the eight hour time frame investigated. Together, these experiments reveal a predominant role of TNFR1 in the recruitment of glomerular leukocytes after exposure to soluble TNF in vivo, consistent with the identified in vitro effects of TNFR1 in TNF-challenged glomeruli. In contrast to neutrophils, TNFR2 contributed to the glomerular infiltration of F4/80+ leukocytes. This demonstrates an additional non-redundant role of TNFR2 specifically in the glomerular recruitment of mononuclear phagocytes, possibly due to its contribution to prolonged expression of inflammatory mediators when glomeruli are exposed to low TNF concentrations.
Figure 11. Contribution of TNFR1 and TNFR2 signaling to TNF-mediated infiltration of glomerular leukocytes.

Compartment-specific flow cytometry was performed on glomerular tissue prepared from wildtype, Tnfr1−/−, Tnfr2−/−, and Tnfr1,2−/− mice 8 hours after intraperitoneal injection of 5 µg TNF. Leukocyte subpopulations were identified as follows: A: CD45+ leukocytes, B: CD45+ Ly6G+ F4/80− neutrophils, C: CD45+ F4/80+ Ly6G− mononuclear phagocytes, and D: CD45+ CD3ε+ T cells. Leukocyte numbers are expressed as percentage of all glomerular cells. ∗ p<0.05, ∗∗ p<0.01 versus wildtype.
Discussion
The functional role of TNF in glomerular inflammation has been shown by a variety of experimental studies [1], [2]. More recent data suggested distinct roles of the two TNF receptors in mediating glomerular inflammation, especially in the setting of immune complex GN [20] or collapsing glomerulopathies [32]. Tnfr1-deficiency delayed the onset of GN, but absent TNFR2 signaling in intrinsic renal cells protected from nephritis [20]. These studies demonstrate that resident glomerular cells differently respond to TNFR1 and TNFR2 activation during local, e.g. immune complex-mediated injury. To better understand underlying TNFR-specific inflammatory pathways in parenchymal glomerular cells that would explain the observed in vivo phenotypes in Tnfr-deficient mice, we analysed isolated intact glomeruli stimulated with soluble TNF ex vivo. This approach excluded additional changes of glomerular gene expression by leukocytes, which, as we show, readily accumulated in glomeruli following TNF exposure in vivo.
In comparison of wildtype and Tnfr1,2-deficient glomeruli without TNF signaling capacity, the results of our microarray experiments demonstrate that TNF induced expression of many innate immune effectors in intrinsic glomerular cells. All of these have been shown to be essential for glomerular leukocyte recruitment and activation, including adhesion molecules, chemokines and cytokines [33], [34], [35], [36], components of the complement system [37], [38], and metalloproteinases [39], [40]. Many inflammatory functions of TNF are induced via activation of the NF-κB signaling cascade. TNF treatment induced expression of several NF-κB transcription family members in wildtype glomeruli, as compared to Tnfr1,2−/− glomeruli, creating pathogenic feed-forward NF-κB loops in TNF-stimulated glomeruli.
When we compared TNF-induced gene expression between wildtype and Tnfr1-deficient glomeruli, differentially expressed genes and functional groups were similar to Tnfr1,2−/− glomeruli, indicating a predominant role of TNFR1 signaling in TNF-mediated glomerular effects. However, overall less genes were significantly regulated in Tnfr1−/− than Tnfr1,2−/− glomeruli, which suggests a contribution of TNFR2 to the inflammatory response in glomeruli when TNFR1 is present. This TNFR1-dependent synergistic effect of TNFR2 was also suggested by the qRT-PCR studies that examined expression profiles of selected chemokines and adhesion molecules in glomeruli challanged with increasing TNF concentrations up to 24 hours. In these experiments, low TNF concentrations did not induce mRNAs of these molecules in both Tnfr1−/− and Tnfr2−/− glomeruli as they did in wildtype glomeruli. In contrast, when glomeruli were stimulated with higher concentrations up to 50 ng/ml, expression of adhesion molecules and chemokines was induced in Tnfr2−/−, but not Tnfr1−/− glomeruli. Thus, in the presence of TNFR1, activation of TNFR2 may enhance proinflammatory TNF signaling when cells are stimulated with low concentrations of TNF for a prolonged time. This may result from the formation of receptor heterocomplexes [41] or a process referred to as ligand passing in which TNFR2-bound TNF increases the local TNF concentration in the vicinity of TNFR1 [42].
Surprisingly, our microarray analysis identified only 4 genes that were differentially regulated in Tnfr2−/− glomeruli compared to wildtype. These genes were not differentially expressed in Tnfr1-deficient glomeruli. The only well-characterized gene within this group was Rab6B, a small intracellular GTPase. Subsequent qRT-PCR analysis revealed that Rab6B expression was constitutively suppressed in Tnfr2−/− and Tnfr1,2−/−, but not Tnfr1−/− glomeruli, and was not induced after TNF stimulation. The reason for the TNFR2-dependent expression of Rab6B is not known, nor have functions of renal cell-expressed Rab6B been described. Proteins of the Rab family are important intracellular regulators of vesicular traffic in the secretory and endocytotic pathways [43], [44]. To this end, deficient Rab6B expression in Tnfr2−/− glomeruli may compromise the cellular capacity to secrete TNF-induced inflammatory mediators and contribute to reduced chemokine secretion, which we observed in TNF-stimulated Tnfr2−/− glomeruli, despite comparable mRNA expression levels with wildtype. Importantly, the latter finding identified a novel posttranscriptional role of glomerular TNFR2 which contributes to local chemokine production.
The lack of exclusively TNFR2-dependently expressed genes in our expression analysis was unexpected. Our data suggest that proinflammatory effects in parenchymal glomerular cells stimulated with soluble TNF are almost exclusively mediated via TNFR1 signaling, with only minor contributions of TNFR2 when TNFR1 is present. Our microarray data were confirmed by qRT-PCR analysis of TNF-stimulated glomeruli and mesangial cells, and importantly correlate in vivo with completely abrogated glomerular leukocyte infiltration in Tnfr1-, but not Tnfr2-deficient mice after intraperitoneal challenge with soluble TNF. In contrast, Bruggeman et al. recently reported a TNFR2-mediated NF-κB-dependent inflammatory response in immortalized podocytes stimulated with soluble TNF in vitro, without requirement for TNFR1 [32]. Although our data on isolated intact glomeruli do not rule out a contribution of podocyte-expressed TNFR2 to the proinflammatory TNFR2-dependent glomerular effects revealed in this study, we could not identify a TNFR1-independent function of TNFR2 in intact glomeruli ex vivo or TNF-stimulated mice in vivo. TNFR activation, however, may be different in podocytes located within intact glomeruli and immortalized podocytes cultures in vitro. Moreover, as all intrinsic glomerular cells (glomerular endothelial and mesangial cells as shown in this study, and podocytes [32]) can express TNFR1 and TNFR2, their relative contributions to TNF-induced glomerular injury remains to be elucidated analysing mice with cell type-specific deletion of these receptors.
Absence of the TNFR1-mediated proinflammatory effects identified in this study may well explain the delayed onset of GN in Tnfr1-deficient mice in vivo described previously [20]. As stimulation of intact glomeruli and mesangial cells with soluble TNF did not reveal any exclusively TNFR2-mediated inflammatory effects, potential mechanisms leading to protection from GN in Tnfr2-deficient mice [20] may involve activation of renal cell-expressed TNFR2 by membrane-bound TNF. This hypothesis is supported by findings that glomerular as opposed to systemic TNF mediates renal injury in GN, either expressed by activated adjacent glomerular cells [15], [16] or infiltrating macrophages [45]. Moreover, in contrast to soluble TNF which efficiently activates TNFR1 in vitro [46], membrane-bound TNF has been suggested to preferentially activate TNFR2 [47]. The high affinity of soluble TNF for TNFR1 apparently results from a marked stability of ligand receptor complexes, as opposed to transient interactions of soluble TNF with TNFR2 [46]. On the other hand, membrane-bound TNF was superior to soluble TNF in mediating TNFR2-dependent T cell activation, thymocyte proliferation, and granulocyte/macrophage colony-stimulating factor production [47]. Moreover, recent evidence indicates that TNFR2 activation induces degradation of TNF receptor-associated factor 2 (TRAF2), a key mediator of signal transduction of both TNFR1 and TNFR2 which is required for transcriptional activation of target genes [48]. When TNFR1 and TNFR2 were activated simultaneously, TNFR2-induced TRAF2 degradation and the subsequent decrease in NF-κB activation resulted in an enhancement of cytotoxicity triggered by TNFR1 [49], [50]. Importantly, it was shown that TNFR2 is also able to trigger a cell death process that is independent of TNFR1 activation after upregulation of membrane-bound TNF and TNFR2, potentially involving necroptosis [50], [51]. In the context of GN these data suggest that glomerular activation of TNFR2 by membrane-bound TNF may trigger local cell injury which is essential for the induction of nephritis, as shown in the NTN model in vivo [20].
In summary, our glomerular expression analysis and in vivo studies identified TNFR1 as the major receptor mediating proinflammatory effects of soluble TNF in intrinsic glomerular cells. In the presence of TNFR1, TNFR2 contributes to this inflammatory response when glomeruli are stimulated with low TNF concentrations and promotes chemokine secretion. The potential contribution of membrane-bound TNF to TNFR2-dependent glomerular inflammation remains to be elucidated.
Supporting Information
Isolation of glomerular and tubulointerstitial tissue fractions from mouse kidneys. A: Appearance of mouse glomeruli isolated after paramagnetic bead perfusion. B: The tubulointerstitial tissue fraction obtained during the first wash of the glomerular isolation procedure contained tubular fragments, tubular cells and polymorphic interstitial cells. Original magnification×100 (A–B). C: mRNA expression of the glomerular marker gene nephrin and D: FXYD2, the γ-subunit of the tubular Na,K-ATPase, in glomerular and tubulointerstitial tissue from wildtype mice as analyzed by quantitative real-time PCR. Values were normalized to rRNA expression used as reference gene. ∗ p<0.05.
(TIF)
Representative genes differentially expressed in TNF-stimulated Tnfr1,2−/−, Tnfr1−/− and Tnfr2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr1,2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr1−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Enriched functional groups of differentially expressed genes in TNF-stimulated Tnfr1,2−/− glomeruli compared to wildtype as identified by DAVID.
(PDF)
Enriched functional groups of differentially expressed genes in TNF-stimulated Tnfr1−/− glomeruli compared to wildtype as identified by DAVID.
(PDF)
Acknowledgments
We thank Patricia Lemnitzer and Dan Draganovici for excellent technical assistance, and Werner Vielhauer for designing the pressure-controlled perfusion device used in these studies. Andreas Blutke, Institute for Veterinary Pathology, Ludwig-Maximilians-University Munich is gratefully acknowledged for his help in the set up the glomerular isolation procedure. We thank Rainer Hoffmann, Munich, for support in performing the microarray studies, and Clemens Cohen, Zurich, for help with microarray data analysis. Portions of this work were prepared by A.T. and M.S. as part of their doctoral thesis at the Faculty of Medicine, Ludwig-Maximilians-University Munich.
Funding Statement
This study was funded by the Deutsche Forschungsgemeinschaft DFG (VI 231/2-1, V.V.) and the Medical Faculty of the Ludwig-Maximilians-University Munich (M.S. and V.V.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vielhauer V, Mayadas TN (2007) Functions of TNF and its receptors in renal disease: distinct roles in inflammatory tissue injury and immune regulation. Semin Nephrol 27: 286–308. [DOI] [PubMed] [Google Scholar]
- 2. Ernandez T, Mayadas TN (2009) Immunoregulatory role of TNFα in inflammatory kidney diseases. Kidney Int 76: 262–276. [DOI] [PubMed] [Google Scholar]
- 3. Takemura T, Yoshioka K, Murakami K, Akano N, Okada M, et al. (1994) Cellular localization of inflammatory cytokines in human glomerulonephritis. Virchows Arch 424: 459–464. [DOI] [PubMed] [Google Scholar]
- 4. Neale TJ, Ruger BM, Macaulay H, Dunbar PR, Hasan Q, et al. (1995) Tumor necrosis factor-α is expressed by glomerular visceral epithelial cells in human membranous nephropathy. Am J Pathol 146: 1444–1454. [PMC free article] [PubMed] [Google Scholar]
- 5. Noronha IL, Kruger C, Andrassy K, Ritz E, Waldherr R (1993) In situ production of TNF-α, IL-1α and IL-2R in ANCA-positive glomerulonephritis. Kidney Int 43: 682–692. [DOI] [PubMed] [Google Scholar]
- 6. Aten J, Roos A, Claessen N, Schilder-Tol EJ, Ten Berge IJ, et al. (2000) Strong and selective glomerular localization of CD134 ligand and TNF receptor-1 in proliferative lupus nephritis. J Am Soc Nephrol 11: 1426–1438. [DOI] [PubMed] [Google Scholar]
- 7. Baud L, Oudinet JP, Bens M, Noe L, Peraldi MN, et al. (1989) Production of tumor necrosis factor by rat mesangial cells in response to bacterial lipopolysaccharide. Kidney Int 35: 1111–1118. [DOI] [PubMed] [Google Scholar]
- 8. Gomez-Guerrero C, Lopez-Armada MJ, Gonzalez E, Egido J (1994) Soluble IgA and IgG aggregates are catabolized by cultured rat mesangial cells and induce production of TNF-α and IL-6, and proliferation. J Immunol 153: 5247–5255. [PubMed] [Google Scholar]
- 9. Chan LY, Leung JC, Tsang AW, Tang SC, Lai KN (2005) Activation of tubular epithelial cells by mesangial-derived TNF-α glomerulotubular communication in IgA nephropathy. Kidney Int 67: 602–612. [DOI] [PubMed] [Google Scholar]
- 10. Wuthrich RP, Glimcher LH, Yui MA, Jevnikar AM, Dumas SE, et al. (1990) MHC class II, antigen presentation and tumor necrosis factor in renal tubular epithelial cells. Kidney Int 37: 783–792. [DOI] [PubMed] [Google Scholar]
- 11. Jevnikar AM, Brennan DC, Singer GG, Heng JE, Maslinski W, et al. (1991) Stimulated kidney tubular epithelial cells express membrane associated and secreted TNFα. Kidney Int 40: 203–211. [DOI] [PubMed] [Google Scholar]
- 12. Bertani T, Abbate M, Zoja C, Corna D, Perico N, et al. (1989) Tumor necrosis factor induces glomerular damage in the rabbit. Am J Pathol 134: 419–430. [PMC free article] [PubMed] [Google Scholar]
- 13. Tomosugi NI, Cashman SJ, Hay H, Pusey CD, Evans DJ, et al. (1989) Modulation of antibody-mediated glomerular injury in vivo by bacterial lipopolysaccharide, tumor necrosis factor, and IL-1. J Immunol 142: 3083–3090. [PubMed] [Google Scholar]
- 14. Le Hir M, Haas C, Marino M, Ryffel B (1998) Prevention of crescentic glomerulonephritis induced by anti-glomerular membrane antibody in tumor necrosis factor-deficient mice. Lab Invest 78: 1625–1631. [PubMed] [Google Scholar]
- 15. Timoshanko JR, Sedgwick JD, Holdsworth SR, Tipping PG (2003) Intrinsic renal cells are the major source of tumor necrosis factor contributing to renal injury in murine crescentic glomerulonephritis. J Am Soc Nephrol 14: 1785–1793. [DOI] [PubMed] [Google Scholar]
- 16. Timoshanko JR, Kitching AR, Iwakura Y, Holdsworth SR, Tipping PG (2004) Leukocyte-derived interleukin-1β interacts with renal interleukin-1 receptor I to promote renal tumor necrosis factor and glomerular injury in murine crescentic glomerulonephritis. Am J Pathol 164: 1967–1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lan HY, Yang N, Metz C, Mu W, Song Q, et al. (1997) TNF-α up-regulates renal MIF expression in rat crescentic glomerulonephritis. Mol Med 3: 136–144. [PMC free article] [PubMed] [Google Scholar]
- 18. Karkar AM, Smith J, Pusey CD (2001) Prevention and treatment of experimental crescentic glomerulonephritis by blocking tumour necrosis factor-α. Nephrol Dial Transplant 16: 518–524. [DOI] [PubMed] [Google Scholar]
- 19. Khan SB, Cook HT, Bhangal G, Smith J, Tam FW, et al. (2005) Antibody blockade of TNF-α reduces inflammation and scarring in experimental crescentic glomerulonephritis. Kidney Int 67: 1812–1820. [DOI] [PubMed] [Google Scholar]
- 20. Vielhauer V, Stavrakis G, Mayadas TN (2005) Renal cell-expressed TNF receptor 2, not receptor 1, is essential for the development of glomerulonephritis. J Clin Invest 115: 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takemoto M, Asker N, Gerhardt H, Lundkvist A, Johansson BR, et al. (2002) A new method for large scale isolation of kidney glomeruli from mice. Am J Pathol 161: 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akis N, Madaio MP (2004) Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int 65: 2223–2227. [DOI] [PubMed] [Google Scholar]
- 23. Satriano JA, Banas B, Luckow B, Nelson P, Schlöndorff DO (1997) Regulation of RANTES and ICAM-1 expression in murine mesangial cells. J Am Soc Nephrol 8: 596–603. [DOI] [PubMed] [Google Scholar]
- 24. Allam R, Lichtnekert J, Moll AG, Taubitz A, Vielhauer V, et al. (2009) Viral RNA and DNA trigger common antiviral responses in mesangial cells. J Am Soc Nephrol 20: 1986–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, et al. (2003) Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics 4: 249–264. [DOI] [PubMed] [Google Scholar]
- 26. Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98: 5116–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57. [DOI] [PubMed] [Google Scholar]
- 28. Huang DW, Sherman BT, Lempicki RA (2009) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hosack DA, Dennis G Jr, Sherman BT, Lane HC, Lempicki RA (2003) Identifying biological themes within lists of genes with EASE. Genome Biol 4: R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vielhauer V, Anders HJ, Pérez de Lema G, Luckow B, Schlöndorff D, et al. (2003) Phenotyping renal leukocyte subsets by four-color flow cytometry: characterization of chemokine receptor expression. Nephron Exp Nephrol 93: e63. [DOI] [PubMed] [Google Scholar]
- 32. Bruggeman LA, Drawz PE, Kahoud N, Lin K, Barisoni L, et al. (2011) TNFR2 interposes the proliferative and NF-κB-mediated inflammatory response by podocytes to TNF-α. Lab Invest 91: 413–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mulligan MS, Johnson KJ, Todd RF, 3rd, Issekutz TB, Miyasaka M, et al (1993) Requirements for leukocyte adhesion molecules in nephrotoxic nephritis. J Clin Invest 91: 577–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vielhauer V, Eis V, Schlöndorff D, Anders HJ (2004) Identifying chemokines as therapeutic targets in renal disease: lessons from antagonist studies and knockout mice. Kidney Blood Press Res 27: 226–238. [DOI] [PubMed] [Google Scholar]
- 35. Segerer S, Schlöndorff D (2007) Role of chemokines for the localization of leukocyte subsets in the kidney. Semin Nephrol 27: 260–274. [DOI] [PubMed] [Google Scholar]
- 36. Tipping PG, Holdsworth SR (2007) Cytokines in glomerulonephritis. Semin Nephrol 27: 275–285. [DOI] [PubMed] [Google Scholar]
- 37. Seelen MA, Daha MR (2006) The role of complement in autoimmune renal disease. Autoimmunity 39: 411–415. [DOI] [PubMed] [Google Scholar]
- 38. Vieyra MB, Heeger PS Novel aspects of complement in kidney injury. Kidney Int 77: 495–499. [DOI] [PubMed] [Google Scholar]
- 39. Tveita A, Rekvig OP, Zykova SN (2008) Glomerular matrix metalloproteinases and their regulators in the pathogenesis of lupus nephritis. Arthritis Res Ther 10: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ronco P, Chatziantoniou C (2008) Matrix metalloproteinases and matrix receptors in progression and reversal of kidney disease: therapeutic perspectives. Kidney Int 74: 873–878. [DOI] [PubMed] [Google Scholar]
- 41. Pinckard JK, Sheehan KC, Schreiber RD (1997) Ligand-induced formation of p55 and p75 tumor necrosis factor receptor heterocomplexes on intact cells. J Biol Chem 272: 10784–10789. [DOI] [PubMed] [Google Scholar]
- 42. Tartaglia LA, Pennica D, Goeddel DV (1993) Ligand passing: the 75-kDa tumor necrosis factor (TNF) receptor recruits TNF for signaling by the 55-kDa TNF receptor. J Biol Chem 268: 18542–18548. [PubMed] [Google Scholar]
- 43. Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103: 11821–11827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grigoriev I, Splinter D, Keijzer N, Wulf PS, Demmers J, et al. (2007) Rab6 regulates transport and targeting of exocytotic carriers. Dev Cell 13: 305–314. [DOI] [PubMed] [Google Scholar]
- 45. Tipping PG, Leong TW, Holdsworth SR (1991) Tumor necrosis factor production by glomerular macrophages in anti-glomerular basement membrane glomerulonephritis in rabbits. Lab Invest 65: 272–279. [PubMed] [Google Scholar]
- 46. Grell M, Wajant H, Zimmermann G, Scheurich P (1998) The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proc Natl Acad Sci USA 95: 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Grell M, Douni E, Wajant H, Lohden M, Clauss M, et al. (1995) The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83: 793–802. [DOI] [PubMed] [Google Scholar]
- 48. Wu CJ, Conze DB, Li X, Ying SX, Hanover JA, et al. (2005) TNF-α induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J 24: 1886–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rodríguez M, Cabal-Hierro L, Carcedo MT, Iglesias JM, Artime N, et al. (2011) NF-κB signal triggering and termination by tumor necrosis factor receptor 2. J Biol Chem 286: 22814–22824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cabal-Hierro L, Lazo PS (2012) Signal transduction by tumor necrosis factor receptors. Cell Signal 24: 1297–1305. [DOI] [PubMed] [Google Scholar]
- 51. Biragyn A, Coscia M, Nagashima K, Sanford M, Young HA, et al. (2008) Murine β-defensin 2 promotes TLR-4/MyD88-mediated and NF-kB-dependent atypical death of APCs via activation of TNFR2. J Leukoc Biol 83: 998–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Isolation of glomerular and tubulointerstitial tissue fractions from mouse kidneys. A: Appearance of mouse glomeruli isolated after paramagnetic bead perfusion. B: The tubulointerstitial tissue fraction obtained during the first wash of the glomerular isolation procedure contained tubular fragments, tubular cells and polymorphic interstitial cells. Original magnification×100 (A–B). C: mRNA expression of the glomerular marker gene nephrin and D: FXYD2, the γ-subunit of the tubular Na,K-ATPase, in glomerular and tubulointerstitial tissue from wildtype mice as analyzed by quantitative real-time PCR. Values were normalized to rRNA expression used as reference gene. ∗ p<0.05.
(TIF)
Representative genes differentially expressed in TNF-stimulated Tnfr1,2−/−, Tnfr1−/− and Tnfr2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr1,2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr1−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Differentially expressed genes in TNF-stimulated Tnfr2−/− glomeruli compared to wildtype as identified by microarray profiling.
(PDF)
Enriched functional groups of differentially expressed genes in TNF-stimulated Tnfr1,2−/− glomeruli compared to wildtype as identified by DAVID.
(PDF)
Enriched functional groups of differentially expressed genes in TNF-stimulated Tnfr1−/− glomeruli compared to wildtype as identified by DAVID.
(PDF)