

Abstract
In this study, the role of sucrose non-fermenting–related kinase (SNRK) in white adipocyte biology was investigated. SNRK is abundantly expressed in adipose tissue, and the expression level is decreased in obese mice. SNRK expression is repressed by inflammatory signals but increased by insulin sensitizer in cultured adipocytes. In vivo, adipose tissue SNRK expression can be decreased by lipid injection but enhanced by macrophage ablation. Knocking down SNRK in cultured adipocytes activates both JNK and IKKβ pathways as well as promotes lipolysis. Insulin-stimulated Akt phosphorylation and glucose uptake are impaired in SNRK knockdown adipocytes. Phosphoproteomic analysis with SNRK knockdown adipocytes revealed significantly decreased phosphorylation of 49 proteins by 25% or more, which are involved in various aspects of adipocyte function with a clear indication of attenuated mTORC1 signaling. Phosphorylation of 43 proteins is significantly increased by onefold or higher, among which several proteins are known to be involved in inflammatory pathways. The inflammatory responses in SNRK knockdown adipocytes can be partially attributable to defective mTORC1 signaling, since rapamycin treatment activates IKKβ and induces lipolysis in adipocytes. In summary, SNRK may act as a suppressor of adipocyte inflammation and its presence is necessary for maintaining normal adipocyte function.
Sedentary lifestyle and excessive energy intake have caused obesity epidemics. Extensive studies have demonstrated that obesity-related insulin resistance and type 2 diabetes are associated with a low degree of inflammation in adipose tissue (1). Obese adipose tissue secretes a variety of inflammatory markers, cytokines, and chemokines at elevated levels. Some of these factors, such as tumor necrosis factor (TNF)-α, interleukin (IL)-6, IL-1β, and MCP-1, have been reported to impair insulin signaling (2–5). Dysregulation of adipocyte lipolysis, induced by increased expression of adipose inflammatory cytokines, contributes to systemic insulin resistance through elevated circulating free fatty acid (FFA) levels. Multiple types of immune cells have been identified to regulate inflammatory pathways in obese adipose tissue, such as macrophages, neutrophils, T cells, and mast cells (6,7). Potent anti-inflammatory effects, such as suppression of adipose macrophage gene expression in vitro and in vivo and inhibition of proinflammatory mononuclear cells, have been reported for the only class of insulin sensitizer, thiazolidinediones (8–11). Curbing inflammation of obese patients with salsalate, a prodrug of salicylate for treating arthritis, has been reported to improve glycemic control (12). These results indicate that obesity-related adipose inflammation plays an important role in the development of insulin resistance. However, the molecular pathways involved in the development of adipose inflammation in response to overnutrition are not fully understood.
Being the most extensively studied member of the family, AMP kinase (AMPK) has been described as a master energy-sensing enzyme activated by increased AMP-to-ATP ratio. Activation of AMPK turns on catabolic pathways that generate ATP while switching off ATP-consuming anabolic pathways. Recent studies also indicate an anti-inflammatory role for AMPKα1 in macrophages through diminishing inflammasome formation and activation of sirtuin 1 (SIRT1) (13,14). Extensive research efforts have established a clear role for AMPK in energy metabolism in muscle, liver, and macrophages. In contrast, limited and contradictory information is available on the role of AMPK in adipocytes. AMPK phosphorylates Ser565 of hormone-sensitive lipase, which was proposed to exert an antilipolytic effect through preventing phosphorylation of Ser563 by protein kinase A (PKA) (15). However, the significance of this mechanism has been questioned because of the recent finding that Ser563 is not essential for hormone-sensitive lipase activation (16). In addition, both pro- and antilipolytic effects have been described for AMPK in adipocytes (17,18). Although the AMPK activator AICAR stimulates glucose uptake in both muscle cells and adipocytes, AMPK only appears to mediate AICAR-induced glucose uptake in muscle cells—not in adipocytes (19). AMPKα2 knockout mice have unchanged fat mass despite impaired glucose tolerance when fed a normal chow diet, suggesting that AMPKα2 is not important for energy metabolism in adipose tissue of normal mice (20). Interestingly, AMPKα2 knockout mice develop adipocyte hypertrophy when fed a high-fat diet, indicating that AMPKα2 may be able to prevent excess lipogenesis in states of nutrition surplus (21). However, it is unclear whether this is because of the deficiency of AMPKα2 in adipocyte or because of the secondary effects of AMPKα2 deficiency in liver or muscle because AMPKα2 is expressed at a low level in fat. AMPKα1 has been reported to be the major isoform of AMPK in adipose tissue; yet, global AMPKα1 knockout mice derived from the same founders were reported to increase adiposity from one laboratory and decrease adiposity from another (18,22). These results raise questions regarding the importance of AMPKα1 in adipocyte function.
The biological functions of many AMPK-related family members have not been clearly defined. In this study, we describe the potential role of an AMPK-related kinase, sucrose non-fermenting–related kinase (SNRK), as a potential suppressor of inflammation in adipocytes. As a family member of AMPK-related kinases, SNRK and its function have not previously been studied in adipose tissue. SNRK was initially cloned from fat cell cDNA library (23). The NH2-terminal catalytic domain has a low homology to AMPKα, but the noncatalytic domain is unique (23). SNRK can be activated by liver kinase B (LKB)1, the same upstream kinase that activates AMPK (24). Gene knockdown study in zebrafish indicates that SNRK may play a role in angioblast development (25,26). A recent study reported that SNRK inhibits colon cancer cell proliferation (27). It is worthy to note that SNRK is a completely different protein from sucrose nonfermenting AMPK-related kinase (SNARK), which is also a member of the AMPK/SNF1 family and can be activated by LKB1 (28,29).
RESEARCH DESIGN AND METHODS
Cells, reagents, and treatments.
3T3-L1 cells were obtained from American Type Culture Collection. 3T3-L1 coxsackie virus and adenovirus receptor (CAR) cells, a 3T3-L1 subline stably expressing the truncated adenovirus receptor, were provided by Dr. David Orlicky (University of Colorado Health Sciences Center) (30). Preadipocytes were differentiated as previously described (31). FFA mixture, dexamethasone, insulin, and isobutylmethylxanthine were purchased from Sigma. Liposyn II was purchased from Webster Veterinary. Jun NH2-terminal kinase (JNK), acetyl CoA carboxylase (ACC), and IκB kinase (IKK)β antibodies were purchased from Santa Cruz Biotechnology. Phosphorylated (phospho-) JNK and phospho-IKKβ antibodies were purchased from Cell Signaling Technology. SNRK antibody was purchased from University of Dundee (24). Tubulin antibody was purchased from Abcam. Phospho-ACC antibody was purchased from Millipore. LC3 antibody was purchased from Novus Biologicals. Raptor expression constructs and antibodies were provided by Dr. Diane Finger (University of Michigan Medical School). Clodronate liposomes were purchased from clodronateliposomes.org (Vrije Universiteit, Amsterdam, the Netherlands) as previously described (32). Stable isotope labeling with amino acids in cell culture (SILAC) medium was purchased from Thermo Scientific. Rapamycin was purchased from Calbiochem. Adenoviruses expressing inducible myr-Akt1 and the tetracycline transactivator (tTA) were purchased from Cell Biolabs.
Mouse models.
Male ob/ob mice and littermate controls were purchased from The Jackson Laboratory. These mice were fed standard chow and killed at 9 weeks of age for tissue collection. For DIO mice, C57BL/6J mice were purchased from The Jackson Laboratory at 3 weeks of age, acclimated for a week, and fed on either a chow diet (5% kcal from fat) or a high-fat diet (60% kcal from fat, D12492; Research Diets) for up to 20 weeks. For macrophage ablation experiment, mice were injected with clodronate liposomes at the dose of 110 mg/kg. At the end of the study, mice were killed by CO2 inhalation and epididymal fat pads were collected. All animal experiments were approved by the Institutional Animal Care and Use Committee of Rhode Island Hospital.
Isolation of primary adipocytes.
Epididymal white fat pads from DIO mice were excised, weighed, and rinsed in isolation buffer. Fat pads were then cut into small pieces in isolation buffer supplemented with 1 mg/mL type I collagenase and digested at 37°C in shaking-water bath at 100 rpm for 45 min. Digested tissues were filtered through 400 μmol/L mesh to get single cell suspension. After centrifugation, floating adipocytes were rinsed twice with isolation buffer.
Adenovirus construction.
For construction of adenoviral vector for mouse SNRK, the coding sequence was amplified by PCR, cloned into the entry vector, and sequence confirmed. The coding sequence was then recombined into the Gateway-based pAd-CMV DEST vector (Invitrogen) according to the manufacturer’s instructions. For construction of adenoviral vector for short hairpin interfering RNA against SNRK, six short hairpin oligonucleotides and complementary strands were designed to target SNRK and ligated into the Gateway-based pENTR/U6 vector (Invitrogen) and recombined into the BLOCK-iT RNAi system after sequence confirmation. As a negative control, adenovirus expressing short hairpin (sh)GFP was used as previously described (33). Amplification of recombinant adenovirus was performed according to the manufacturer’s instructions using human embryonic kidney 293A cells.
RNA isolation and real-time PCR analysis.
RNA samples were extracted using the TRIZOL reagent. Gene expression was measured using real-time PCR analysis. Random hexamers were used for reverse transcription. Real-time PCR analysis was performed in a 15-μL reaction on 96-well clear plate using a Power SYBR Green RT-PCR Reagents kit on ABI Prism thermal cycler model StepOnePlus. The relative mRNA expression levels were normalized to expression of β-actin or 28S rRNA. The sequences of primers are shown in Supplementary Table 1.
Immunoprecipitation and Western blot analysis.
For immunoprecipitation of endogenous SNRK from adipose tissue, thirty microliters of A/G plus matrix (Santa Cruz Biotechnology) slurry were used to preclear 1 mg protein lysates at 4°C for 30 min. Then, 3 μg SNRK antibody was added for 1 h followed by 40 μL A/G plus matrix slurry for overnight incubation at 4°C. For immunoblot analysis, immunoprecipitated protein or 100 μg protein lysates from each sample was used. After PAGE on 4–12% gel (Bio-Rad Laboratories), the resolved proteins were transferred onto polyvinylidene fluoride membranes. Membranes were blocked in 1% BSA/1× Tris-buffered saline with Tween (TBST) or 5% milk/1× TBST for 1 h and then incubated with the appropriate primary antibodies in the presence of 1% BSA/1× TBST or 5% milk/1× TBST followed by incubation with appropriate horseradish peroxidase–linked secondary antibodies for 1 h in 5% milk/ 1× TBST. Protein bands were detected by enhanced chemiluminescence (ECL) Western blotting detection reagent (Perkin Elmer).
Phosphoproteomic study.
3T3-L1 CAR preadipocytes were cultured in SILAC medium for eight doublings and then seeded on sixwell plates for differentiation in SILAC medium. One-half of the cells were labeled with light Arg and Lys and infected with adenovirus expressing shSNRK. The other half were labeled with heavy Arg and Lys and infected with adenovirus expressing shGFP. For each sample, 2.5 mg light Arg- and Lys-labeled protein lysates was mixed with 2.5 mg heavy Arg- and Lys-labeled protein lysates. Samples were reduced with 45 mmol/L dithiothreitol at 60°C for 20 min, alkylated with 100 mmol/L iodoacetimide for 15 min in the dark, and digested overnight with L-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK)-treated trypsin. Peptides were then desalted using Sep-Pak C18 columns and enriched for phosphopeptides using Titansphere Phos-Tio kit before loading to Orbitrap Velos ETD mass spectrometer. Results from four biological replicates were used for data analysis. Q values for multiple hypothesis tests were also calculated based on the determined P values using the R package QVALUE as previously described (34).
Statistical analysis.
Student t test was used to analyze experiments involving two groups. Two-way ANOVA and Bonferroni posttests were used to analyze multiple experimental groups. Error bars represent means ± SE.
RESULTS
Expression and regulation patterns of SNRK in adipocyte and adipose tissue.
Through study of tissue distribution patterns of >300 human kinases including all family members of AMPK/SNF1-related kinases, SNRK gene was found to be abundantly and predominantly expressed in white adipose tissue in human and in both white and brown adipose tissue in mouse (Fig. 1A and B). SNRK protein expression pattern confirmed its predominant presence in adipose tissue (Fig. 1C). AMPKα1 has been shown to be the major isoform of AMPK in adipose tissue (18). However, the relative mRNA expression level of SNRK is 74% higher than that of AMPKα1 in human white adipose tissue and 2.1-fold higher than that of AMPKα1 in mouse adipose tissue. Expression levels of AMPKα2 are very low in both human and mouse adipose tissue, supporting the hypothesis that AMPKα2 may not play an important role in adipocyte energy metabolism. SNRK has a nuclear localization signal and has been shown to be localized in nucleus of cultured rat cerebellar granule neurons by immunohistochemistry (35). For localization of SNRK in adipocyte, SNRK-GFP fusion protein was expressed via adenoviral-mediated gene transfer and revealed a large overlapping with lysosomal tracker (Fig. 1D), indicating that SNRK may have different cellular localization patterns in different cell types. SNRK expression in white adipose tissue is significantly reduced in both ob/ob and DIO mice at both mRNA and protein levels (Fig. 2A–C). Further analysis indicates that the decrease of SNRK gene expression in adipose tissue of DIO mice occurs in adipocyte, not in stromal vascular, cells (Fig. 2D). Interestingly, SNRK mRNA and protein levels can be induced by fasting in adipose tissue of lean mice, but this regulation is lost in DIO mice (Fig. 2A and C), suggesting that SNRK may be involved in regulating metabolism in response to fasting in lean mice.
FIG. 1.

Tissue distribution of SNRK and AMPKα. A: Relative mRNA expression levels of SNRK, AMPKα1, and AMPKα2 in human tissues. RNA samples from pooled human tissues were purchased from Clonetech (stomach cat. no. 636126, small intestine 636125, pancreas 636119, white adipose tissue 636162, lung 636105, heart 636113, testis 636115, liver 636101, kidney 636118, brain 636102, spleen 636121, and skeletal muscle 636120). B: Relative mRNA expression levels of SNRK, AMPKα1, and AMPKα2 in mouse tissues. Tissues from four male C57BL/6 mice were pooled for RNA preparation. C: SNRK protein levels in mouse tissues. Tissues from four male C57BL/6 mice were pooled for protein preparation. SNRK was immunoprecipitated from 1 mg protein lysates. D: SNRK localization. The SNRK-GFP fusion protein was expressed in 3T3-L1 CAR adipocytes via adenovirus-mediated gene transfer. Forty-eight hours after infection, adipocytes were stained with Lyso tracker (10,000×, cat. no. L-7528; Invitrogen) (red) for presence of lysosomes and DAPI (final concentration at 1 μg/mL) (blue) for presence of nucleus during a 2-h incubation. The images were overlaid, and the orange color indicates overlapping of SNRK-GFP and Lyso tracker. BAT, brown adipose tissue; S., small; WAT, white adipose tissue.
FIG. 2.

Regulation of SNRK expression in adipose tissue. A: Regulation of SNRK gene expression in adipose tissue of ob/ob mice (top panel) (n = 5 per group) and high-fat diet DIO mice (bottom panel) (n = 4 per group). B: Regulation of SNRK protein expression in adipose tissue of ob/ob mice (n = 3 per group). C: Regulation of SNRK protein expression in adipose tissue of DIO mice (n = 4 per group). D: Regulation of SNRK gene expression in adipocytes and stromal vascular cells isolated from chow-fed and DIO mice (fat pads were pooled from 6–8 mice in each group). Adi, adipocytes; HF, high fat; SV, stromal vascular cells. *P < 0.05. Error bars stand for mean ± SE.
It is well established that FFA release and production of inflammatory factors are increased in adipose tissue of obese state and contribute to the development of insulin resistance and hyperglycemia. For exploration of whether these factors are potentially responsible for downregulating SNRK expression in adipose tissue, FFAs and proinflammatory factor TNF-α were used to treat cultured adipocytes. As shown in Fig. 3A and B, SNRK gene expression was significantly reduced by treatment with both FFA and TNF-α, indicating that activation of inflammatory pathways suppresses SNRK expression. The constitutively active form of IκB kinase β (IKKβ SE), a master kinase that turns on transcription of many inflammatory genes, was also overexpressed in 3T3-L1 CAR adipocytes by adenovirus-mediated gene transfer. IKKβ SE significantly repressed SNRK gene expression (Fig. 3C). In addition, significantly decreased SNRK gene expression is observed in isolated adipocytes prepared from lean mice treated with intravenous injection of liposyn II (Fig. 3D), suggesting that the decreased SNRK expression in obese adipose tissue is likely due to the occurrence of obesity-induced adipose inflammation and/or lipid toxicity. In contrast, treatment with rosiglitazone, an insulin sensitizer with potent anti-inflammation effect, increased SNRK gene expression in cultured adipocytes (Fig. 3E). Ablation of adipose macrophages, an important source of inflammatory factors in obesity, by clodronate liposomes significantly increased SNRK gene expression in adipose tissue of DIO mice (Fig. 3F). Further experiments confirmed that SNRK protein expression levels are also decreased by inflammatory signals and increased by rosiglitazone in cultured adipocytes (Fig. 3G–J).
FIG. 3.
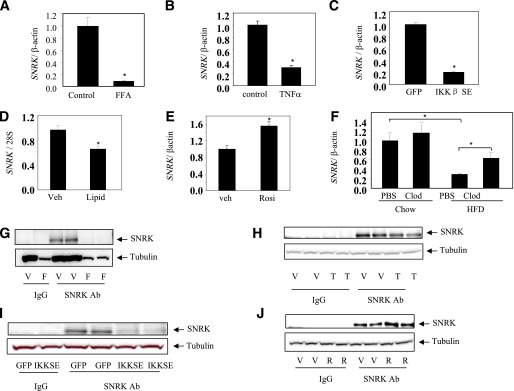
Effect of proinflammatory signals on SNRK expression. A: Effect of FFA on SNRK gene expression in cultured adipocytes. FFA mixture (cat. no. F7050; Sigma) was used at a final concentration of 0.25 μmol/L for 6 h. B: Effect of TNF-α on SNRK gene expression in cultured adipocytes. TNF-α was used at a final concentration of 25 ng/mL for 6 h. C: Effect of overexpressing the constitutively active IKKβ (IKKβ SE) on SNRK gene expression in L1-CAR adipocytes. D: Effect of intravenous lipid injection in lean mice on SNRK gene expression in isolated adipocytes. Epididymal fat pads were pooled from three mice in each group for adipocyte isolation 30 min after injection with liposyn II at 3 mL/kg/h. E: Effect of rosiglitazone (Rosi) treatment on SNRK gene expression in cultured adipocytes. Rosiglitazone was used at a final concentration of 10 μmol/L for 6 h. F: Effect of adipose tissue macrophage depletion on SNRK gene expression in lean and DIO mice (n = 4 in each group). Mice were 23 weeks old and on a high-fat diet for 19 weeks. Clodronate or PBS liposomes were injected into peritoneal cavity at the dose of 110 mg/kg. G: Effect of FFA on SNRK protein expression in cultured adipocytes. H: Effect of TNF-α on SNRK protein expression in cultured adipocytes. I: Effect of overexpressing IKKβ SE on SNRK protein expression in L1-CAR adipocytes. J: Effect of rosiglitazone treatment on SNRK protein expression in cultured adipocytes. *P < 0.05. For cell-based experiments, triplicate samples were used in gene expression and duplicate samples were used in protein expression. Results shown are representative from three independent experiments. Error bars stand for mean ± SE. Ab, antibody; Clod, clodronate liposomes; F, FFA; Lipid, liposyn II; PBS, PBS liposomes; R, rosiglitazone; T, TNF-α; V, vehicle; Veh, vehicle. (A high-quality color representation of this figure is available in the online issue.)
Effect of SNRK knockdown in cultured adipocytes.
For understanding of the function of SNRK, adenovirus-mediated short hairpin interfering RNA was used to knock down SNRK expression in 3T3-L1 CAR adipocytes. Expression levels of SNRK mRNA and protein were significantly reduced by interfering RNA against SNRK compared with control adipocytes expressing interfering RNA against GFP (Fig. 4A). Reduction of SNRK expression in 3T3-L1 CAR adipocytes activated IKKβ and JNK pathways, as shown by increased phosphorylation (Fig. 4B), which is accompanied by increased basal lipolysis as reflected by elevated glycerol release (Fig. 4C). Consistent with activation of inflammatory pathways, markedly increased expression levels of proinflammatory cytokines, including TNF-α and IL-6, were observed in SNRK knockdown 3T3-L1 CAR adipocytes compared with control cells (Fig. 4D–F). TNF-α is a potent inducer of lipolysis, which may contribute to increased lipolysis. It has been previously reported that multiple monocyte chemotactic factors are upregulated in adipose tissue and adipocytes of DIO mice (31). Reduction of SNRK expression in cultured adipocytes also led to increased gene expression of three monocyte chemotactic factors, MCP-1, MCP-2, and MCP-3, indicating that decreased SNRK expression in adipocytes may potentially enhance macrophage infiltration in obesity. Reduced SNRK expression also increased expression of chop gene. In contrast, mRNA expression levels of insulin-responsive glut4 and insulin-sensitizing adiponectin are significantly decreased upon SNRK knockdown (Fig. 4G). Insulin-stimulated Akt phosphorylation on Ser473 and glucose uptake are significantly reduced (Fig. 4H–I). Expression levels of very-long-chain acyl-coA dehydrogenase and medium-chain acyl-coA dehydrogenase are also significantly reduced in SNRK knockdown adipocytes, suggesting impaired fatty acid oxidation. Furthermore, Atg12 gene was significantly decreased, suggesting deficiency in autophagy.
FIG. 4.

Effect of SNRK knockdown on adipocyte biology. A: SNRK knockdown by adenovirus-mediated shRNA on mRNA (top panel) and protein (bottom panel) levels. B: Effect of SNRK knockdown on phosphorylation of IKKβ and JNK. C: Effect of SNRK knockdown on lipolysis. D: Effect of SNRK knockdown on expression of TNF-α, IL-6, MCP-1, MCP-2, MCP-3, and chop genes. E: Effect of SNRK knockdown on TNF-α secretion. F: Effect of SNRK knockdown on IL-6 secretion. G: Effect of SNRK knockdown on expression of Glut4, Adipo, VLACAD, MACAD, and Atg12 genes. H: Effect of SNRK knockdown on insulin-stimulated Akt phosphorylation. I: Effect of SNRK knockdown on insulin-stimulated glucose uptake. *P < 0.05. Triplicate samples were used in each experiment. Results shown were representative from three independent experiments. Error bars stand for mean ± SE. S, Ser; Veh, vehicle.
Effect of SNRK knockdown on protein phosphorylation in cultured adipocytes.
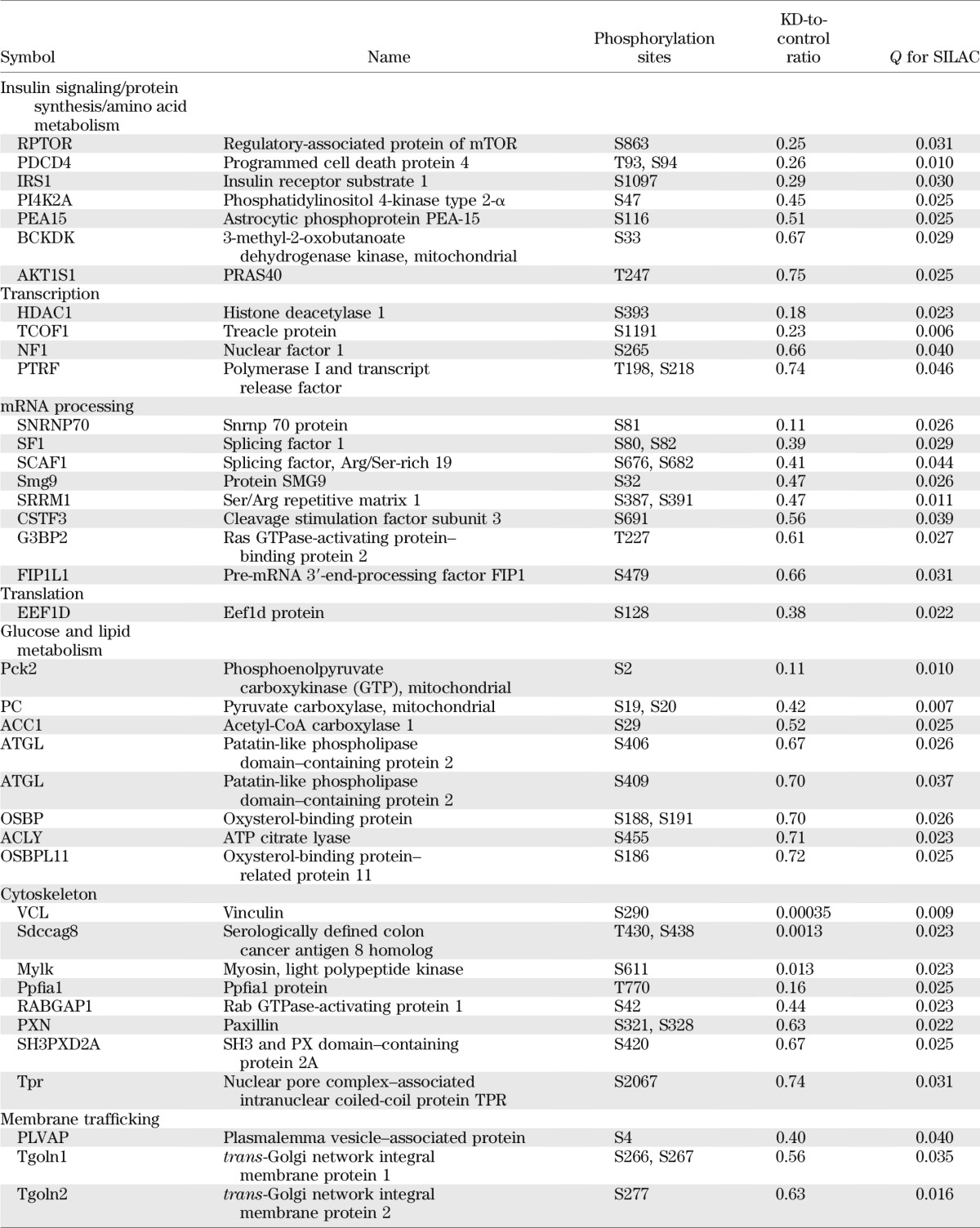
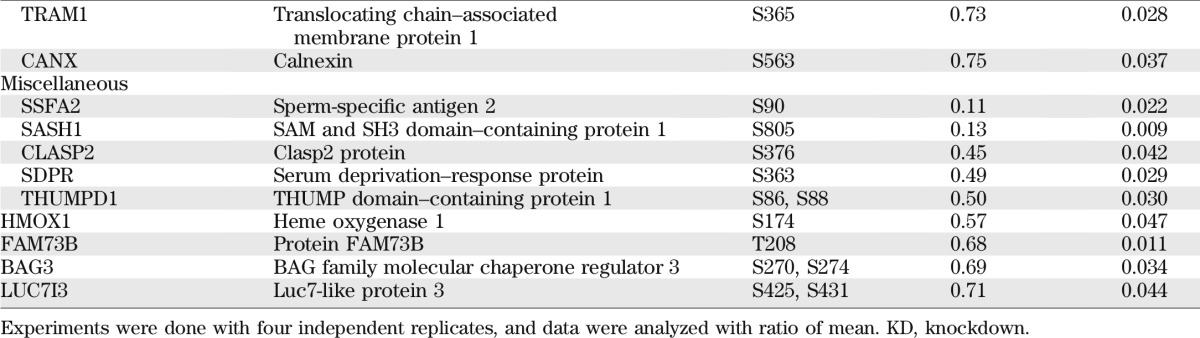
For a broad understanding regarding the function of SNRK in adipocytes and the downstream signaling events, a quantitative phosphoproteomic approach was used to evaluate changes of global phosphorylation in SNRK knockdown adipocytes compared with control adipocytes expressing shGFP. The SILAC method was used to ensure accurate comparison. SNRK knockdown adipocytes were labeled with light (12C or 14N) Arg and Lys, whereas control adipocytes were labeled with heavy (13C or 15N) Arg and Lys. All peptide sequence assignments were filtered down to 1% false discovery rate by a logistic spectral score (36). Results shown in Supplementary Tables 2 and 3 are the statistically significant (q value < 0.05) average peak ratios of light versus heavy amino acids from four biological replicates. Reduction of SNRK expression in adipocytes significantly reduced signals of 55 phosphopeptides by 25% or more, which are derived from 49 proteins involved in various aspects of cell function (Table 1), including transcription, mRNA processing, translation, glucose and lipid metabolism, and cytoskeleton. Most of the phosphosites have not been functionally annotated, but reduced phosphorylation occurs in factors normally required for transcription initiation, RNA processing, and protein synthesis. Interestingly, several components of mTORC1-signaling pathway have reduced phosphorylation, including regulatory-associated protein of mTOR (raptor) on Ser863, PDCD4 on Thr93 and Ser94, insulin receptor substrate (IRS)-1 on Ser1097, and proline-rich Akt1 substrate 1 (PRAS40) on Thr247. Phosphorylation on Ser863 of raptor activates mTORC1 signaling (37). Ser1097 of IRS-1 is reported to be a site for S6 K phosphorylation (38). PRAS40 inhibits mTOR signaling, and Akt phosphorylates PRAS40 on Thr247 to relieve the inhibition. Decreased phosphorylation on raptor Ser863, IRS-1 Ser1097, and PRAS40 Thr247 indicates attenuated mTOR signaling. Phosphorylation of ACC1, a well-characterized AMPK substrate, is decreased on Ser29, which is an uncharacterized site. In addition, SNRK knockdown also decreased phosphorylation of several enzymes involved in glucose and lipid metabolism, including mitochondrial phosphoenolpyruvate carboxykinase, mitochondrial pyruvate carboxylase, patatin-like phospholipase domain–containing protein 2, ATP citrate lyase, oxysterol-binding protein, and oxysterol-binding protein–related protein 11.
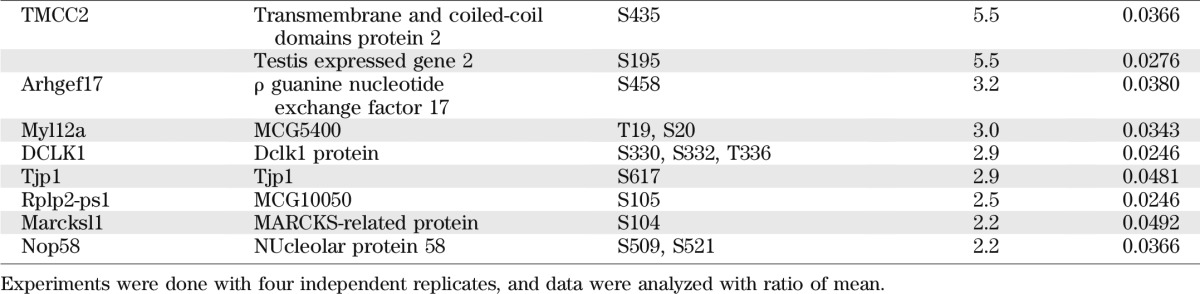
TABLE 1.
Phosphoproteins decreased by 25% or more in SNRK knockdown adipocytes
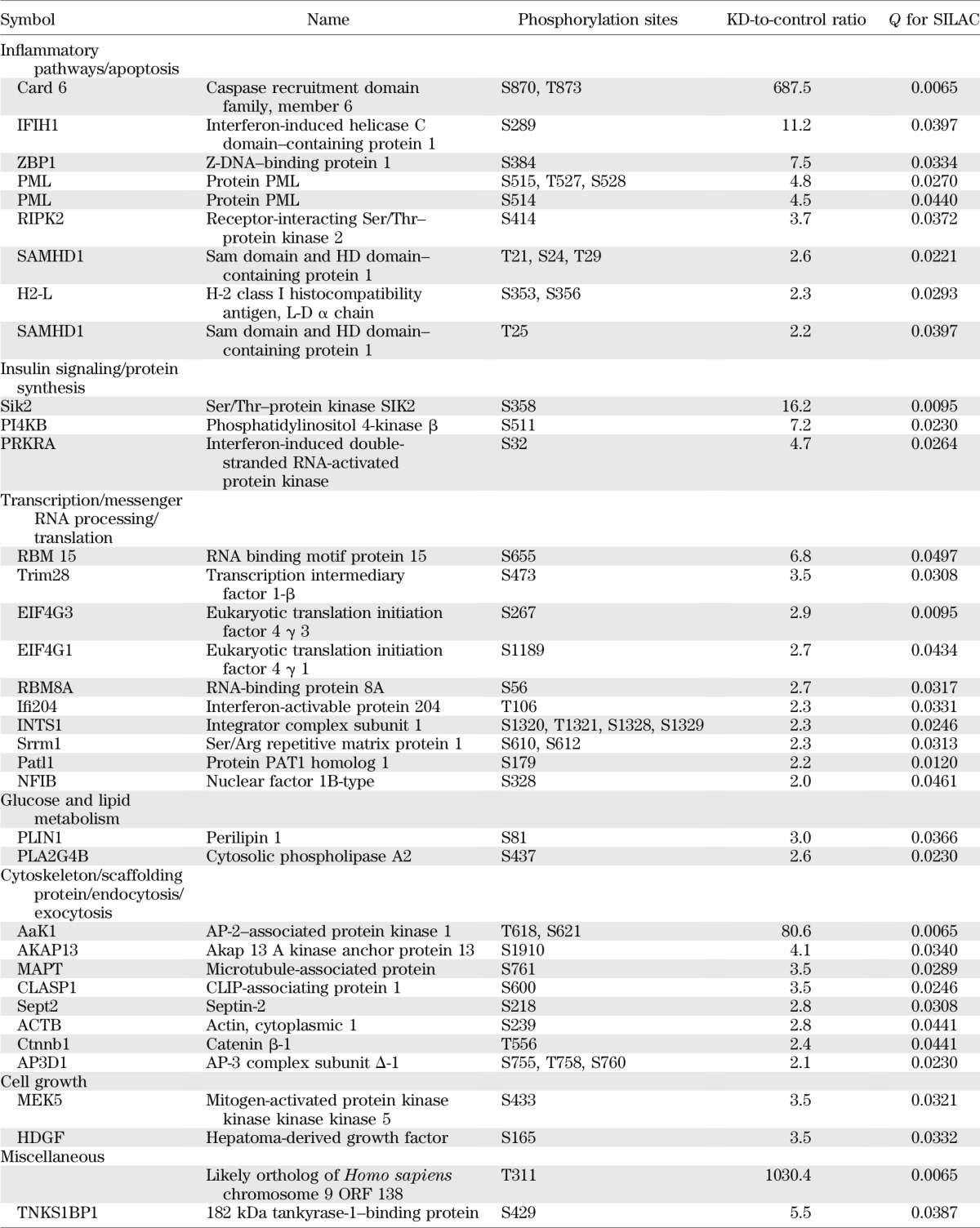
Reduction of SNRK expression in adipocytes also significantly increased signals of 55 phosphopeptides by onefold or more, which are derived from 43 proteins (Table 2). These proteins also affect various aspects of adipocyte function. Distinct from the dataset of decreased phosphorylation, several proteins involved in inflammatory pathways have increased phosphorylation. Among these proteins, caspase recruitment domain family member 6 (Card6), IFIH1, ZBP1, and receptor-interacting Ser/Thr–protein kinase 2 are known to activate nuclear factor-κB transcription factors, the substrate of IKKβ. Interferon-induced double-stranded RNA-activated protein kinase (PRKRA) has been characterized as a critical component of an inflammatory complex that activates JNK and IKKβ pathways and induces insulin resistance (39). Phosphorylation levels of multiple sites of protein PML, a potent growth suppressor and proapoptotic factor, are significantly increased. In addition, increased phosphorylation occurs in proteins normally involved in repression of transcription (Trim 28, Ifi204, and HDGF), promotion of mRNA degradation (IFIH1 and Patl1), and inhibition of protein synthesis (PRKRA). Phosphorylation also increases in proteins involved in hydrolysis of glycerophospholipids (phospholipase A2 on Ser437) and triglycerides (perilipin on Ser81, a PKA phosphorylation site), consistent with observed increase of basal lipolysis. Phosphorylation on Ser358 of SIK2 increased 15-fold, a site that has been reported to be phosphorylated by PKA in response to forskolin and CL316243 in adipocytes (40).
TABLE 2.
Phosphoproteins increased by onefold or more in SNRK knockdown adipocytes
Effect of SNRK overexpression in hepatocytes.
Despite successful SNRK knockdown in 3T3-L1 CAR adipocytes, SNRK protein cannot be overexpressed at a high level in this cell type in spite of hugely overexpressed SNRK mRNA, but SNRK-GFP fusion protein could be expressed at a low level that is observable under fluorescence microscope. One possibility is that the COOH-terminal of SNRK contains two regions of PEST-rich sequences, which are motifs indicative of rapid protein turnover (23). In addition, there may be a mechanism in adipocytes to limit SNRK overload due to abundantly expressed endogenous SNRK protein in this cell type. We therefore used a liver cell line to examine whether SNRK overexpression can increase phosphorylation of raptor and ACC. As shown in Fig. 5A, SNRK protein was overexpressed in Fao hepatoma cells and significantly increased phosphorylation of ACC on Ser79 (an AMPK phosphorylation site) and raptor on Ser863. SNRK kinase assay confirmed that overexpressed SNRK is active in hepatoma cells (Fig. 5B). For determination of whether raptor and SNRK exist in the same protein complex, raptor and SNRK were coexpressed in 293A cells. Immunoprecipitation of SNRK brought down raptor as evidenced by immunoblot analysis (Fig. 5C). SNRK overexpression also suppressed chop gene expression (Fig. 5D), consistent with increased chop gene expression in SNRK knockdown adipocytes. Interestingly, SNRK overexpression significantly increased LC3II-to-LC3I ratio under the fed condition when treated with chloroquine and rapamycin and under the fasted condition when treated with rapamycin, indicating activation of autophagy (Fig. 5E).
FIG. 5.

SNRK overexpression in hepatoma cells. A: SNRK overexpression and phosphorylation on ACC and raptor. B: SNRK activity in hepatoma cells overexpressing GFP or mouse SNRK. For kinase assay, SNRK was immunoprecipitated (IP) and incubated with the reaction mixture (200 μmol/L AMARA peptide; 1 mmol/L ATP; 10 μci γ-32P-ATP; 10 mmol/L magnesium acetate; 50 mmol/L Tris.Cl, pH 7.5; 0.1 mmol/L EGTA; and 0.1% v/v 2-mercaptoethanol) for 30 min at 30°C in a shaking-water bath. Reactions were then loaded on p81 filters. Radioactivities were counted after four washes with 0.5% phosphoric acid and once with water. Background activities were determined using IgG immunoprecipitated samples and subtracted from SNRK antibody immunoprecipitated samples. C: SNRK and raptor coimmunoprecipitation in 293A cells. D: SNRK overexpression and chop gene expression. E: Effect of SNRK overexpression on autophagy. *P < 0.05, Ad-SNRK–infected hepatoma cells vs. Ad-GFP–infected hepatoma cells. Duplicate samples were used in Western blots, and triplicate samples were used in gene expression and kinase activity experiments. Results shown were representative from three independent experiments. Error bars stand for means ± SE. WS, Western blot; Vec, vector; Ab, antibody; S, Ser; WT, wild type.
mTOR pathway and adipocyte inflammation.
The effects of SNRK on autophagy and ACC/raptor phosphorylation were further confirmed in adipocytes. In SNRK knockdown adipocytes, both LC3I and LC3II were significantly reduced after normalization to tubulin control, but LC3II-to-LC3I ratio was not affected (Fig. 6A). Reduction of SNRK also significantly reduced phosphorylation levels of raptor S863 and ACC S79. Defective autophagy has been linked to induction of inflammation and may be partially responsible for the inflammatory phenotype in adipocytes with reduced SNRK expression. In addition, phosphoproteomic study revealed many potential downstream targets of SNRK. The mTORC1 pathway stood out owing to decreased phosphorylation of several components. To explore whether defective mTOR signaling pathway contributes to adipocyte inflammation, 3T3-L1 adipocytes were treated with rapamycin and examined for activations of JNK and IKKβ pathways, lipolysis, and expression of inflammatory cytokines. Treatment with rapamycin increased phosphorylation of IKKβ but not JNK and stimulated lipolysis (Fig. 6B and C). Gene expression levels of proinflammatory factors IL-6, MCP-1, and MCP-3 are also significantly upregulated (Fig. 6D). Consistent with these results, activation of mTOR signaling by overexpressing the constitutively active Akt1 (myr-Akt) repressed lipolysis and decreased expression of proinflammatory factors (Fig. 6E–F). These data indicate that defective mTOR signaling in SNRK knockdown adipocytes may be partially responsible for the inflammatory phenotype and that other pathways are likely involved, since JNK is not activated by rapamycin treatment.
FIG. 6.

mTOR pathway and adipocyte inflammation. A: SNRK knockdown in 3T3-L1 CAR adipocyte and the effect on raptor and ACC phosphorylation. B: Rapamycin activates IKKβ in 3T3-L1 adipocytes. C: Rapamycin induces lipolysis in 3T3-L1 adipocytes. D: Rapamycin increases expression of proinflammatory factors. *P < 0.05, shSNRK-infected adipocytes vs. shGFP-infected adipocytes or rapamycin (Rapa)-treated adipocytes vs. vehicle (Veh)-treated adipocytes. Duplicate samples were used in Western blots, and triplicate samples were used in gene expression and lipolysis experiments. E: Overexpression of the constitutively active Akt1 (myr-Akt) activates mTOR signaling. F: Overexpression of myr-Akt suppresses lipolysis. G: Overexpression of myr-Akt decreases expression of inflammatory genes. *P < 0.05, cells expressing tTA plus myr-Akt vs. cells expressing tTA alone. Error bars stand for mean ± SE. S, Ser.
DISCUSSION
The underlying molecular mechanism responsible for origination of inflammation in adipocyte upon overnutrition is still not fully understood. In this study, we identified SNRK as a potential suppressor of adipocyte inflammation and hypothesize that decreased SNRK expression in adipose tissue may contribute to the development of adipose inflammation commonly observed in obesity. AMPKα has been reported to repress inflammation in macrophages and polarize macrophages to an anti-inflammatory phenotype (13,14,41). AMPKα-deficient bone marrow macrophages are proinflammatory and are sufficient to induce insulin resistance when transplanted to irradiated wild-type mice upon high-fat-diet feeding (22). In addition, AMPKα activity is decreased in adipose tissue of obese and insulin-resistant humans (42), suggesting that AMPKα is potentially another candidate for suppressing obesity-induced adipose tissue inflammation. However, further experiments are necessary to elucidate the anti-inflammatory role of AMPKα in adipose tissue and to determine the cell type of action, since literature data are mostly available with regard to macrophages. Despite the fact that AMPKα1 is the major isoform in adipocytes, contradictory phenotypes of AMPKα1 global knockout mice have been reported. Zhang et al. (22) showed that deficiency of AMPKα1 increases adipocyte size and adipose mass in vivo and AMPKα1 knockdown adipocytes are proinflammatory but accumulate more lipids. In contrast, Daval et al. (18) reported that absence of AMPKα1 reduces adipocyte size and adipose mass, possibly attributed to increased lipolysis. Interestingly, both laboratories obtained AMPKα1 knockout mice from the same source.
It will be interesting to determine whether SNRK and AMPKα have redundant roles in suppressing inflammatory responses in adipocytes. In our study, AMPKα expression levels were not changed in SNRK knockdown adipocytes, and SNRK antibody does not cross-react with AMPKα even under the condition of overexpression (data not shown), indicating that SNRK likely functions independent of AMPKα. This is the first study to elucidate the role of SNRK in adipocyte biology, which is a protein with very little available literature information regarding its function. Phosphoproteomic analysis revealed increased phosphorylation levels of several additional inflammatory proteins in SNRK knockdown adipocytes, many of which are known to activate the nuclear factor-κB pathway. These results suggest that the presence of SNRK in adipocytes is necessary to inhibit inflammatory responses. When SNRK expression is reduced, activation of inflammatory pathways leads to increased lipolysis, impairs insulin signaling, and reduces insulin-stimulated glucose uptake.
Interestingly, phosphoproteomic analysis reveals attenuation of mTOR signaling pathway in SNRK knockdown adipocytes. Phosphorylation of raptor ser863 is critical to activate mTOR (37). We demonstrated that SNRK interacts with raptor and phosphorylates raptor on Ser863, suggesting that raptor may be a target of SNRK. In adipocytes, treatment with rapamycin elicited weak inflammatory responses as reflected by IKKβ activation, increased expression of proinflammatory factors, and elevated lipolysis. However, the extent of IKKβ activation and fold of increase in proinflammatory gene expression are milder than those observed in SNRK knockdown adipocytes, indicating that impairment in mTOR signaling pathway is most likely partially responsible for the effects elicited by reduced SNRK expression. The role of mTOR in linking nutrient sensing and obesity is complex depending on the tissue examined. In adipocytes, acute inhibition of mTOR signaling by rapamycin increases insulin-stimulated glucose uptake, but chronic rapamycin treatment impairs insulin-stimulated glucose uptake by reducing Akt2 activation despite improved PI3K activation (43). Mice with adipose tissue–specific raptor deficiency are protected from diet-induced obesity and have increased energy expenditure (44). However, it is unclear whether adipose inflammation has been examined. Several transgenic models with overexpression of components in the inflammatory pathway prior to obesity development also display reduced adiposity (45–47). However, it is well established that adipose inflammation and impairment of energy homeostasis coexist in obesity.
Another potential mechanism to explain increased inflammation in SNRK knockdown adipocytes is defective autophagy. Autophagy has been demonstrated to play a role in repressing adipocyte inflammation (48). Our data indicate that SNRK is localized to lysosomes in adipocyte, an important site for degradation of large intracellular organelles or protein aggregates through autophagy. In addition, our results support a role of SNRK in promoting autophagy. The potential role of SNRK in linking overnutrition and activation of inflammatory pathways as well as impairment of energy metabolism in adipocytes requires further investigation. Loss-of-function in vivo study in the near future will provide more useful information regarding the role of SNRK in obesity-induced adipose inflammation. In addition to the inflammatory pathway, the global phosphoproteomic study also provided evidence to show that SNRK has profound effects on adipocyte biology and impacts multiple pathways that are common to most cell types, such as transcription, translation, and mRNA processing. The overall changes in protein phosphorylation patterns suggest a role for SNRK in maintaining normal cell growth and function.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the scientist development grant from the American Heart Association (0830190N) and National Institute of Diabetes and Digestive and Kidney Diseases Grant R01 DK 080746 awarded to H.X.
No potential conflicts of interest relevant to this article were reported.
Y.L., Y.N., and Y.H. performed experiments, analyzed data, and contributed to the writing of the manuscript. G.D. was involved in experimental design, data analysis, and manuscript writing. B.F. performed experiments, analyzed data, and contributed to the writing of the manuscript. G.X. and A.S. were involved in experimental design, data analysis, and manuscript writing. H.X. designed and supervised experiments and drafted and finalized the manuscript. H.X. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the the Keystone Symposia: Adipose Tissue Biology, Keystone, Colorado, 27 January–1 February 2013.
The authors thank Dr. Diane Finger (University of Michigan Medical School) for providing expression constructs of raptor and raptor S863A, as well as phospho-raptor and raptor antibodies.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1081/-/DC1.
REFERENCES
- 1.Hotamisligil GS. Inflammation and metabolic disorders. Nature 2006;444:860–867 [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA 1994;91:4854–4858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rotter V, Nagaev I, Smith U. Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 2003;278:45777–45784 [DOI] [PubMed] [Google Scholar]
- 4.Tack CJ, Stienstra R, Joosten LA, Netea MG. Inflammation links excess fat to insulin resistance: the role of the interleukin-1 family. Immunol Rev 2012;249:239–252 [DOI] [PubMed] [Google Scholar]
- 5.Sartipy P, Loskutoff DJ. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc Natl Acad Sci USA 2003;100:7265–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schipper HS, Prakken B, Kalkhoven E, Boes M. Adipose tissue-resident immune cells: key players in immunometabolism. Trends Endocrinol Metab 2012;23:407–415 [DOI] [PubMed] [Google Scholar]
- 7.Sun S, Ji Y, Kersten S, Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annu Rev Nutr 2012;32:261–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79–82 [DOI] [PubMed] [Google Scholar]
- 9.Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest 2000;106:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghanim H, Garg R, Aljada A, et al. Suppression of nuclear factor-kappaB and stimulation of inhibitor kappaB by troglitazone: evidence for an anti-inflammatory effect and a potential antiatherosclerotic effect in the obese. J Clin Endocrinol Metab 2001;86:1306–1312 [DOI] [PubMed] [Google Scholar]
- 11.Mohanty P, Aljada A, Ghanim H, et al. Evidence for a potent antiinflammatory effect of rosiglitazone. J Clin Endocrinol Metab 2004;89:2728–2735 [DOI] [PubMed] [Google Scholar]
- 12.Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, TINSAL-T2D (Targeting Inflammation Using Salsalate in Type 2 Diabetes) Study Team The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann Intern Med 2010;152:346–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wen H, Gris D, Lei Y, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 2011;12:408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Kahn BB, Shi H, Xue BZ. Macrophage alpha1 AMP-activated protein kinase (alpha1AMPK) antagonizes fatty acid-induced inflammation through SIRT1. J Biol Chem 2010;285:19051–19059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garton AJ, Campbell DG, Carling D, Hardie DG, Colbran RJ, Yeaman SJ. Phosphorylation of bovine hormone-sensitive lipase by the AMP-activated protein kinase. A possible antilipolytic mechanism. Eur J Biochem 1989;179:249–254 [DOI] [PubMed] [Google Scholar]
- 16.Anthonsen MW, Rönnstrand L, Wernstedt C, Degerman E, Holm C. Identification of novel phosphorylation sites in hormone-sensitive lipase that are phosphorylated in response to isoproterenol and govern activation properties in vitro. J Biol Chem 1998;273:215–221 [DOI] [PubMed] [Google Scholar]
- 17.Yin W, Mu J, Birnbaum MJ. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis In 3T3-L1 adipocytes. J Biol Chem 2003;278:43074–43080 [DOI] [PubMed] [Google Scholar]
- 18.Daval M, Diot-Dupuy F, Bazin R, et al. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J Biol Chem 2005;280:25250–25257 [DOI] [PubMed] [Google Scholar]
- 19.Sakoda H, Ogihara T, Anai M, et al. Activation of AMPK is essential for AICAR-induced glucose uptake by skeletal muscle but not adipocytes. Am J Physiol Endocrinol Metab 2002;282:E1239–E1244 [DOI] [PubMed] [Google Scholar]
- 20.Viollet B, Andreelli F, Jørgensen SB, et al. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest 2003;111:91–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Villena JA, Viollet B, Andreelli F, Kahn A, Vaulont S, Sul HS. Induced adiposity and adipocyte hypertrophy in mice lacking the AMP-activated protein kinase-alpha2 subunit. Diabetes 2004;53:2242–2249 [DOI] [PubMed] [Google Scholar]
- 22.Zhang W, Zhang X, Wang H, et al. AMP-activated protein kinase α1 protects against diet-induced insulin resistance and obesity. Diabetes 2012;61:3114–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becker W, Heukelbach J, Kentrup H, Joost HG. Molecular cloning and characterization of a novel mammalian protein kinase harboring a homology domain that defines a subfamily of serine/threonine kinases. Eur J Biochem 1996;235:736–743 [DOI] [PubMed] [Google Scholar]
- 24.Jaleel M, McBride A, Lizcano JM, et al. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett 2005;579:1417–1423 [DOI] [PubMed] [Google Scholar]
- 25.Chun CZ, Kaur S, Samant GV, et al. Snrk-1 is involved in multiple steps of angioblast development and acts via notch signaling pathway in artery-vein specification in vertebrates. Blood 2009;113:1192–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pramanik K, Chun CZ, Garnaas MK, et al. Dusp-5 and Snrk-1 coordinately function during vascular development and disease. Blood 2009;113:1184–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rines AK, Burke MA, Fernandez RP, Volpert OV, Ardehali H. Snf1-related kinase inhibits colon cancer cell proliferation through calcyclin-binding protein-dependent reduction of β-catenin. FASEB J 2012;26:4685–4695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koh HJ, Toyoda T, Fujii N, et al. Sucrose nonfermenting AMPK-related kinase (SNARK) mediates contraction-stimulated glucose transport in mouse skeletal muscle. Proc Natl Acad Sci USA 2010;107:15541–15546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rune A, Osler ME, Fritz T, Zierath JR. Regulation of skeletal muscle sucrose, non-fermenting 1/AMP-activated protein kinase-related kinase (SNARK) by metabolic stress and diabetes. Diabetologia 2009;52:2182–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orlicky DJ, DeGregori J, Schaack J. Construction of stable coxsackievirus and adenovirus receptor-expressing 3T3-L1 cells. J Lipid Res 2001;42:910–915 [PubMed] [Google Scholar]
- 31.Jiao P, Chen Q, Shah S, et al. Obesity-related upregulation of monocyte chemotactic factors in adipocytes: involvement of nuclear factor-kappaB and c-Jun NH2-terminal kinase pathways. Diabetes 2009;58:104–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feng B, Jiao P, Nie Y, et al. Clodronate liposomes improve metabolic profile and reduce visceral adipose macrophage content in diet-induced obese mice. PLoS ONE 2011;6:e24358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu H, Wilcox D, Nguyen P, et al. Hepatic knockdown of mitochondrial GPAT1 in ob/ob mice improves metabolic profile. Biochem Biophys Res Commun 2006;349:439–448 [DOI] [PubMed] [Google Scholar]
- 34.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA 2003;100:9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida K, Yamada M, Nishio C, Konishi A, Hatanaka H. SNRK, a member of the SNF1 family, is related to low K(+)-induced apoptosis of cultured rat cerebellar granule neurons. Brain Res 2000;873:274–282 [DOI] [PubMed] [Google Scholar]
- 36.Yu K, Sabelli A, DeKeukelaere L, et al. Integrated platform for manual and high-throughput statistical validation of tandem mass spectra. Proteomics 2009;9:3115–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster KG, Acosta-Jaquez HA, Romeo Y, et al. Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation. J Biol Chem 2010;285:80–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Gao Z, Yin J, Quon MJ, Ye J. S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-(alpha) signaling through IKK2. J Biol Chem 2008;283:35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nakamura T, Furuhashi M, Li P, et al. Double-stranded RNA-dependent protein kinase links pathogen sensing with stress and metabolic homeostasis. Cell 2010;140:338–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henriksson E, Jones HA, Patel K, et al. The AMPK-related kinase SIK2 is regulated by cAMP via phosphorylation at Ser358 in adipocytes. Biochem J 2012;444:503–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sag D, Carling D, Stout RD, Suttles J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol 2008;181:8633–8641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu XJ, Gauthier MS, Hess DT, et al. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J Lipid Res 2012;53:792–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veilleux A, Houde VP, Bellmann K, Marette A. Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes. Mol Endocrinol 2010;24:766–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, Hall MN. Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 2008;8:399–410 [DOI] [PubMed] [Google Scholar]
- 45.Xu H, Hirosumi J, Uysal KT, Guler AD, Hotamisligil GS. Exclusive action of transmembrane TNF alpha in adipose tissue leads to reduced adipose mass and local but not systemic insulin resistance. Endocrinology 2002;143:1502–1511 [DOI] [PubMed] [Google Scholar]
- 46.Tang T, Zhang J, Yin J, et al. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem 2010;285:4637–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiao P, Feng B, Ma J, et al. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology 2012;153:154–165 [DOI] [PubMed] [Google Scholar]
- 48.Jansen HJ, van Essen P, Koenen T, et al. Autophagy activity is up-regulated in adipose tissue of obese individuals and modulates proinflammatory cytokine expression. Endocrinology 2012;153:5866–5874 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.