

Abstract
In mammals, a network of circadian clocks regulates 24-h rhythms of behavior and physiology. Circadian disruption promotes obesity and the development of obesity-associated disorders, but it remains unclear to which extent peripheral tissue clocks contribute to this effect. To reveal the impact of the circadian timing system on lipid metabolism, blood and adipose tissue samples from wild-type, ClockΔ19, and Bmal1−/− circadian mutant mice were subjected to biochemical assays and gene expression profiling. We show diurnal variations in lipolysis rates and release of free fatty acids (FFAs) and glycerol into the blood correlating with rhythmic regulation of two genes encoding the lipolysis pacemaker enzymes, adipose triglyceride (TG) lipase and hormone-sensitive lipase, by self-sustained adipocyte clocks. Circadian clock mutant mice show low and nonrhythmic FFA and glycerol blood content together with decreased lipolysis rates and increased sensitivity to fasting. Instead circadian clock disruption promotes the accumulation of TGs in white adipose tissue (WAT), leading to increased adiposity and adipocyte hypertrophy. In summary, circadian modulation of lipolysis rates regulates the availability of lipid-derived energy during the day, suggesting a role for WAT clocks in the regulation of energy homeostasis.
Living organisms are influenced by rhythmic changes in the environment due to the Earth's rotation around its axis. In an attempt to optimally adapt to such recurring events, most species have evolved circadian clocks, internal timing systems controlling 24-h rhythms of behavior and physiology (1). In mammals, most, if not all, cells harbor their own molecular timer. Internal synchrony and thus overt behavioral and physiological rhythms are regulated via a hierarchical system of central and peripheral clocks. External time information is perceived by a master circadian pacemaker located in the hypothalamic suprachiasmatic nucleus, which relays timing cues to the rest of the body. In the suprachiasmatic nucleus and the periphery, the molecular clock machinery is based on interlocked transcriptional–translational feedback loops comprised of a set of clock genes. The basic helix–loop–helix transcription factors CLOCK and BMAL1 (ARNTL) induce expression of the negative regulators Per1–3 and Cry1/2 through binding to E-box promoter elements. With a delay of several hours, PER/CRY protein complexes enter the nucleus and repress activity of CLOCK/BMAL1 heterodimers, thereby shutting down their own transcription. Further loops interact with this E-box–mediated transcription rhythm and stabilize its characteristic 24-h periodicity. Clock genes further regulate the activity of numerous tissue-specific output genes, thereby translating time-of-day information into physiologically meaningful signals (2).
Both rodent and human studies suggest a tight interaction between circadian clock regulation and energy homeostasis. Circadian disruption, either external (as seen for example in shift workers) or internal (e.g., in Clock gene mutant mice), can lead to obesity and the development of type 2 diabetes and metabolic syndrome (3–6). While appetite regulation appears mostly centrally controlled, recent animal studies indicate an additional role for peripheral tissue clocks in the control of energy metabolism. For instance, local circadian oscillators in liver and pancreas regulate glucose utilization, whereas cardiomyocyte clocks are involved in cardiac repolarization (6–8).
White adipose tissues (WATs) store large amounts of lipids in the form of triglycerides (TGs). During extended periods of energy shortage (e.g., fasting), the release of lipids from WAT mediated by the hydrolysis of TGs to free fatty acids (FFAs) and glycerol (lipolysis) becomes an important energy source for other organs. The timing of FFA release from adipose stores, however, has to be tightly controlled, as excess of circulating lipids may lead to lipotoxicity and promote cardiovascular disorders (9). In contrast, redundant deposition of TGs causes obesity, a risk factor for type 2 diabetes. Previous reports show that adipose tissues exhibit rhythmic clock gene expression in mice and man (10–12). Cell-based and animal studies suggest that clock genes are positive regulators of adipogenesis (13,14). It remains unclear, however, how circadian disruption may lead to increased adipose tissue deposition and obesity, as observed in human shift workers and in various clock gene mutant mice.
In this study, we analyze the role of white adipose clocks in lipid utilization in mice. We show that self-sustained local clocks regulate rhythmic FFA release from WAT stores, thus revealing a novel and peripherally regulated mechanism by which circadian disruption may impinge on energy homeostasis.
RESEARCH DESIGN AND METHODS
Experimental animals.
Male wild-type (C57BL/6), congenic homozygous ClockΔ19 (15), Bmal1−/− (16), and PER2::LUCIFERASE (17) mice of 2–4 months of age were used for all experiments. All animal experiments were done after ethical assessment and licensed by the Office of Consumer Protection and Food Safety of the State of Lower Saxony and in accordance with the German Law of Animal Welfare. Mice were housed in small groups of five or fewer under a 12-h light/12-h dark cycle (LD) or constant darkness (DD) conditions with food and water access ad libitum.
Fat pad cultures.
Wild-type or PER2::LUCIFERASE mice (17) were LD entrained and killed by cervical dislocation at Zeitgeber time (ZT) 9 (i.e., 3 h before lights off). WAT and brown adipose tissue (BAT) samples were isolated and washed with 1× Hanks’ balanced salt solution (PAA Laboratories, Cölbe, Germany). Tissues were divided into 20–30-mg pieces and cultured in colorless Dulbecco's modified Eagle's medium (DMEM; PAA Laboratories) supplemented with 10% FBS (PAA Laboratories) and 100 nmol/L luciferin sodium salt (Biosynth, Staad, Switzerland). For RNA extraction fat pads were removed at 6-h intervals, and total RNA was extracted and processed as described below. Bioluminescence recordings were performed using a Lumicycle luminometer (Actimetrics, Willmette, IL). Period and damping rate were calculated using the Lumicycle Analysis software (Actimetrics).
Quantitative real-time PCR.
Mice were entrained to LD for at least 2 weeks, released into DD, and killed at 37, 43, 49, 55, and 61 h after lights off (corresponding to circadian times 1, 7, 13, and 19, respectively). For LD cohorts, mice were kept under LD and killed at ZT1, 7, 13, and 19. Epididymal fat was isolated, and total RNA was extracted using TRIzol reagent (Life Technologies, Darmstadt, Germany). cDNA synthesis was performed by High Capacity cDNA Reverse Transcription Kit (Life Technologies) using random hexamer primers. Quantitative real-time PCR was performed using iQ SYBR Green Supermix on an CFX96 thermocycler (Bio-Rad, Munich, Germany) according to the manufacturer’s protocol. Relative gene expression was quantified using a ∆∆ threshold cycle method and Eef1α as a reference gene (18). Primer sequences are listed in Supplementary Table 1.
Cloning of adipose TG lipase and hormone-sensitive lipase promoters and PCR mutagenesis.
A 4.3-kb fragment containing the murine adipose TG lipase (Atgl) upstream sequence and the first intron were PCR-amplified from genomic DNA using Advantage 2 polymerase mix (Clontech, Mountain View, CA). PCR products were digested with HindIII (New England Biolabs, Ipswich, MA) and XhoI (New England Biolabs) and cloned into HindIII/XhoI–digested pGL4 vector (Promega, Madison, WI). A 4.5-kb fragment of the murine hormone-sensitive lipase (Hsl) promoter was PCR-amplified as described for Atgl. PCR products were cloned into the pGL3 vector by In-Fusion Cloning (Clontech). Inserts were confirmed by sequencing. E-box mutations to GGATCC were performed using a PCR mutagenesis kit (Agilent Technologies, Santa Clara, CA) and confirmed by sequencing. Primer sequences are listed in Supplementary Table 2.
Luciferase promoter assays.
HEK293A and NIH-3T3 cells were maintained in DMEM (PAA Laboratories) supplemented with 10% FBS (PAA Laboratories) and antibiotics. One day prior to transfection, cells were plated onto 24-well plates at a density of 105 cells/well. HEK cells were cotransfected using PEI (Sigma-Aldrich, Hamburg, Germany) and 50 ng of pGL3/4 reporter plasmid in the presence of 200 ng of Bmal1, Clock, or Cry1 constructs. Empty pcDNA3.1 vector was used to make up the total amount of DNA to 0.7 μg/well. A total of 10 ng of a pRL-CMV Renilla luciferase reporter vector (Promega) was added to each reaction as internal control. Two days later, cells were harvested and assayed using the Dual-Luciferase Reporter Assay System (Promega) on a TriStar LB941 luminometer (Berthold Technologies, Wildbach, Germany). NIH-3T3 cells were transfected using X-fect (Clontech) with 1 μg pGL3-Hsl, pGL4-Atgl, or Bmal1-luc (19) reporter plasmids. Two days later, cells were synchronized by 50% serum shock for 2 h. For luminescence recordings, cells were kept in colorless DMEM, 10% FBS, and 100 nmol/L luciferin sodium salt (Life Technologies).
Chromatin immunoprecipitation.
Epididymal fat pads from wild-type and Bmal1−/− mice were isolated at ZT7 and ZT19, homogenized, and immediately cross linked in 1% formaldehyde. Chromatin was sonicated to obtain an average DNA length of ∼400 bp (15-s on/20-s off cycles for 22 min) using a Bioruptor (Diagenode Inc.). Samples were incubated overnight at 4°C with BMAL1 antibody (N-20; Santa Cruz Biotechnology). After clearing, samples were incubated with A/G agarose beads (Thermo Scientific) for 1 h at 4°C followed by intensive washings. Afterward, samples were boiled for 10 min in 10% Chelex (Bio-Rad) with Proteinase K (150 µg/mL) and spun down. DNA-containing supernatant was collected for PCR. Quantitative real-time PCR was performed as described above, and values were normalized to percentage of input. Primer sequences are listed in Supplementary Table 3.
Lipolysis assays.
Epididymal fat pads from were dissected at the given time points, cut into 10–25 mg samples, and incubated at 37°C in DMEM and 10% FBS with antibiotics. Glycerol release was measured from media aliquots using Free Glycerol Reagent (Sigma-Aldrich) and normalized to fat pad dry weight.
Blood glycerol and FFA measurements.
Trunk blood was collected at circadian times/ZT 1, 7, 13, and 19 and was allowed to clot. Subsequently serum was isolated by centrifugation at 2,000g for 20 min at 4°C. Serum FFAs, TG/glycerol, and cholesterol levels were determined using a NEFA kit (Zen-Bio, Research Triangle Park, NC), serum TG determination kit (Sigma-Aldrich), and cholesterol assay kit (Cayman Chemical) according to the manufacturers' protocols.
Adipocyte histology.
Freshly isolated epididymal WAT samples were fixed with 4% paraformaldehyde/PBS overnight. Samples were dehydrated with ascending alcohol concentrations and embedded in paraffin. Sections were cut at 6 μm and stained with hematoxylin-eosin. Adipocyte size was determined with Image J software (National Institutes of Health, Bethesda, MD).
Statistical analysis.
All results are expressed as a mean ± SEM. For statistical comparison unpaired two-tailed Student t tests or two-way ANOVAs with Bonferroni posttests were performed using Prism 5 software (GraphPad Software). Analysis of circadian gene expression was performed with CircWave software (20) downloaded at http://www.euclock.org. P values <0.05 were considered significant.
RESULTS
Circadian ClockΔ19 mutants show increased adiposity and blunted FFA and glycerol rhythms in blood.
To gain more insights into the circadian regulation of lipid metabolism, we compared diurnal profiles of various lipid parameters in serum between circadian clock–deficient ClockΔ19 and wild-type mice. Consistent with a previous report (4), ClockΔ19 mice had significantly higher cholesterol levels in the blood, albeit no significant circadian variation was observed in either genotype (Fig. 1A). In contrast, serum TG levels were rhythmic, but indistinguishable between genotypes (Fig. 1B). Interestingly, FFA serum levels showed a robust circadian profile in wild-type animals (P = 0.005). This rhythm was abolished, and overall levels were decreased in ClockΔ19 mutants (P = 0.802) (Fig. 1C), suggesting an involvement of the circadian clock machinery in the regulation of fatty acid release from TG stores. In line with this hypothesis, serum glycerol concentrations showed similar variations in wild-type serum, whereas in ClockΔ19 mutants, levels were nonrhythmic and overall low (P = 0.003 vs. P = 0.678) (Fig. 1D). Comparable effects on FFA and glycerol levels were also observed under LD conditions (Fig. 1E and F) and in another clock-deficient mouse model, Bmal1−/− (Fig. 1G and H). Surprisingly, while blood TG and FFA levels were unaltered or low, ClockΔ19 mutants at the same time exhibited higher overall body weight gain with increased adiposity (Fig. 2A and B and Supplementary Fig. 1A) together with hyperphagy, but unaltered overall activity (Supplementary Fig. 1B and C) (4). Histological analysis of WAT paraffin sections revealed increased WAT lipid accumulation and adipocyte hypertrophy in ClockΔ19 mutant mice (Fig. 2C), suggesting that the Clock mutation either promotes lipid accumulation or inhibits lipid mobilization in WAT.
FIG. 1.
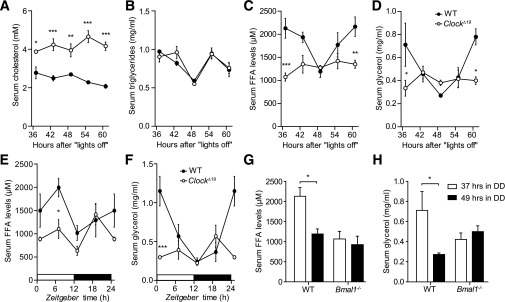
Serum lipids change in ClockΔ19 mice. Diurnal profiles of cholesterol (A), TGs (B), FFAs (C), and glycerol (D) in the serum of wild-type (WT; closed circles) and ClockΔ19 (open circles) mice in DD (n = 3/time point). Time points indicate hours spent in DD after the last lights off. Diurnal profiles of FFAs (E) and glycerol (F) in the serum of wild-type (closed circles) and ClockΔ19 (open circles) mice in LD conditions (n = 3–5/time point). Time-of-day dependent variations in serum FFA and glycerol levels in WT (black bars) and Bmal1−/− (white bars) animals kept in DD (n = 3–5/time point). All data are shown as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by two-way ANOVA with Bonferroni posttest.
FIG. 2.

Increased body weight and adiposity in ClockΔ19 mice. Body weight (A) and adiposity (epididymal fat to body weight ratio) (B) in wild-type (WT; black bars) and ClockΔ19 (white bars) mice fed standard chow for 10 weeks (n = 14). C: Representative sections and adipocyte size of epididymal WAT after 10 weeks of standard diet (scale bars, 100 μm; n = 3). *P < 0.05; **P < 0.01; ***P < 0.001 by unpaired t test. All data are shown as means ± SEM.
Circadian regulation of genes involved in WAT lipid metabolism.
Circadian clocks regulate local cellular physiology via transcriptional programs involving large numbers of tissue-specific clock-controlled genes (21). To test if molecular clocks are involved in regulating WAT physiology, we analyzed circadian variations in mRNAs of genes involved in WAT lipid metabolism. Genes were selected using the Gene Ontology database (http://www.geneontology.org) and expression data assembled on BioGPS (http://biogps.org) and compared with published literature (Supplementary Fig. 2A). Epididymal WAT transcript levels were assessed at the beginning of the rest phase (37 h in DD) and in the early subjective night (49 h in DD). For genes which showed significant regulation between both time points, we determined full circadian transcriptional profiles in ClockΔ19 and wild-type mice. Of the genes associated with FFA transport and TG biosynthesis (i.e., the conversion of FFAs for storage in adipose lipid droplets), Caveolin2, the acyl-CoA synthetases Acsl1 and Acsl4 and the phosphatidate phosphatase Lpin1 showed significant differences in expression between 37 and 49 h in DD (Fig. 3A). In ClockΔ19 mutants, FFA transport/TG biosynthesis mRNAs were either unaffected (Acsl4, Lpl) or dampened and overall downregulated when compared with wild-type controls (Fig. 3B and C and Supplementary Fig. 2B). In contrast, the mRNAs of two rate-limiting lipolytic enzymes, Atgl (or Pnpla2) and Hsl (or Lipe), exhibited circadian variations in wild-type animals (Fig. 3A), while in ClockΔ19 mutants, Atgl and Hsl mRNA levels (together with that of Mgll) were significantly reduced and arrhythmic (Fig. 4A) (P < 0.001 vs. P = 0.85 [Atgl] and P < 0.001 vs. P = 0.63 [Hsl], respectively), strongly correlating with the reduced and arrhythmic FFA/glycerol serum levels observed in these mice (Fig. 1). To test TG breakdown from WAT stores more directly, we performed lipolysis assays on fat pad explants at four different time points in DD and LD. In wild-types, glycerol excretion showed a rhythmic pattern, whereas dampened rhythmicity and a general downregulation of basal lipolysis rates were observed in explants of ClockΔ19 mice (Fig. 4B and C) (P = 0.001 vs. P = 0.95 [DD] and P < 0.001 vs. P = 0.63 [LD], respectively). Of note, on the second day in DD, Clock mutants retained a rhythmic, though dampened, feeding profile (Supplementary Fig. 3A). Moreover, when animals were subjected to nighttime restricted feeding (i.e., food access between ZT12 and ZT24) under LD conditions, daytime Atgl and Hsl levels remained reduced in Clock mutant mice, indicating that Atgl/Hsl transcription does not merely reflect feeding rhythms (Supplementary Fig. 3B). Analogously, Bmal1−/− mice showed lower expression levels of Atgl and Hsl (Fig. 4D) and Bmal1-deficient fat pads displayed lower lipolysis rates (Fig. 4E)
FIG. 3.
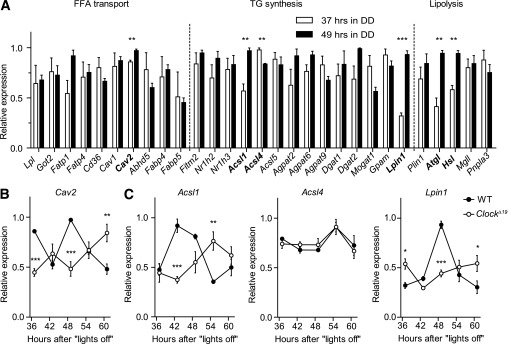
Circadian clock controls TG metabolism in WAT. A: mRNA levels of genes involved in adipocyte TG metabolism from wild-type (WT) WAT samples isolated at two opposite circadian time points, 37 h (open bars) and 49 h (closed bars) in DD (n = 3 per time point [*P < 0.05; **P < 0.01 by unpaired t test]). Circadian expression profiles of candidate genes from (A) involved in FFA transport (B) and TG synthesis (C) in WAT samples from WT (closed circles) and ClockΔ19 (open circles) mice in DD (n = 3/time point [*P < 0.05; **P < 0.01; ***P < 0.001 by two-way ANOVA with Bonferroni posttest]). All data are shown as means ± SEM.
FIG. 4.
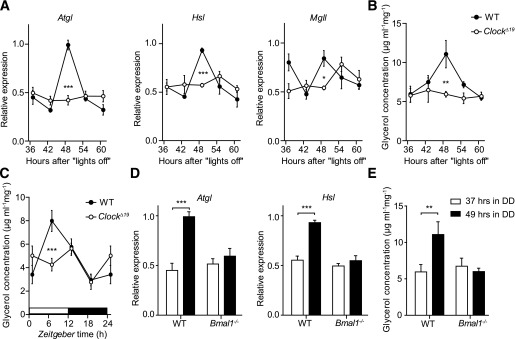
Circadian clock regulates lipid mobilization in WAT. A: Circadian expression profiles of genes involved in lipolysis in adipose tissue from wild-type (WT; closed circles) and ClockΔ19 (open circles) mice in DD (n = 3–4/time point). Profiles of glycerol excretion from WT (closed circles) and ClockΔ19 (open circles) epididymal WAT fat-pad explants harvested in DD (B) and LD (C) (n = 6–14/time point). D: Expression of Atgl and Hsl in WAT of WT (black bars) and Bmal1−/− (white bars) animals at 37 and 49 h after lights off (n = 2–3/time point). E: Changes in glycerol excretion from WT (black bars) and Bmal1−/− (white bars) epididymal WAT fat-pad explants harvested at 37 and 49 h after lights off (n = 8/time point). All data are shown as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by two-way ANOVA with Bonferroni posttest.
Adipose peripheral clocks regulate rhythmic expression of Atgl and Hsl.
To further characterize the circadian regulation of adipose physiology, we analyzed WAT clock gene regulation in DD. Expression of Bmal1, Per2, and Dbp genes exhibited robust circadian variations in wild-type animals, which were significantly downregulated and arrhythmic (Per2, Dbp) or dampened (Bmal1) in ClockΔ19 mutant mice (Fig. 5A). To characterize the sustainability of molecular circadian rhythms in WAT and compare clock function between different fat depots, we cultured epididymal, perirenal, peritoneal, subcutaneous white, as well as intrascapular BAT fat explants from PER2::LUCIFERASE circadian reporter mice. All cultures showed sustained bioluminescence rhythms in the circadian range for several days (Fig. 5B and Supplementary Fig. 4A) (17). The phases of luminescent oscillations were comparable between different depots and to those reported from other peripheral tissue explants (17,22) (Supplementary Fig. 4B). Periodogram analysis revealed endogenous periodicities of ∼25 h; statistically significant differences in period were only observed between epididymal white fat and BAT (25.8 ± 0.2 vs. 24.7 ± 0.3 h; Supplementary Fig. 4C). Moreover, rhythm dampening was comparable among all adipose tissues tested (Supplementary Fig. 4D). To test if Atgl and Hsl transcription is clock-driven at the tissue level, we kept epididymal fat explants in culture for 36 h. We observed rhythmic Atgl and Hsl mRNA expression in wild-type fat pads (P < 0.001 and P = 0.006, respectively). Similar to what was observed in vivo, transcript variations were abolished, and overall levels were reduced in explants from ClockΔ19 mice (P > 0.8 for both genes) (Fig. 5C). Notably, the expression rhythms of the two lipolytic genes were in phase coherence with E-box–regulated genes such as Per2 and in antiphase to Bmal1 (compare Fig. 5C and D). Taken together, these data suggested a direct control of Atgl and Hsl expression by CLOCK and BMAL1.
FIG. 5.

Local WAT clocks regulate expression of Atgl and Hsl. A: Expression profiles of Bmal1, Per2, and Dbp mRNAs in WAT of wild-type (WT; closed circles) and ClockΔ19 (open circles) mice in DD (n = 3/time point). B: Representative baseline-subtracted luminescence recordings from epididymal, peritoneal, and subcutaneous WAT explants of PER2::LUCIFERASE circadian reporter mice. C: Rhythmic expression of Atgl and Hsl transcripts in WT (closed circles), but not ClockΔ19 (open circles), cultured fat-pad explants (n = 3/time point). D: Antiphasic expression of Per2 (open circles) and Bmal1 (closed circles) mRNAs in cultured WT fat-pad explants (n = 3/time point). All data are shown as means ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 by two-way ANOVA with Bonferroni posttest. CPS, counts per second.
BMAL1 and CLOCK regulate expression of Atgl and Hsl via E-boxes.
We identified two canonical E-box sequences (CACGTG) in the first intron of Atgl (chr7: 148642099–148642104, 148642860–148642866) and in the upstream region of Hsl (chr7: 26181156–26181161, 26181424–26181430). Genomic DNA fragments containing these cis-regulatory elements were used for luciferase-based transactivation assays in HEK293A cells (Fig. 6A). Cotransfection with Clock and Bmal1 increased the activity of both Atgl and Hsl promoters by 6.5- and 2.3-fold, respectively (Fig. 6B). Activation was inhibited by cotransfection with Cry1, a negative E-box regulator. Moreover, mutation of the E-box proximal to the second exon of the Atgl gene (chr7: 148642860–148642866) abolished transcriptional activation by CLOCK/BMAL1 (Fig. 6B, left). Similar results were obtained upon mutation of both upstream E-boxes in the Hsl promoter (Fig. 6B, right). To confirm direct BMAL1 promoter binding in vivo, we performed chromatin immunoprecipitation (ChIP) on epididymal adipose tissue sampled at two different time points. ChIP analysis revealed time-of-day dependent occupancy of BMAL1 on Dbp, Atgl, and Hsl E-boxes, but not on 500-bp downstream regions in wild-type mice. In Bmal1-deficient animals, binding signal was markedly reduced (Fig. 6C). Finally, we transfected Atgl-luc and Hsl-luc constructs into NIH-3T3 cells and recorded bioluminescence for 48 h after serum shock synchronization. We observed a rhythmic bioluminescence signal for Atgl-luc and Hsl-luc antiphasic to Bmal1-luc (Supplementary Fig. 4E and F), comparable to what has been reported for other E-box controlled genes such as Per2 (23). Together, these results strongly suggest that circadian Atgl and Hsl transcription is directly regulated by CLOCK/BMAL1 via E-box activation at the adipose tissue level.
FIG. 6.

CLOCK and BMAL1 drive rhythmic transcription of Atgl and Hsl via E-boxes. A: Maps of the 5′ regions of the genomic loci of murine Atgl (top) and Hsl (bottom) on chromosome 7 (Chr.7). Putative E-box enhancers are indicated by ovals. Black arrows depict the genomic sequences cloned for promoter studies. Mutated E-boxes are indicated by gray arrowheads. B: Luciferase reporter assays in HEK293 cells for wild-type (WT) and mutated Atgl (left) and Hsl (right) promoters in response to cotransfection with Clock/Bmal1 and Cry1 (n = 3; ***P < 0.001 by one-way ANOVA with Bonferroni posttest). C: Time-dependent BMAL1 binding to Dbp, Atgl, and Hsl E-boxes and 500-bp downstream regions as identified by ChIP in WAT (n = 3/time point [*P < 0.05; **P < 0.01; ***P < 0.001 by two-way ANOVA with Bonferroni posttest]). All data are shown as means ± SEM.
Defective lipolysis leads to fasting intolerance in ClockΔ19 mutant mice.
Lipolysis becomes an important energy source during the fasting (i.e., rest) phase of the day. In line with this, reduced FFA mobilization provokes aberrant physiological responses under fasting conditions (24). To test if fasting responses are impaired in ClockΔ19 mutants, we food-deprived mutant and wild-type mice starting at the end of the light phase (ZT12). FFA and glycerol levels were upregulated in serum in response to 12- and 24-h starves (starting at the end of the normal rest phase, at ZT12) in wild-types, whereas the lipolytic response was severely dampened in ClockΔ19 animals (Fig. 7A and B). Given that FFA release, and, thus, the availability of lipids as energy source, appeared impaired, we speculated that under fasting conditions, mutants may rely more heavily on carbohydrate or protein utilization (24). In line with this, liver glycogen stores in ClockΔ19 mice were depleted much faster than in wild-types under fasting conditions (Fig. 7C). Moreover, after food removal, mutants showed lower rectal temperature when compared with wild-type controls, implying decreased thermogenesis due to general energy shortage (Fig. 7D).
FIG. 7.
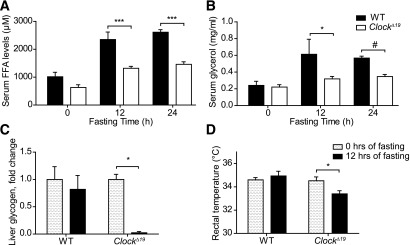
Aberrant fasting responses in ClockΔ19 mice. Serum FFA (A) and glycerol (B) levels after 12 and 24 h of food deprivation in wild-type (WT; closed bars) and ClockΔ19 (open bars) mice (n = 3–5/time point). C: Normalized changes in liver glycogen content of WT and ClockΔ19 mice during 12-h fasting (n = 3 to 4). D: Rectal temperature in WT and ClockΔ19 animals during 12-h fasting (n = 6–12). All data are shown as means ± SEM. *P < 0.05; ***P < 0.001 by two-way ANOVA with Bonferroni posttest; #P < 0.001 by unpaired t test.
DISCUSSION
Circadian physiological rhythms are generated by interplay between systemic time cues (e.g., rhythmic hormones or metabolites) and the orchestration of transcriptional programs by local tissue oscillators. As a result many physiological parameters that exhibit diurnal variations are altered in mice with perturbed molecular clocks (5,6). In this article, we report that blood FFA and glycerol concentrations show strong variations across the day. This rhythm appears to not merely reflect rhythmic food intake and seems to critically depend on the presence of functional CLOCK/BMAL1. Moreover, in humans, it has been shown that FFA and glycerol are still rhythmic under constant routine conditions (25). During fasting conditions (e.g., during the daily rest phase), adipose-released FFAs become an important energy source (26,27). Therefore, the mobilization of FFAs may be critically involved in the regulation of metabolic homeostasis.
In line with previous studies in rats (28,29), we showed circadian rhythms in baseline WAT lipolysis rates and FFA levels in the blood of wild-type mice. In ClockΔ19 and Bmal1−/− mutants, both rhythms were flattened and downregulated [our data and (30,31)], indicating defects in lipid mobilization. In agreement with this, it has previously been demonstrated that young Bmal1−/− mice show increased adiposity (8,32,33), while C57BL/6 ClockΔ19 animals are obese and are more sensitive to high-fat diet (4). Rhythmic clock gene expression has been detected in many tissues in rodents and humans including various adipose depots (10–12). Using PER2::LUCIFERASE fat-pad explants cultures, we showed that adipose clocks display similar endogenous rhythm sustainment as described for other peripheral oscillators. We identified Atgl and Hsl, which encode for two lipolysis pacemaker enzymes responsible for >95% of TG hydrolysis activity (34), as targets of the local WAT clock machinery. Atgl/Hsl transcriptional rhythms were accompanied by rhythmic lipolytic activity in explant cultures and rhythmic elevation of the lipolysis products FFAs and glycerol in the blood. Of note, increased Atgl/Hsl mRNA expression preceded the elevation of lipolytic products in the blood of wild-type animals by several hours, probably reflecting a delay between the initiation of the lipolytic response and the emergence of a measurable physiological effect. This delay results in high glycerol and FFA levels during the rest phase when both are used for energy production. Hsl-deficient mice show hypertrophic adipocytes, reduced lipolysis rates, and decreased FFA levels, though their body weight is normal (35). Atgl conventional and adipocyte-specific mutants display the same phenotype together with increased body weight (36–39). Although the precise mechanism by which impaired lipolysis contributes to the development of obesity is not well-understood, cell-based studies suggest that reduced expression of Atgl and Hsl leads to TG accumulation in lipid droplets in adipocytes (40,41). Consistent with this hypothesis, our data suggest that dampened circadian lipolysis rhythms, as seen in Clock and Bmal1 mutant mice, may promote TG accumulation in WAT. Moreover, reduced availability of FFAs as peroxisome proliferator–activated receptor cofactors may disrupt oxidative metabolism in mitochondria in other organs (39,42). Of note, expression of both enzymes was reported to be decreased in adipose tissue in obesity, thus leaving a possibility that downregulation of Atgl and Hsl transcript levels in ClockΔ19 and Bmal1−/− animals might be rather the consequence than the cause of their adiposity. Our in vitro data suggest that Atgl and Hsl are direct targets of CLOCK/BMAL1, independent of metabolic state, but further studies are needed to clarify the impact of body weight and composition in this context (43,44). Moreover, we cannot exclude that disrupted feeding rhythms in ClockΔ19 (Supplementary Fig. 2) mice may impinge on lipolysis rates via hormonal regulation (e.g., via leptin or insulin, which are known to induce and inhibit lipolysis, respectively) (45,46). However, circadian feeding profiles were not totally arrhythmic in Clock mutant mice, and Atgl/Hsl expression remained dampened in rhythmically fed Clock mutants. Further, reduced pancreatic secretion of insulin and hyperleptinemia have been reported in ClockΔ19 mice (4,6), both of which appear insufficient to restore blood FFA and glycerol levels. Instead, our results argue that rhythmic lipolysis is, at least in part, locally controlled by adipose circadian clocks. In line with this, oscillations of Atgl and Hsl mRNAs are sustained in fat pad explants ex vivo. Reporter gene and ChIP assays further indicate that CLOCK/BMAL1 directly and rhythmically bind to E-boxes in the promoters of Atgl and Hsl.
Mice with defective lipolysis exhibit impaired metabolic compensation during food deprivation (24). Consistently, our data reveal that ClockΔ19 mutants show aberrant fasting responses. Once external energy supplies are disrupted, glycogen stores become quickly depleted, while mice are unable to fully engage their lipid stores in WAT to deliver further energy for maintaining stable body temperature. Alternatively, the development of brown adipocytes may be affected in ClockΔ19 mice, or the ClockΔ19 mutation may directly impair brown adipose thermogenesis (11,39).
During the inactive phase, wild-type mice switch their energy substrate preference to lipids, which corresponds to a lower respiratory exchange ratio (47). Hsl- and Atgl-deficient animals, however, continue to primarily use carbohydrates during that time due to impaired FFA release into the blood (38). Remarkably, a similar respiratory exchange ratio phenotype was described for Clock−/− and Bmal1−/− mice (48,49), which, as our data suggest, could be explained by impaired lipid use. In contrast, animals lacking the transcriptional repressor and negative regulator of Bmal1 REV-ERBα (NR1D1) show increased expression of both Clock and Bmal1 and use lipids as the predominant energy source through their efficient FFA mobilization and membrane transport (50).
So far, the mechanistic link between circadian disruption and associated metabolic defects is not clearly understood. In this study, we show that adipose-tissue clocks may directly affect diurnal lipid homeostasis by regulating FFA/glycerol mobilization from WAT stores via transcriptional regulation of the lipolytic machinery. By this, adipocyte clock function may directly impinge on energy homeostasis, thus representing a potential new target for pharmacological resetting of circadian physiology (51,52). Based on the evidence obtained from ClockΔ19 and Bmal1−/− mice, we suggest that impaired lipid mobilization may lead to an imbalance in energy homeostasis, promoting increased adiposity. It remains to be clarified, however, to which extent WAT lipolysis defects contribute to the obese phenotype seen in ClockΔ19 mutant mice since other alterations such as changed feeding patterns may further promote the development of obesity after circadian disruption (4,53,54).
Supplementary Material
ACKNOWLEDGMENTS
A.S. is supported by the Max Planck Society. H.O. is an Emmy Noether Fellow of the Deutsche Forschungsgemeinschaft and a Lichtenberg Fellow of the Volkswagen Foundation.
No potential conflicts of interest relevant to this article were reported.
A.S. and H.O. planned the study, researched data, and wrote the manuscript. J.M.-K. researched data. H.O. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Dr. Ana Martinez Hernandez (Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany) for critical reading of the manuscript, and Dr. Moritz Rossner (Max-Planck-Institute for Experimental Medicine, Göttingen, Germany) and Dr. Steve A. Brown (University of Zürich, Zürich, Switzerland) for providing DNA constructs.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db12-1449/-/DC1.
See accompanying commentary, p. 2175.
REFERENCES
- 1.Harmer SL, Panda S, Kay SA. Molecular bases of circadian rhythms. Annu Rev Cell Dev Biol 2001;17:215–253 [DOI] [PubMed] [Google Scholar]
- 2.Storch KF, Lipan O, Leykin I, et al. Extensive and divergent circadian gene expression in liver and heart. Nature 2002;417:78–83 [DOI] [PubMed] [Google Scholar]
- 3.Bray MS, Young ME. Circadian rhythms in the development of obesity: potential role for the circadian clock within the adipocyte. Obes Rev 2007;8:169–181 [DOI] [PubMed] [Google Scholar]
- 4.Turek FW, Joshu C, Kohsaka A, et al. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 2005;308:1043–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudic RD, McNamara P, Curtis AM, et al. BMAL1 and CLOCK, two essential components of the circadian clock, are involved in glucose homeostasis. PLoS Biol 2004;2:e377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 2010;466:627–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jeyaraj D, Haldar SM, Wan X, et al. Circadian rhythms govern cardiac repolarization and arrhythmogenesis. Nature 2012;483:96–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lamia KA, Storch KF, Weitz CJ. Physiological significance of a peripheral tissue circadian clock. Proc Natl Acad Sci USA 2008;105:15172–15177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta 2010;1801:209–214 [DOI] [PubMed]
- 10.Ando H, Yanagihara H, Hayashi Y, et al. Rhythmic messenger ribonucleic acid expression of clock genes and adipocytokines in mouse visceral adipose tissue. Endocrinology 2005;146:5631–5636 [DOI] [PubMed] [Google Scholar]
- 11.Zvonic S, Ptitsyn AA, Conrad SA, et al. Characterization of peripheral circadian clocks in adipose tissues. Diabetes 2006;55:962–970 [DOI] [PubMed] [Google Scholar]
- 12.Otway DT, Mäntele S, Bretschneider S, et al. Rhythmic diurnal gene expression in human adipose tissue from individuals who are lean, overweight, and type 2 diabetic. Diabetes 2011;60:1577–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grimaldi B, Bellet MM, Katada S, et al. PER2 controls lipid metabolism by direct regulation of PPARγ. Cell Metab 2010;12:509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shimba S, Ishii N, Ohta Y, et al. Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci USA 2005;102:12071–12076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vitaterna MH, King DP, Chang AM, et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 1994;264:719–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell 2000;103:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoo SH, Yamazaki S, Lowrey PL, et al. PERIOD2:LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USA 2004;101:5339–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oster H, Damerow S, Kiessling S, et al. The circadian rhythm of glucocorticoids is regulated by a gating mechanism residing in the adrenal cortical clock. Cell Metab 2006;4:163–173 [DOI] [PubMed] [Google Scholar]
- 19.Brown SA, Ripperger J, Kadener S, et al. PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 2005;308:693–696 [DOI] [PubMed] [Google Scholar]
- 20.Oster H, Damerow S, Hut RA, Eichele G. Transcriptional profiling in the adrenal gland reveals circadian regulation of hormone biosynthesis genes and nucleosome assembly genes. J Biol Rhythms 2006;21:350–361 [DOI] [PubMed] [Google Scholar]
- 21.Panda S, Antoch MP, Miller BH, et al. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 2002;109:307–320 [DOI] [PubMed] [Google Scholar]
- 22.Pezuk P, Mohawk JA, Yoshikawa T, Sellix MT, Menaker M. Circadian organization is governed by extra-SCN pacemakers. J Biol Rhythms 2010;25:432–441 [DOI] [PubMed] [Google Scholar]
- 23.Meng QJ, McMaster A, Beesley S, et al. Ligand modulation of REV-ERBalpha function resets the peripheral circadian clock in a phasic manner. J Cell Sci 2008;121:3629–3635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu JW, Wang SP, Casavant S, Moreau A, Yang GS, Mitchell GA. Fasting energy homeostasis in mice with adipose deficiency of desnutrin/adipose triglyceride lipase. Endocrinology 2012;153:2198–2207 [DOI] [PubMed] [Google Scholar]
- 25.Dallmann R, Viola AU, Tarokh L, Cajochen C, Brown SA. The human circadian metabolome. Proc Natl Acad Sci USA 2012;109:2625–2629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. Regulation of lipolysis in adipocytes. Annu Rev Nutr 2007;27:79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmadian M, Duncan RE, Sul HS. The skinny on fat: lipolysis and fatty acid utilization in adipocytes. Trends Endocrinol Metab 2009;20:424–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tsutsumi K, Inoue Y, Kondo Y. The relationship between lipoprotein lipase activity and respiratory quotient of rats in circadian rhythms. Biol Pharm Bull 2002;25:1360–1363 [DOI] [PubMed] [Google Scholar]
- 29.Benavides A, Siches M, Llobera M. Circadian rhythms of lipoprotein lipase and hepatic lipase activities in intermediate metabolism of adult rat. Am J Physiol 1998;275:R811–R817 [DOI] [PubMed] [Google Scholar]
- 30.Oishi K, Atsumi G, Sugiyama S, et al. Disrupted fat absorption attenuates obesity induced by a high-fat diet in Clock mutant mice. FEBS Lett 2006;580:127–130 [DOI] [PubMed] [Google Scholar]
- 31.Kennaway DJ, Owens JA, Voultsios A, Boden MJ, Varcoe TJ. Metabolic homeostasis in mice with disrupted Clock gene expression in peripheral tissues. Am J Physiol Regul Integr Comp Physiol 2007;293:R1528–R1537 [DOI] [PubMed] [Google Scholar]
- 32.Guo B, Chatterjee S, Li L, et al. The clock gene, brain and muscle Arnt-like 1, regulates adipogenesis via Wnt signaling pathway. FASEB J 2012;26:3453–3463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemmeryckx B, Himmelreich U, Hoylaerts MF, Lijnen HR. Impact of clock gene Bmal1 deficiency on nutritionally induced obesity in mice. Obesity (Silver Spring) 2011;19:659–661 [DOI] [PubMed] [Google Scholar]
- 34.Schweiger M, Schreiber R, Haemmerle G, et al. Adipose triglyceride lipase and hormone-sensitive lipase are the major enzymes in adipose tissue triacylglycerol catabolism. J Biol Chem 2006;281:40236–40241 [DOI] [PubMed] [Google Scholar]
- 35.Osuga J, Ishibashi S, Oka T, et al. Targeted disruption of hormone-sensitive lipase results in male sterility and adipocyte hypertrophy, but not in obesity. Proc Natl Acad Sci USA 2000;97:787–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Haemmerle G, Lass A, Zimmermann R, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 2006;312:734–737 [DOI] [PubMed] [Google Scholar]
- 37.Hoy AJ, Bruce CR, Turpin SM, Morris AJ, Febbraio MA, Watt MJ. Adipose triglyceride lipase-null mice are resistant to high-fat diet-induced insulin resistance despite reduced energy expenditure and ectopic lipid accumulation. Endocrinology 2011;152:48–58 [DOI] [PubMed] [Google Scholar]
- 38.Huijsman E, van de Par C, Economou C, et al. Adipose triacylglycerol lipase deletion alters whole body energy metabolism and impairs exercise performance in mice. Am J Physiol Endocrinol Metab 2009;297:E505–E513 [DOI] [PubMed] [Google Scholar]
- 39.Ahmadian M, Abbott MJ, Tang T, et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab 2011;13:739–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep 2006;7:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmermann R, Strauss JG, Haemmerle G, et al. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 2004;306:1383–1386 [DOI] [PubMed] [Google Scholar]
- 42.Haemmerle G, Moustafa T, Woelkart G, et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med 2011;17:1076–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oliver P, Caimari A, Díaz-Rúa R, Palou A. Diet-induced obesity affects expression of adiponutrin/PNPLA3 and adipose triglyceride lipase, two members of the same family. Int J Obes (Lond) 2012;36:225–232 [DOI] [PubMed] [Google Scholar]
- 44.Langin D, Dicker A, Tavernier G, et al. Adipocyte lipases and defect of lipolysis in human obesity. Diabetes 2005;54:3190–3197 [DOI] [PubMed] [Google Scholar]
- 45.Morimoto C, Tsujita T, Okuda H. Antilipolytic actions of insulin on basal and hormone-induced lipolysis in rat adipocytes. J Lipid Res 1998;39:957–962 [PubMed] [Google Scholar]
- 46.Wang MY, Lee Y, Unger RH. Novel form of lipolysis induced by leptin. J Biol Chem 1999;274:17541–17544 [DOI] [PubMed] [Google Scholar]
- 47.Satoh Y, Kawai H, Kudo N, Kawashima Y, Mitsumoto A. Time-restricted feeding entrains daily rhythms of energy metabolism in mice. Am J Physiol Regul Integr Comp Physiol 2006;290:R1276–R1283 [DOI] [PubMed] [Google Scholar]
- 48.Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci USA 2012;109:5541–5546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimba S, Ogawa T, Hitosugi S, et al. Deficient of a clock gene, brain and muscle Arnt-like protein-1 (BMAL1), induces dyslipidemia and ectopic fat formation. PLoS ONE 2011;6:e25231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delezie J, Dumont S, Dardente H, et al. The nuclear receptor REV-ERBα is required for the daily balance of carbohydrate and lipid metabolism. FASEB J 2012;26:3321–3335 [DOI] [PubMed] [Google Scholar]
- 51.Solt LA, Wang Y, Banerjee S, et al. Regulation of circadian behaviour and metabolism by synthetic REV-ERB agonists. Nature 2012;485:62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hirota T, Lee JW, St John PC, et al. Identification of small molecule activators of cryptochrome. Science 2012;337:1094–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatori M, Vollmers C, Zarrinpar A, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metab 2012;15:848–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barclay JL, Husse J, Bode B, et al. Circadian desynchrony promotes metabolic disruption in a mouse model of shiftwork. PLoS ONE 2012;7:e37150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.