

Abstract
The twin arginine transport (Tat) system transports folded proteins across the prokaryotic cytoplasmic membrane and the plant thylakoid membrane. In Escherichia coli three membrane proteins, TatA, TatB and TatC, are essential components of the machinery. TatA from Providencia stuartii is homologous to E. coli TatA but is synthesized as an inactive pre-protein with an N-terminal extension of eight amino acids. Removal of this extension by the rhomboid protease AarA is required to activate P. stuartii TatA. Here we show that P. stuartii TatA can functionally substitute for E. coli TatA provided that the E. coli homologue of AarA, GlpG, is present. The oligomerization state of the P. stuartii TatA pro-protein was compared with that of the proteolytically activated protein and with E. coli TatA. The pro-protein still formed small homo-oligomers but cannot form large TatBC-dependent assemblies. In the absence of TatB, E. coli TatA or the processed form of P. stuartii TatA form a complex with TatC. However, this complex is not observed with the pro-form of P. stuartii TatA. Taken together our results suggest that the P. stuartii TatA pro-protein is inactive because it is unable to interact with TatC and cannot form the large TatA complexes required for transport.
Introduction
Protein transport across the cytoplasmic membrane of prokaryotes and the thylakoid membrane of plant chloroplasts proceeds by one of two general mechanisms. The Sec pathway uses the energy of ATP hydrolysis and the transmembrane proton electrochemical gradient (Δp) to drive the transport of unfolded proteins across the membrane (Driessen and Nouwen, 2008). By contrast, the Tat pathway transports folded proteins, in a reaction powered solely by Δp (Cline and Theg, 2007; Palmer et al., 2010b). Proteins are directed to each of these pathways by the presence of cleavable N-terminal signal peptides. The signal peptides of Tat substrates, unlike Sec signal peptides, contain a highly conserved twin arginine motif which is essential for efficient targeting of substrates to the Tat transport machinery (Berks, 1996; Stanley et al., 2000).
In Gram-negative bacteria, such as Escherichia coli, the Tat machinery is made up of three membrane-bound proteins, TatA, TatB and TatC (Bogsch et al., 1998; Sargent et al., 1998; 1999; Weiner et al., 1998). In E. coli a fourth protein, TatE, is a functional homologue of TatA and forms a minor component of the Tat machinery (Sargent et al., 1998; Jack et al., 2001). In E. coli and closely related bacteria TatD is encoded by the fourth gene in the tatABCD operon, but is a cytoplasmic nuclease with no role in Tat transport (Wexler et al., 2000; Lindenstrauss et al., 2010). The three major Tat components form two different types of multimeric complexes in E. coli membranes. A complex of TatB and TatC can be isolated which contains each protein in a 1:1 molar ratio (Bolhuis et al., 2001). The exact number of TatB and TatC proteins within this complex is unknown, but it is probably between six and eight of each subunit (Bolhuis et al., 2001; Richter and Bruser, 2005; Tarry et al., 2009). Low levels of TatA co-purify with the TatBC complex when all three proteins are overproduced, but the complex lacks TatA when purified from cells expressing tatABC at native levels (McDevitt et al., 2005; 2006). The TatBC complex functions as a receptor for Tat substrates, with the twin arginine motif of the substrate signal peptide being recognized by TatC (Cline and Mori, 2001; de Leeuw et al., 2002; Alami et al., 2003; Tarry et al., 2009).
The TatA protein comprises a single transmembrane helix, followed by an amphipathic helix and an unstructured C-terminal tail (Porcelli et al., 2002; Lee et al., 2006). The N-terminus of TatA is located at the periplasmic side of the membrane (Lee et al., 2006; Koch et al., 2012). Purified TatA forms a series of large homo-oligomers. Analysis of these TatA complexes by negative stain electron microscopy reveals ring-shaped structures with a range of different diameters in which a large central cavity is enclosed at one end (Gohlke et al., 2005). The variation in diameter results from differences in the number of TatA subunits while the presence of an enclosed cavity suggests that TatA complexes form transport channels of different internal size (Gohlke et al., 2005). Large assemblies of TatA have also been seen in vivo when a C-terminally YFP-tagged variant of TatA was produced at native levels (Leake et al., 2008). However, in the absence of TatB or TatC, the large assemblies of YFP-tagged TatA were not seen. Instead, fluorescence recovery after photobleaching (FRAP) experiments are consistent with TatA–YFP being arranged as small, possibly tetrameric units (Leake et al., 2008). Cross-linking studies of the chloroplast TatA orthologue Tha4 have also been interpreted as showing this protein in a tetramer state in resting thylakoid membranes (Dabney-Smith and Cline, 2009).
The requirement for TatBC in the assembly of large TatA–YFP oligomers indicates at least transient interactions between TatA with TatBC. This inference is supported by cross-linking experiments in thylakoids which detected Tha4 interactions with the thylakoid equivalent of the TatBC complex in the presence of substrate and a Δp (Mori and Cline, 2002). These cross-links were no longer observed once the substrate had passed across the membrane indicating that TatA-TatBC interactions are transitory and occur only during active protein transport (Mori and Cline, 2002). Nevertheless, a recent cross-linking study suggests that monomeric TatA may associate with TatBC even in the absence of substrate. Contacts were observed between the TatA transmembrane helix and TatC, and between the TatA amphipathic helix and TatB (Frobel et al., 2011).
The Tat machineries of some Gram-positive bacteria and of archaea are comprised only of TatA and TatC proteins in contrast to the three-component TatB-containing systems found in Gram-negative bacteria and plant chloroplasts (Jongbloed et al., 2004; Dilks et al., 2005). However, protein purification studies indicate that the two-component Tat system of the Gram-positive bacterium Bacillus subtilis still forms two distinct membrane-bound complexes, one of which contains TatA and TatC proteins, and the second of which contains only TatA (Barnett et al., 2008; 2009). The physiological relevance of a large cytoplasmic aggregate of B. subtilis TatA protein that is seen in addition to membrane-bound TatA in the native organism, and also upon heterologous production in E. coli, is not clear (Westermann et al., 2006; Barnett et al., 2009). It is likely that the two- and three-component Tat machineries operate by a similar mechanism despite their differences in subunit composition. This hypothesis is supported by the isolation of point mutations in tatA that permit function of the E. coli Tat machinery in the absence of TatB (Blaudeck et al., 2005).
Recently it was demonstrated that the TatA protein from the enterobacterium Providencia stuartii, is synthesized as an inactive pro-protein with an N-terminal extension of eight amino acids (Stevenson et al., 2007). Activation of this TatA protein requires the regulated removal of the N-terminal extension by the membrane-bound rhomboid protease, AarA (Stevenson et al., 2007; Strisovsky et al., 2009). The active site of the rhomboid protease localizes close to the periplasmic side of the membrane, consistent with the N-out topology of TatA (Wang et al., 2006; Maegawa et al., 2007). P. stuartii is a close relative of E. coli and the P. stuartii TatA protein can replace the function of E. coli TatA and TatE in E. coli strains lacking these Tat components (Stevenson et al., 2007). GlpG is the E. coli homologue of the AarA rhomboid protease and has been shown to process P. stuartii TatA in vitro (Strisovsky et al., 2009).
In this study we have taken advantage of the cross-functionality of the P. stuartii and E. coli TatA proteins to address how the N-terminal extension of the P. stuartii TatA pro-protein impedes Tat function. Our results suggest that the extension inhibits the formation of large assemblies of TatA by preventing interaction between TatA and TatC.
Results
The TatA protein of P. stuartii requires N-terminal processing by GlpG to function in E. coli
It was previously reported that the P. stuartii TatA protein (hereafter TatAPs) can functionally replace E. coli TatA and TatE in phenotypic tests (Stevenson et al., 2007). However, the efficiency of this complementation, and whether the activity of TatAPs in E. coli was dependent upon the presence of the E. coli rhomboid protease GlpG, has not been assessed. To provide this information we deleted glpG in E. coli strain JARV16 (ΔtatAΔtatE) and then used this genetic background to express plasmid-borne alleles coding for either C-terminally His-tagged E. coli TatA (TatAEc), TatAPs, or a truncated TatAPs variant (TatAPsΔ2–8) lacking the rhomboid-sensitive N-terminal extension (deletion of amino acids two to eight; shown schematically in Fig. 1A). We cultured the strains anaerobically in the presence of trimethylamine-N-oxide (TMAO) and then assayed the activity of the Tat substrate TMAO reductase in the periplasmic fraction.
Fig. 1.

P. stuartii TatA is functional in E. coli but requires processing by GlpG for activity. A. Alignment of the N-termini of the E. coli and P. stuartii TatA proteins (TatAEc and TatAPs respectively), along with the truncated variant of P. stuartii TatA (TatAPsΔ2–8) used in this study. Identical amino acids are shaded in black, conserved amino acids are shaded grey. The cleavage site for the rhomboid proteases AarA and GlpG is indicated with an arrow. B. P. stuartii TatA is active in E. coli in a glpG+ background. TMAO reductase activities were measured from the periplasmic fractions of the indicated strains carrying either pQE60 (labelled Vector), or pQE60 encoding C-terminally His-tagged variants of each of E. coli TatA (TatAEc), P. stuartii TatA (TatAPs) or a genetically truncated variant of P. stuartii TatA where codons 2–8 were lacking (TatAPsΔ2–8). One hundred per cent activity is that determined from the periplasmic fraction of MC4100 harbouring pQE60 and corresponds to an activity of 4.7 µM benzyl viologen oxidized per min per mg protein. The error bars represent standard error of the mean (n = 5–8). C. P. stuartii TatA is processed in vivo by E. coli GlpG. Crude membrane fractions were prepared from E. coli strains JARV16-P (ΔtatAΔtatE) and H43FF-P (ΔtatAΔtatEΔglpG) producing C-terminally His-tagged variants of the full-length or genetically truncated P. stuartii TatA proteins. Samples (1 µg membrane protein) were separated by SDS-PAGE (using a 10–20% Tris-tricine gradient gel) electroblotted and protein detected with a horseradish-peroxidase-conjugated penta-histidine antibody.
E. coli strain JARV16 expressing TatAPs had a periplasmic TMAO reductase activity that was indistinguishable from the Tat+E. coli parental strain (Fig. 1B) indicating that TatAPs can support a high level of Tat transport activity. In contrast, when glpG was also deleted from the ΔtatAΔtatE mutant strain, expression of TatAPs no longer complemented the ΔtatAΔtatE mutant phenotype. This was confirmed by the inability of this strain and plasmid combination to grow anaerobically on minimal media with TMAO as sole electron acceptor, or on media containing SDS [sensitivity to which arises due to the inability to export two Tat-dependent amidases involved in cell wall remodelling (Bernhardt and de Boer, 2003; Ize et al., 2003); data not shown]. As expected, plasmid-borne tatAEc restored periplasmic export of TMAO reductase to the ΔtatAΔtatE mutant strain, JARV16, regardless of whether GlpG was present or not, confirming that the activity of E. coli TatA does not depend upon processing by GlpG. The truncated TatAPsΔ2–8 variant restored TMAO reductase export in E. coli strains lacking native TatA and TatE, and this complementation was independent of GlpG. However, it should be noted that TatAΔ2–8 supported a considerably lower periplasmic TMAO reductase activity than found for the wild-type E. coli strain. This suggests that the genetically truncated P. stuartii TatA variant is less active than the GlpG-processed protein. This difference in activity may be ascribed to the initiator methionine found only on the genetic truncation.
The results presented in Fig. 1B show that the Tat-dependent activity of TatAPs in E. coli strains depends on the presence of GlpG. To ascertain whether this was associated with processing of TatAPs to a shorter form, Western blot analysis of P. stuartii TatA was performed using the C-terminal His-tag as an epitope. These experiments were carried out using the same E. coliΔtatAΔtatE mutant strains described above but additionally containing the pcnB1 allele to restrict plasmid copy number and avoid saturation of GlpG protease activity by overproduced TatAPs. In the glpG+ background, JARV16-P, the TatAPs protein has the same mobility as the genetically truncated TatAPsΔ2–8 variant (Fig. 1C). By contrast, in the glpG mutant strain, H43FF-P, the TatAPs protein migrated with a slightly lower mobility than the truncated form. These results indicate that all of the TatAPs is fully cleaved by GlpG, and that GlpG is the only protease in E. coli that can process the pro-protein. These findings are fully consistent with in vitro experiments showing that the transmembrane domain of P. stuartii TatA can be cleaved by purified GlpG (Stevenson et al., 2007; Strisovsky et al., 2009).
We next tested whether the tatAPs gene showed genetic dominance, i.e. whether the inactive TatAPs protein that is produced in the absence of GlpG interferes with the function of wild-type E. coli TatA. To this end, plasmid-borne tatAPs was expressed in both wild-type and ΔglpG strains (MC4100 and H1FF respectively). No reduction in periplasmic TMAO reductase activity was observed in either strain (Fig. S1). We therefore conclude that the pro-form of P. stuartii TatA protein does not interfere with the function of the native E. coli Tat machinery.
The N-terminal extension of P. stuartii TatA does not prevent homo-oligomerization
One of the key features of TatA proteins is their ability to self-interact. Chemical cross-linking has been used to detect homo-oligomers of E. coli TatA, and of the thylakoid TatA orthologue Tha4, in membrane fractions (De Leeuw et al., 2001; Dabney-Smith et al., 2006; Greene et al., 2007). We therefore used chemical cross-linking to ascertain whether oligomerization of the P. stuartii TatA protein could be observed and whether this was affected by the presence of the N-terminal amino acid extension. Plasmid-encoded His-tagged TatAPs was produced in E. coliΔtatAΔtatE mutant strains that were either glpG+ (JARV16) or deleted for glpG (H43FF). Membrane fractions were then prepared from these strains, and from control strains producing His-tagged TatAEc, and treated with the bifunctional chemical cross-linker disuccinimidyl suberate (DSS) which cross-links exposed amine residues.
Cross-linked species up to apparent homo-tetramers could be detected for both TatAPs and TatAEc (Fig. 2), consistent with an earlier study of DSS cross-linking of TatAEc (De Leeuw et al., 2001). Importantly, cross-linked oligomers of TatAPs were observed regardless of whether GlpG was present or absent, demonstrating that the N-terminal extension on the P. stuartii TatA protein does not prevent homo-oligomerization of the protein to at least the level of tetramer.
Fig. 2.

The N-terminal extension present on P. stuartii TatA does not prevent homo-oligomer formation. Crude membrane fractions were prepared from E. coli strains JARV16 (ΔtatAΔtatE) and H43FF (ΔtatAΔtatEΔglpG) producing C-terminally His-tagged variants of either E. coli or P. stuartii TatA. Membrane fractions (30 µg protein) were treated with 2 mM DSS at pH 7.4 for 30 min. Samples (2 µg per lane) were subsequently separated by Tris-glycine SDS-PAGE (12.5% acrylamide), electroblotted and protein was detected with a horseradish-peroxidase-conjugated penta-histidine antibody. The positions of TatA multimers are indicated to the right-hand side and of molecular weight markers to the left-hand side of each blot.
The N-terminal extension of P. stuartii TatA inhibits the formation of large TatA complexes in vivo
It has previously been reported that a C-terminal YFP fusion of TatAEc forms distinct fluorescent foci containing tens of TatAEc–YFP molecules in E. coli cells when both TatB and TatC are present (Leake et al., 2008). In the absence of TatB or TatC, these large TatA assemblies were not detected and instead TatAEc–YFP was present in smaller clusters containing an average of four molecules. Since our data reported above supported the idea that the pro-form of TatAPs was still able to self-interact to form at least small oligomers, we next sought to test whether the formation of the large assemblies of TatA were prevented by the presence of the N-terminal amino acid extension. To achieve this, we constructed a TatAPs–YFP fusion and expressed this under the control of the E. coli tatA promoter from the lambda phage attachment site of selected E. coli strains. Figure 3A shows the periplasmic TMAO reductase activity of E. coliΔtatAΔtatE strain JARV16, producing either TatAEc, or TatAPs, or TatAPs–YFP from a single gene copy at the lambda attachment site. The strain producing TatAPs–YFP has almost the same level of periplasmic TMAO reductase activity as the same strain producing the non-YFP-tagged P. stuartii TatA protein. If glpG was also absent from the ΔtatAΔtatE strain, the TatAPs–YFP protein was, as expected, not functional, with the periplasmic TMAO reductase activity measured from this strain being indistinguishable from that of the negative control. Taken together these results indicate that the YFP-tagged variant of TatAPs retains good Tat transport activity which is fully dependent upon N-terminal processing by GlpG.
Fig. 3.
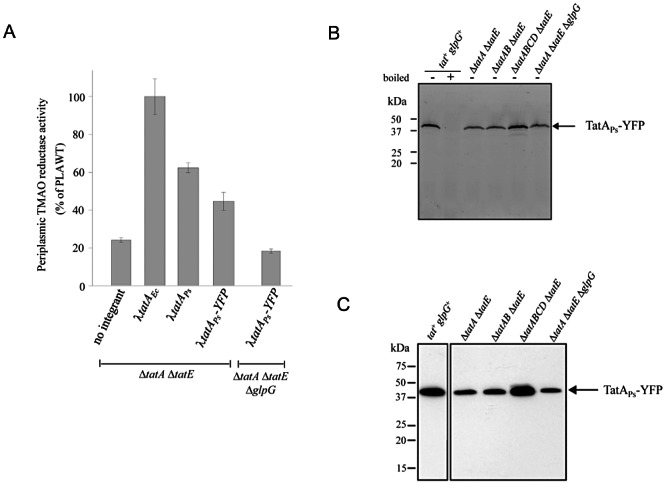
A P. stuartii TatA–YFP fusion protein is stably produced in E. coli and has TatA activity. A. P. stuartii TatA fused to YFP is active in E. coli. Periplasmic TMAO reductase activity was measured from strains deleted for the chromosomal tatA and tatE genes and expressing from the attB site: E. coli tatA (λtatAEc; strain PLAWT), P. stuartii tatA (λtatAPs; strain JARV16 λTatAPs) or a P. stuartii tatA–YFP fusion (λtatAPs–YFP; strain JARV16 λAPsALYFP). In addition activity was also measured from the strain producing the P. stuartii tatA–YFP fusion that was lacking tatA, tatE and glpG (strain H43FF λAPsALYFP). Activities shown are relative to that of the periplasmic fraction of strain PLAWT which corresponds to 3.3 µM benzyl viologen oxidized per min per mg protein. Error bars represent standard error of the mean (n = 3). B. The P. stuartii TatA–YFP fusion protein is fluorescent. Cell lysates (approximately 30 µg protein) of the strains MC4100 λAPsALYFP (tat+, glpG+), JARV16 λAPsALYFP (ΔtatAΔtatE), BEAD λAPsALYFP (ΔtatABΔtatE), DADE λAPsALYFP (ΔtatABCDΔtatE) and H43FF λAPsALYFP (ΔtatABCDΔtatEΔglpG) each producing the P. stuartii TatA–YFP fusion encoded at the attB site were separated by SDS-PAGE. The samples were not boiled prior to analysis with the exception of one of the MC4100 λAPsALYFP (tat+glpG+) samples, as indicated. Following SDS-PAGE the gel was excited with a laser at 473 nm and the fluorescent image was captured. C. The P. stuartii TatA–YFP fusion protein is stable. Un-boiled cell lysates of the indicated strains producing the P. stuartii TatA–YFP fusion protein were separated by SDS-PAGE, electroblotted and detected using an anti-GFP antibody. In (B) and (C) the molecular weight marker is shown to the left-hand side of the gel. An arrow at the right-hand side of the gel indicates the position of the P. stuartii TatA–YFP fusion.
Before the in vivo behaviour of the TatAPs–YFP fusion protein was analysed, we first confirmed that the fusion protein was folded and stable. A single fluorescent species was detected in whole cell samples analysed by semi-native PAGE (Fig. 3B). Analysis of the same samples by denaturing SDS-PAGE and Western blotting with an anti-GFP antibody revealed a single immunoreactive band corresponding to the size of the fusion protein with no evidence of degradation to smaller forms (Fig. 3C). More fusion protein was present in the tat+ and ΔtatABCDΔtatE backgrounds than in the other strains. The reason for this is unclear because all of the strains were constructed in an identical manner. Analysis of the subcellular localization of the fusion protein indicated that it was found exclusively in the membrane fraction in all strains (data not shown).
We next analysed the oligomerization behaviour of the TatAPs–YFP fusion protein. When the fusion was produced in the E. coli JARV16 (ΔtatAΔtatE) strain background, several large foci of fluorescence could be seen in each cell (Fig. 4). The TatAPs–YFP protein, therefore, shows similar behaviour to the E. coli TatA–YFP fusion, which also forms large fluorescent foci when TatB and TatC are present (Leake et al., 2008). In the DADE (which lacks all E. coli Tat components) or BEAD (which produces only E. coli TatC) strain backgrounds, fluorescent foci of TatAPs–YFP were not seen, and instead diffuse fluorescence could be seen all around the cell periphery, again as observed previously with E. coli TatA–YFP (Leake et al., 2008). Similarly a strain lacking all chromosomal tat genes and producing TatAPs–YFP did not form fluorescent foci when TatB or TatC alone were produced from an inducible plasmid (Fig. S2). It should be noted that cells of these strains show a chaining morphology due to the failure to export Tat-dependent cell wall amidases (Bernhardt and de Boer, 2003; Ize et al., 2003). Importantly when the TatAPs–YFP protein was observed in an E. coliΔtatAΔtatE background that was additionally deleted for glpG, the large fluorescent foci were also no longer seen, with only disperse fluorescence visible around the cell periphery. Taken together these results strongly suggest that the N-terminal extension on P. stuartii TatA inactivates Tat function by preventing the formation of large TatA assemblies in vivo.
Fig. 4.

The N-terminal extension of P. stuartii TatA prevents the formation of large TatA assemblies in vivo. Fluorescence microscopy of strains JARV16 λAPsALYFP (ΔtatAΔtatE), H43FF λAPsALYFP (ΔtatABCDΔtatEΔglpG), BEAD λAPsALYFP (ΔtatABΔtatE) and DADE λAPsALYFP (ΔtatABCDΔtatE) producing the P. stuartii TatA–YFP fusion encoded at the attB site. The top images show cells in differential interference contrast (DIC) and bottom images show fluorescence of the P. stuartii TatA–YFP fusion protein. Scale bars correspond to 5 µm.
As a control, we also examined the behaviour of the genetically truncated TatAPsΔ2–8 variant fused to YFP. As expected, this YFP fusion protein formed foci in the ΔtatAΔtatE background regardless of whether glpG was present or absent, and also corrected the chain-forming phenotype of the strain in a glpG-independent manner (Fig. S3).
E. coli TatAC complexes can be isolated in the absence of TatB
In current models for Tat transport binding of a substrate to the TatBC complex in energized membranes primes TatBC to bind and polymerize TatA (e.g. Mori and Cline, 2002; Alami et al., 2003). Thus, one possible explanation for the failure of the pro-form of TatAPs to form large complexes may be that it is unable to interact with one or more components of the TatBC complex. Several lines of evidence indicate that TatA is likely to bind to the TatC component. However, to date a complex containing only E. coli TatA and TatC proteins has not been isolated.
We investigated whether E. coli TatC and TatA were able to form complexes by co-producing TatA and hexahistidine-tagged TatC (TatChis) in the absence of other Tat components. Cell membranes were solubilized with 1% digitonin and the solubilized material was applied to a Ni2+-affinity column. TatChis was eluted from the column using an imidazole gradient. Fractions containing TatChis were pooled and subjected to gel filtration chromatography. Immunoblotting revealed TatA co-migrating with the affinity-purified TatChis across a broad peak centred on an apparent molecular mass of approximately 500 kD (Fig. 5). These data indicate that E. coli TatA can interact directly with TatC in the absence of TatB. Instead of forming a single discrete complex, as seen for TatBChis (Orriss et al., 2007), the TatAChis complexes are heterogeneous. The yield of purified TatAChis complexes was too low to allow further analysis. However, SDS-PAGE analysis of the concentrated affinity-purified fractions loaded on the gel filtration column revealed only two Coomassie Blue-staining bands corresponding to TatA and TatChis (Fig. 5, inset). The relative staining intensities of the two bands suggest that TatA is not present at a significant molar excess over TatChis in these complexes. Analysis of the unbound fraction from the Ni2+-affinity column showed that the vast majority of the TatA present in the soluble extract was not retained by the column and thus was not stably associated with TatChis (data not shown).
Fig. 5.

E. coli TatA co-purifies with TatC in the absence of TatB. TatA and TatChis were co-produced in E. coli strain DADE (ΔtatABCDΔtatE). Membrane proteins were solubilized in digitonin. His-tagged TatC and associated TatA was purified using nickel IMAC chromatography and visualized by Coomassie Blue staining after SDS-PAGE (inset). Purified TatAChis complexes were subjected to gel filtration chromatography on a Superose 6 column and eluted as a broad peak (main image). Beta-amylase (200 kDa), apoferritin (443 kDa) and thyroglobulin (669 kDa) were used for molecular weight calibration (arrows). TatA and TatC were detected in the gel filtration fractions (indicated by lines) by Western blotting. All analysed fractions contained both TatA and TatC (bottom).
The N-terminal extension of P. stuartii TatA inhibits complex formation with TatC
To investigate whether the unprocessed N-terminal extension of P. stuartii TatA impaired interaction with TatC we developed a small-scale co-purification assay to allow parallel analysis of proteins expressed in multiple background strains. To this end, E. coli TatChis was co-produced with either TatAPs or TatAEc, membrane proteins were extracted with the detergent C12E9, and histidine-tagged TatC was purified from the solubilized material using Ni2+-affinity resin. The presence of TatA protein in the bound sample was then detected by Western blotting.
For these experiments it was necessary to provide P. stuartii TatA with a C-terminal epitope to allow detection, since our E. coli TatA antiserum does not detect TatAPs. Initially we added a C-terminal haemagglutinin (HA) tag to give construct TatAPsHA. However, periplasmic TMAO reductase activity assays (Fig. 6A) show that this modification blocked Tat transport (and indeed the same tag also completely inactivated E. coli TatA; data not shown). Therefore, as an alternative approach, we tagged TatAPs with the last 10 C-terminal amino acids of E. coli TatA (forming TatAPsEc) since we had shown previously that this was the major epitope recognized by the E. coli TatA antiserum (Lee et al., 2002). The E. coliΔtatAΔtatE mutant strain, JARV16-P, producing TatAPsEc had periplasmic TMAO reductase activity levels that were as high as those of cells expressing the corresponding constructs with E. coli TatA or untagged P. stuartii TatA (Fig. 6A). Additionally, it is clear from the Western blot shown in Fig. 6B that fusing the last 10 amino acids of E. coli TatA to the C-terminus of P. stuartii TatA results in strong recognition of the tagged P. stuartii TatA protein by the anti-E. coli TatA antiserum.
Fig. 6.

The N-terminal extension of P. stuartii TatA inhibits interaction with TatC. A. P. stuartii TatA with a C-terminal epitope from E. coli TatA is active in the E. coli Tat system. Periplasmic TMAO reductase activity was measured from E. coli strain JARV16-P (tatAΔtatE pcnB1) harbouring plasmids producing His-tagged E. coli TatChis in tandem with either E. coli TatA (TatAEc), P. stuartii TatA (TatAPs), P. stuartii TatA with a C-terminal haemagglutinin epitope (TatAPsHA) or P. stuartii TatA with a C-terminal epitope comprising the last 10 amino acids from E. coli TatA (TatAPsEc). The same strain harbouring plasmid pQE60 (vector) was used as a negative control. Activities shown are relative to that of the periplasmic fraction of JARV16/pQE TatAEc-TatChis and correspond to 5.7 µM benzyl viologen oxidized per min per mg protein. Error bars represent standard error of the mean (n = 3). B. P. stuartii TatA with a C-terminal epitope of E. coli TatA is detected by the anti-E. coli TatA antiserum. Cell lysates of JARV16-P containing pQE60 (vector) or producing His-tagged E. coli TatC in tandem with either E. coli TatA (TatAEc), P. stuartii TatA (TatAPs) or P. stuartii TatA with a C-terminal epitope from E. coli TatA (TatAPsEc) were separated by SDS-PAGE (15% acrylamide). Proteins were electroblotted and detected with an E. coli TatA antiserum. Arrows at the right-hand side of the blot indicate the positions of protein bands of TatAEc and TatAPsEc. Molecular weight markers are shown to the left-hand side of the blot. C and D. E. coli TatA or processed P. stuartii TatA co-purify with His-tagged E. coli TatC. Crude membrane fractions of the E. coli strain DADE (ΔtatABCDΔtatE; left-hand panels in C and D) or H0FF (ΔtatABCDΔtatE, ΔglpG; right-hand panels in C and D) both harbouring pREP4 (Zamenhof and Villarejo, 1972) and over-producing either E. coli TatA (TatAEc) or epitope-tagged P. stuartii TatA (TatAPsEc) in tandem with hexa-histidine-tagged E. coli TatC (TatChis) were solubilized with detergent and the TatChis protein purified using nickel-charged beads as described in Experimental procedures. Proteins that eluted from the beads were separated by SDS-PAGE (15% acrylamide), electroblotted and immunoreactive bands were detected with anti-tetra-histidine antibody (TatC blot, top) or anti-E. coli TatA antiserum (TatA blot, bottom). Samples are crude membrane fraction (CM), solubilized membrane fraction (SM), unbound fraction (U), wash (W), elution (E).
The co-purification assay was applied to E. coli TatA and TatChis co-produced in a ΔtatABCDΔtatE background using strain DADE. Blotting the eluted fraction with anti-TatA or anti-histag antibodies revealed that TatAEc and TatChis were both present in the eluate confirming that the assay can detect the interaction between these two E. coli proteins (Fig. 6C). As expected, E. coli TatA co-purified with TatChis regardless of whether the proteins were isolated from a glpG+ or a glpG- strain (compare Fig. 6C left-hand and right-hand panels).
When the same experiments were repeated with epitope-tagged P. stuartii TatA protein, TatAPsEc (Fig. 6D), the P. stuartii TatA protein co-purified with His-tagged E. coli TatC when the proteins were isolated from a glpG+ strain (left-hand panel). However, when the proteins were isolated from a glpG strain, none of the P. stuartii TatA protein was found in the eluted fraction, even though His-tagged TatC was clearly eluted from the resin (Fig. 6D, right-hand panel). To confirm that the binding of the P. stuartii TatA protein to the Ni2+-charged resin was due to interaction with TatC, and not to unspecific binding to the resin, control experiments were carried out where a non-tagged variant of E. coli TatC was used. In this case neither TatC nor the epitope-tagged P. stuartii TatA protein bound to the column, regardless of whether they were produced in a glpG+ or a glpG background (Fig. S4). Furthermore, we observed the same GlpG-dependent interaction of the (transport inactive) HA-tagged P. stuartii TatA protein with E. coli TatChis (data not shown), indicating that the interaction is not dependent upon the nature of epitope tag. Taken together these results demonstrate that the N-terminal extension found on the P. stuartii TatA protein inhibits interaction with TatC.
As an additional control we assessed whether the genetically truncated variant of TatAPsEc, which lacked codons 2–8 could interact with E. coli TatC in a GlpG-independent manner. We found that this truncated TatA variant did not detectably co-purify with His-tagged E. coli TatC (Fig. S5). This is consistent with the observation that the genetically truncated TatAPs could not fully complement the Tat transport defect of the E. coli tatA/E mutant strain (Fig. 1) and suggests that the presence of the N-terminal methionine (which is lacking in the rhomboid-cleaved TatAPs variant) alters the stability of the TatAC complex so that it cannot survive detergent extraction or salt washing.
Discussion
In this study we have used the inactive TatA protein from P. stuartii, which harbours a short N-terminal extension, as a tool to probe the Tat transport process. This protein is able to complement the Tat deficiency of an E. coliΔtatAΔtatE mutant strain provided that the E. coli rhomboid protease GlpG is present to cleave off the extension. This cross-complementation allowed us to take advantage of the range of E. coli genetic backgrounds and expression systems to probe the origin of the Tat defect associated with the unprocessed amino acid extension on TatA and to use this information to make inferences about the mechanism of Tat transport.
Previous studies have indicated that the E. coli TatA protein is able to self-interact to form complexes of varying size ranging from putative tetramers to much larger assemblies containing tens of subunits (De Leeuw et al., 2001; Porcelli et al., 2002; Gohlke et al., 2005; Oates et al., 2005; Leake et al., 2008). Similar behaviour has been inferred for the thylakoid TatA orthologue Tha4 (Dabney-Smith et al., 2006; Dabney-Smith and Cline, 2009). Our in vitro cross-linking experiments with the unprocessed form of TatAPs have shown that the N-terminal extension does not stop the TatA protein from forming small (tetrameric) homo-oligomers. However, in vivo imaging of a TatAPs–YFP fusion revealed that the N-terminal extension prevented the formation of the large assemblies of TatA that were observed for the processed form of the protein and that have been previously reported for the E. coli TatA protein. We found that formation of large TatAPs–YFP assemblies by the processed form of TatA requires the TatB and TatC proteins, again as previously observed for E. coli TatA (Leake et al., 2008). This raised the possibility that the unprocessed form of TatAPs was inactive because it was unable to interact with the TatBC complex.
The precise nature of the interaction of TatA with TatBC during Tat transport is not known. The purification of affinity-tagged TatA from cells overproducing all three E. coli proteins results in the co-purification of a low level of TatB, but no detectable TatC is co-purified (Sargent et al., 2001; de Leeuw et al., 2002). However, almost all of the TatB protein is found in a complex with TatC when affinity-tagged TatC is purified (e.g. Bolhuis et al., 2001; McDevitt et al., 2006). Very little TatA is associated in this TatBC complex, and it is not known with which of the proteins in the TatBC complex that TatA interacts. On the basis that variant E. coli TatA proteins have been isolated that are bifunctional and support a low level of Tat transport activity in the absence of TatB (Blaudeck et al., 2005), and that some Gram-positive Tat systems lack a TatB component (Jongbloed et al., 2006), we reasoned that TatA might interact directly with TatC during the Tat transport process rather than via TatB. Indeed, we were able to show that a stable interaction between E. coli TatC and TatA occurs in the absence of TatB. This observation is consistent with bimolecular fluorescence complementation data that indicate interaction between TatA and TatC in the absence of TatB (Kostecki et al., 2010) and site-specific cross-linking experiments that suggest that the transmembrane helix of TatA is close to TatC but not TatB (Frobel et al., 2011). Earlier analyses of E. coli strains expressing TatA and TatC in the absence of TatB failed to observe distinct TatC-containing complexes in solubilized membrane extracts (Barrett et al., 2007; Behrendt et al., 2007). This was ascribed to TatA interfering with the formation of the otherwise stable TatC complex or the formation of unstable TatAC complexes. Nevertheless, we have succeeded in isolating TatAC complexes. These complexes are apparently heterogeneous in composition and we speculate that this is why they were not observed in the earlier studies which were attempting to identify discrete TatC species.
Although the processed form of TatAPs was able to form a complex with TatC, similar to E. coli TatA, the unprocessed form of the protein could no longer be co-purified with TatC. This indicates that the N-terminal extension on TatA disrupts the interaction between TatA and TatC. Combining this idea with the observation that unprocessed TatAPs is also defective in the formation of large TatA complexes in vivo leads to the inference that substrate-induced polymerization of TatA is mediated by direct contact of TatC with TatA. Since strains co-producing either E. coli or P. stuartii TatA–YFP with TatC in the absence of TatB are unable to form large TatA complexes (Fig. 4; Leake et al., 2008) even though we are able to purify TatAC complexes containing small amounts of TatA (Figs 5 and 6C and D) we infer that without the assistance of TatB TatC can only bind a nucleus of TatA and is unable to drive full TatA polymerization. Our observations highlight the N-terminal region of TatA as playing a key role in the interaction with TatC. This is consistent with the findings of Blaudeck et al. (2005) who found that mutations falling within the first five N-terminal amino acids of TatA permitted the E. coli Tat pathway to function in the absence of TatB. It remains an open question whether the homologous TatA and TatB proteins have distinct binding sites on TatC or whether they alternately occupy the same site at different points during translocation.
Experimental procedures
Bacterial strains and growth conditions
Strains used in this study are listed in Tables 1 and S1. In general, E. coli strains were grown aerobically at 37°C in Luria–Bertani (LB) medium (Sambrook and Russell, 2001). Antibiotics were used at the following final concentrations; ampicillin – 100 µg ml−1, chloramphenicol – 25 µg ml−1, kanamycin – 50 µg ml−1, apramycin – 100 µg ml−1. Cultures (50 ml) for TMAO reductase activity measurements were grown anaerobically overnight at 37°C in LB containing 0.4% (w/v) TMAO and 0.5% (v/v) glycerol.
Table 1.
Key strains and plasmids used in this study
| Bacterial strain | Genotype | Reference |
|---|---|---|
| MC4100 | F-, [araD139]B/r, Δ(argF-lac)U169, λ-, e14-, flhD5301, Δ(fruK-yeiR)725(fruA25), relA1, rpsL150(StrR), rbsR22, Δ(fimB-fimE)632(::IS1), deoC1 | Casadaban (1976) |
| JARV16 | MC4100, ΔtatA, ΔtatE | Sargent et al. (1999) |
| JARV16-P | MC4100, ΔtatA, ΔtatE, pcnB1 zad-981::Tn10d (KanR) | Sargent et al. (1999) |
| BEAD | MC4100, ΔtatAB,ΔtatE | Lee et al. (2002) |
| DADE | MC4100, ΔtatABCD,ΔtatE | Wexler et al. (2000) |
| H1FF | MC4100, ΔglpG::aac(3)IV (ApraR) | This study |
| H43FF | MC4100, ΔtatA, ΔtatE, ΔglpG::aac(3)IV (ApraR) | This study |
| H43FF-P | MC4100, ΔtatA, ΔtatE, pcnB1 zad-981::Tn10d (KanR), ΔglpG::aac(3)IV (ApraR) | This study |
| H0FF | MC4100, ΔtatABCD,ΔtatE,ΔglpG::aac(3)IV (ApraR) | This study |
| PLAWT | MC4100, ΔtatA, ΔtatE, attB::PtatA(tatA+E. coli) | Lee et al. (2002) |
| JARV16 λtatAPs | MC4100, ΔtatA, ΔtatE, attB::PtatA(tatA+P. stuartii) | This study |
| H43FF λtatAPs | MC4100, ΔtatA, ΔtatE, ΔglpG::aac(3)IV (ApraR), attB::PtatA(tatA+P. stuartii) (KanR) | This study |
| MC4100 λAPsALYFP | MC4100, attB::PtatA(tatAP. stuartii-tatAE. coli50–89–YFP) (KanR) | This study |
| JARV16 λAPsALYFP | MC4100, ΔtatA, ΔtatE, attB::PtatA(tatAP. stuartii-tatAE. coli50–89–YFP) (KanR) | This study |
| BEAD λAPsALYFP | MC4100, ΔtatAB,ΔtatE, attB::PtatA(tatAP. stuartii-tatAE. coli50–89–YFP) (KanR) | This study |
| DADE λAPsALYFP | MC4100, ΔtatABCD,ΔtatE, attB::PtatA(tatAP. stuartii-tatAE. coli50–89–YFP) (KanR) | This study |
| H43FF λAPsALYFP | MC4100, ΔtatA, ΔtatE, ΔglpG::aac(3)IV (ApraR), attB::PtatA(tatAP. stuartii-tatAE. coli50–89–YFP) (KanR) | This study |
| Plasmid | Characteristics | Reference |
|---|---|---|
| pQE60PsTatA | pQE60 encoding C-terminally His-tagged P. stuartii TatA | This study |
| pQE60PsTatAΔ2–8 | pQE60 encoding C-terminally His-tagged P. stuartii TatA lacking codons 2–8 inclusive | This study |
| pFAT584 | pQE60 encoding C-terminally His-tagged E. coli TatA | De Leeuw et al. (2001) |
| pQEA(ΔB)C | Overexpression of E. coli tatA and tatC under control of T5 promoter and lac operator; TatC produced without a His-tag | This study |
| pQEA(ΔB)Chis | Overexpression of E. coli tatA and tatC under control of T5 promoter and lac operator; codons for C-terminal His-tag on tatC | This study |
| pQEAPsHA(ΔB)Chis | Overexpression of P. stuartii tatA and E. coli tatC under control of T5 promoter and lac operator; codons for C-terminal His-tag on tatC and C-terminal haemagglutinin tag on tatA | This study |
| pQEAPsEc(ΔB)C | Overexpression of P. stuartii tatA and E. coli tatC under control of T5 promoter and lac operator; codons 79–89 of E. coli as C-terminal epitope on P. stuartii tatA. TatC produced without a His-tag | This study |
| pQEAPsEc(ΔB)Chis | Overexpression of P. stuartii tatA and E. coli tatC under control of T5 promoter and lac operator; codons for C-terminal His-tag on tatC and codons 79–89 of E. coli as C-terminal epitope on P. stuartii tatA | This study |
Note that this is an abridged table and a full list of strains and plasmids can be found in Tables S1 and S2 respectively.
For chromosomal deletions of glpG the apramycin resistance cassette of plasmid pIJ790 (Gust et al., 2003) was amplified by PCR using primers glpGup and glpGdown (all oligonucleotides used in this study are listed in Table S3). This resulted in a PCR product where the apramycin cassette was flanked by 36 bp of sequence homologous to the up and downstream regions of glpG, including the start and stop codons respectively. Homologous recombination of the PCR product with the chromosomal glpG+ allele was carried out in E. coli strain BW25113 as described previously (Datsenko and Wanner, 2000) using the lambda Red recombinase expression plasmid pIJ773 (Gust et al., 2003). The disrupted glpG allele harbouring the apramycin resistance cassette was transferred from strain BW25113 ΔglpG to other E. coli strains by P1-mediated transduction using standard procedures (Miller, 1972).
Plasmids
Plasmids used or constructed in this study are listed in Tables 1 and S2. Constructs pQE60PsTatA and pQE60PsTatAΔ2–8 harbour full-length P. stuartii tatA and a genetically truncated variant lacking codons 2–8, respectively, in pQE60. Full-length P. stuartii tatA was amplified by PCR using primers PStatANco and PstatABgl and the genetically truncated version using primers fwPs_NO_2–8 and rvPS_NO_2–8 with plasmid pBC.TatAPs (Stevenson et al., 2007) as a template. The PCR products were cloned into the multiple-cloning site of pQE60 using restriction enzymes NcoI and BglII.
The E. coli tatA promoter was fused to sequence of P. stuartii tatA as follows. The tatA promoter was amplified by PCR using primers UNIREP1 and coli/stuartiiblunt from MC4100 genomic DNA and digested with EcoRI. P. stuartii tatA was amplified using primers StuartiiTatAEcoRV and stuartirev2 with plasmid pBC.TatAPs (Stevenson et al., 2007) as template and digested with EcoRV and BamHI. The blunt end of the digested EcoRV site in the P. stuartii tatA PCR product was ligated directly to the undigested end of the PCR product with the E. coli tatA promoter and the resulting fragment was cloned into pLitmus28 (NEB) with EcoRI and BamHI, giving rise to construct pLitPstuaTatA. Plasmid pLitPstuaTatAstop was constructed by amplifying a PCR product with primers UNIREP1 and LitPstuatatAstop1 using plasmid pLitPstuatatA as template, and the product was cloned into pLitmus28 after digestion with EcoRI and BamHI. The P. stuartii tatA gene under control of the E. coli tatA promoter was excised from pLitPstuaTatAstop by digestion with EcoRI and BamHI and cloned into similarly digested pRS552 (Simons et al., 1987), resulting in construct pRSTatAPs. The tatA allele on this construct was subsequently integrated into the lambda attachment site on the chromosome of E. coli strains as described previously (Simons et al., 1987).
To construct P. stuartii TatA fused via an E. coli TatA linker region to YFP, first the E. coli tatA linker–YFP construct was assembled. Plasmid pTatA-NOSTOP2 contains the E. coli tatA gene with its native promoter in pBluescript (Leake et al., 2008). DNA covering E. coli tatA codons 50–89 fused to YFP was amplified using primers fwTATALINK and rvTATALINK using pTatA-NOSTOP2 as template. The PCR product was digested with EcoRI and BamHI and cloned into Bluescript, giving rise to plasmid pCTermTatAYFP. Subsequently the P. stuartii tatA gene under control of the E. coli tat promoter was amplified by PCR with primers UNIREP1 and TatAPsNsiI using pLitPstuaTatAstop as a template, and the PCR product was cloned into pCTermTatAYFP following digestion with EcoRI and NsiI, to give plasmid pAPsALYFP. The tatA–YFP gene fusion present in pAPsALYFP was subsequently excised by digestion with EcoRI and BamHI, and cloned into similarly digested pRS552, resulting in construct pRSAPsALYFP. Finally the tatA–YFP fusion was integrated into the lambda attachment site of E. coli strains according to the method of Simons et al. (1987).
To construct equivalent clones producing the genetically truncated variant lacking codons 2–8, a synthetic construct, pBSK-TatAPsD2–8, was purchased from Dundee Cell Products (Dundee, UK) comprising the promoter region, ribosome binding site and start codon of E. coli tatA and P. stuartii TatA starting from codon 8. The synthetic DNA fragment was cloned into pBluescript as an EcoRI–BamHI fragment and had an NsiI site introduced just prior to the P. stuartii tatA stop codon (sequence of this synthetic construct available upon request). A fragment covering the E. coli tat promoter-P. stuartii tatA truncated gene was excised by digestion with EcoRI and NsiI and cloned into similarly digested pAPsALYFP, thus replacing the full-length P. stuartii tatA gene on this construct with a fragment covering the genetic truncation, giving plasmid pAPsΔ2–8ALYFP. The truncated tatA–YFP gene fusion present in pAPsΔ2–8ALYFP was cloned into pRS552 as an EcoRI–BamHI fragment, giving construct pRSAPsΔ2–8ALYFP and integrated into the lambda attachment site of E. coli strains as before (Simons et al., 1987).
Plasmids for coexpression of E. coli or P. stuartii tatA along with E. coli tatC in vector pQE60 were constructed as follows. DNA covering E. coli tatA to the start of tatB was amplified using primers CANDIDA (Sargent et al., 1998) and TatBdelupXhoI2, with MC4100 chromosomal DNA as template, and the product was digested with EcoRI and XhoI. DNA covering the last few codons of E. coli tatB along with the entire tatC gene were amplified using TatBdeldownXho and either TatCBam (Lee et al., 2006; this primer includes the tatC stop codon) or TatCH2 (de Leeuw et al., 2002; this primer lacks the tatC stop codon) and MC4100 chromosomal DNA as template. These two DNA fragments were digested with XhoI and either BamHI (for the product generated with TatCBam) or BglII (for the product generated with TatCH2). The tatB deletion alleles were assembled by three-way ligation into pQE60 that had been digested with EcoRI and either BglII (for the fragment containing tatC lacking its stop codon) or BamHI (for tatC with its stop codon) to give plasmids pQEA(ΔB)Chis and pQEA(ΔB)C respectively. The E. coli tatA genes in plasmids pQEA(ΔB)C and pQEA(ΔB)Chis were replaced with variants of the P. stuartii tatA genes. For plasmid pQEAPs(ΔB)Chis, the wild-type P. stuartii tatA allele was amplified using primers TatA5 (Sargent et al., 1998) and TatAPsBEcXhoI and pLitPstuaTatAstop as template, the PCR product was digested with EcoRI and XhoI cloned first into pBluescript as an EcoRI–XhoI fragment and then excised as an EcoRI–EcoRV fragment and cloned into similarly digested pQEA(ΔB)Chis. Plasmid pQEAPsHA(ΔB)Chis was constructed by amplifying P. stuartii tatA using primers TatA5 (Sargent et al., 1998) and TatAPsHABEcXhoI (which includes DNA coding for a C-terminal haemagglutinin tag) with pLitPstuaTatAstop as a template and the PCR product was digested with EcoRI and XhoI and cloned first into pBluescript as an EcoRI–XhoI fragment and then excised as an EcoRI–EcoRV fragment and cloned into similarly digested pQEA(ΔB)Chis. Plasmids pQEAPsEc(ΔB)C and pQEAPsEc(ΔB)Chis contain the P. stuartii tatA gene fused to DNA coding for a C-terminal tag of the last 10 codons of E. coli tatA. The P. stuartii tatA gene was amplified using primers TatA5 (Sargent et al., 1998) and TatAPsEcBEcXhoI, with pLitPstuaTatAstop as a template. The resulting PCR product was digested with EcoRI and XhoI and cloned first into pBluescript as an EcoRI–XhoI fragment and then excised as an EcoRI–EcoRV fragment and cloned into each of pQEA(ΔB)C and pQEA(ΔB)Chis that had been similarly digested.
To construct a control plasmid producing the N-terminally truncated variant of P. stuartii TatA (with a C-terminal E. coli TatA epitope) along with histagged E. coli TatC in vector pQE60, DNA encoding the truncated P. stuartii TatA variant was amplified by PCR using primers CANDIDA and atAPsEcBEcXhoI with pBSK-TatAPsD2–8 as template. The DNA was first cloned into pBluescript as an EcoRI–XhoI fragment and subsequently excised as an EcoRI–EcoRV fragment and cloned into similarly digested pQEA(ΔB)Chis to give pQEΔ2–8ApsEc(ΔB)Chis.
Protein methods
Periplasmic fractions were prepared using EDTA/lysozyme treatment and TMAO:benzyl viologen oxidoreductase activity in the periplasmic fraction was measured as described previously (Silvestro et al., 1988; Palmer et al., 2010a). SDS-PAGE and immunoblotting were carried out according to the methods of Laemmli (1970) and Towbin et al. (1979) respectively. Tris-Tricine gels were purchased from Bio-Rad. Antisera against E. coli TatC or E. coli TatA were used as described (Sargent et al., 2001; Alami et al., 2002) and immunoreactive bands were visualized with a chemiluminescent horseradish peroxidase substrate (Millipore). TatA–YFP fusion proteins were detected by immunoblotting using monoclonal Anti-Green Fluorescent Protein (GFP) antibody (Sigma-Aldrich, cat-Nr. G1546) and hexa-histidine-tagged TatA and TatC proteins were detected with anti-histidine antibodies (QIAGEN, Cat. No. 34670 and 34660). In-gel fluorescence of TatA–YFP fusion proteins after SDS-PAGE was detected by excitation with a laser at 473 nm in fluorescent image analyser FLA-5100 (Fujifilm) using filter LPB.
Protein cross-linking
Crude membrane preparations and chemical protein cross-linking using DSS were performed as described by de Leeuw et al. (2001). Briefly, cell cultures (400 ml) were harvested and washed in 20 mM Na-MOPS (pH 7.2), 200 mM NaCl. The cells were disrupted with a French pressure cell at 8000 p.s.i. and after a short centrifugation step to remove unbroken cells, the crude membrane fraction was recovered by centrifugation at 200 000 g for 90 min. The pelleted membranes were resuspended in 1 ml of 20 mM Na-MOPS (pH 7.4), 200 mM NaCl. Protein (30 µg) in the crude membrane fraction was treated with 2 mM DSS (Sigma) in 20 mM K-HEPES (pH 7.4), 20 mM KCl, 250 mM sucrose, 1 mM EDTA at 25°C for 30 min and the reaction was stopped with 1 M Tris-HCl (pH 7.5) to a final concentration of 90 mM.
Fluorescence microscopy
Cultures for fluorescence microscopy were grown aerobically at 37°C to an OD600 of 0.3. Cells were harvested by centrifugation, washed and resuspended in M9 minimal medium to one fifth of the original volume (Sambrook and Russell, 2001). Strain DADE-A λAPsALYFP expressing tatB alone, tatC alone or tatB and tatC together from plasmids pBAD33-tatB, pBAD24-tatC (both a kind gift from George Georgiou, University of Texas, Austin, USA) or pBAD-BC (Leake et al., 2008), respectively, was initially grown in LB medium supplemented with 0.2% glucose to repress transcription from the araBAD promoter (to prevent toxicity resulting from overexpression of tatB alone; Sargent et al., 1999). Cells were harvested, washed and resuspended as above in M9 minimal medium supplemented with 0.02% l-arabinose and incubated for 30 min to induce expression of plasmid encoded tatB and tatC. Cells were immobilized on coverslips using CellTak (BD Biosciences) and analysed by wide-field fluorescence microscopy (150× plan-apochromat objective, filter set 38He, Imager M1 microscope; Zeiss). Digital images were taken with a CCD camera (AxioCam MRm; Zeiss) and analysed using digital imaging software (Axio Vision LE 4.8; Zeiss).
Purification of TatAC
A total of 4 × 750 ml of LB medium containing 0.4% (w/v) glycerol in baffled 2.5 l flasks was inoculated with 12.5 ml each of an overnight culture of E. coli DADE/pREP4/pQEA(ΔB)Chis and grown at 37°C to an OD600 of 0.7. IPTG was then added to a final concentration of 0.5 mM. After a further 3 h growth, the cells were harvested by centrifugation at 9000 g for 20 min. The cell pellet was resuspended in 20 ml of 20 mM MOPS/NaOH pH 8.0, 200 mM NaCl and broken by two passages through a French pressure cell at 14 000 p.s.i. Cell debris was pelleted by centrifugation at 12 000 g for 15 min before the supernatant was centrifuged at approximately 200 000 g for 1 h at 4°C to pellet the membrane fraction. The pellet was resuspended in 20 ml of 20 mM MOPS/NaOH, 200 mM NaCl before an equal volume of buffer containing 2% digitonin was added. After incubation at room temperature with gentle agitation for 3 h, the supernatant was cleared by centrifugation at 200 000 g for 1 h.
Imidazole was added to the supernatant to a final concentration of 20 mM and the resulting solution was applied to a 1 ml HisTrap (GE) column. The column was then washed with 10 column volumes of 20 mM MOPS pH 8, 200 mM NaCl, 20 mM imidazole, 0.15% digitonin. A linear gradient of 20–500 mM imidazole in 15 column volumes of the same buffer was then applied and protein-containing fractions were collected. After addition of 10 mM EDTA to scavenge nickel ions that may have leached from the resin, fractions containing TatAC complexes were pooled and concentrated to a volume of 0.5 ml using a centrifugal concentrator with a 100 kDa cut-off (Amicon). The concentrate was then applied to a Superose 6 10/300 (GE Healthcare) gel filtration column equilibrated with 20 mM MOPS pH 8, 200 mM NaCl, 0.15% digitonin. Fractions were analysed by Western blotting.
Protein co-purification assay
Escherichia coli strains DADE/pREP4 or H0FF/pREP4 were used for co-purification assays of TatA and TatC proteins produced from plasmids pQEA(ΔB)Chis, pQEAPsEc(ΔB)C or pQEAPsEc(ΔB)Chis. Freshly transformed cells were grown in 500 ml of LB medium at 37°C with shaking to an OD600 of 0.6 and protein expression was induced with 2 mM IPTG at 25°C for 16 h. Cells were harvested by centrifugation, washed and resuspended in 25 ml of resuspension buffer (20 mM Na-HEPES (pH 7.2), 200 mM NaCl) supplemented with 1 mM PMSF (Sigma). Cells were disrupted with a French pressure cell (Thermo) at 8000 psi and the crude membrane fraction was pelleted by centrifugation at 200 000 g for 90 min at 4°C. The crude membrane pellet was resuspended in 1 ml of wash buffer (20 mM Na-HEPES (pH 7.2), 200 mM NaCl, 15 mM imidazole, 0.01% C12E9). Membrane proteins in the crude membrane fraction were solubilized at 4°C for 1 h with 1% C12E9 at a total protein concentration of 5 mg ml−1 and unsolubilized material was pelleted by centrifugation at 270 000 g for 30 min at 4°C. Nine hundred microlitres of solubilized membrane fraction was mixed with 100 µl of Profinity IMAC Ni-Charged resin (Bio-Rad) and gently pelleted by centrifugation at 400 g for 1 min at 4°C. The supernatant was retained as the unbound sample and the resin was washed four times with 1 ml wash buffer followed by gentle centrifugation as above. The supernatant after the last wash was retained and the resin was resuspended in 100 µl of elution buffer [20 mM Na-HEPES (pH 7.2), 200 mM NaCl, 300 mM imidazole, 0.01% C12E9]. The resin was pelleted by centrifugation at 400 g for 1 min at 4°C and the supernatant was kept as the eluted fraction. Subsequently, samples of the crude membrane fraction, the solubilized membrane fraction, the unbound fraction, the last wash fraction and the final eluted fraction were mixed 1:1 with 2× Laemmli sample buffer. Samples for Western blots against TatA were diluted 200-fold due to higher expression of tatA compared with tatC from the constructs. Finally, 5 µl of each sample was separated by SDS-PAGE and subsequently subjected to Western blotting using anti-TatA antiserum and anti-tetra-histidine tag antibodies, or anti-TatC antiserum in the case of untagged TatC.
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council through a PhD studentship (to M.F.) and by Grant BB/F02150X/1, and by the Wellcome Trust through Studentship Grant 072681/Z/03/Z (to M.T.). We thank Nicholas Greene for advice on the construction of TatA–YFP fusions and Meike Baumgart for her contributions to the early stages of this work. Dr Philip Rather is acknowledged for kindly providing us with plasmid pBC.TatAPs, and Prof. George Georgiou is thanked for providing us with pBAD33-tatB and pBAD24-tatC plasmids.
Supporting information
Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alami M, Trescher D, Wu LF, Muller M. Separate analysis of twin-arginine translocation (Tat)-specific membrane binding and translocation in Escherichia coli. J Biol Chem. 2002;277:20499–20503. doi: 10.1074/jbc.M201711200. [DOI] [PubMed] [Google Scholar]
- Alami M, Luke I, Deitermann S, Eisner G, Koch HG, Brunner J, Muller M. Differential interactions between a twin-arginine signal peptide and its translocase in Escherichia coli. Mol Cell. 2003;12:937–946. doi: 10.1016/s1097-2765(03)00398-8. [DOI] [PubMed] [Google Scholar]
- Barnett JP, Eijlander RT, Kuipers O, Robinson C. A minimal Tat system from a gram-positive organism: a bifunctional TatA subunit participates in discrete TatAC and TatA complexes. J Biol Chem. 2008;283:2534–2542. doi: 10.1074/jbc.M708134200. [DOI] [PubMed] [Google Scholar]
- Barnett JP, van der Ploeg R, Eijlander RT, Nenninger A, Mendel S, Rozeboom R, et al. The twin-arginine translocation (Tat) systems from Bacillus subtilis display a conserved mode of complex organization and similar substrate recognition requirements. FEBS J. 2009;276:232–243. doi: 10.1111/j.1742-4658.2008.06776.x. [DOI] [PubMed] [Google Scholar]
- Barrett CM, Freudl R, Robinson C. Twin arginine translocation (Tat)-dependent export in the apparent absence of TatABC or TatA complexes using modified Escherichia coli TatA subunits that substitute for TatB. J Biol Chem. 2007;282:36206–36213. doi: 10.1074/jbc.M704127200. [DOI] [PubMed] [Google Scholar]
- Behrendt J, Lindenstrauss U, Bruser T. The TatBC complex formation suppresses a modular TatB-multimerization in Escherichia coli. FEBS Lett. 2007;581:4085–4090. doi: 10.1016/j.febslet.2007.07.045. [DOI] [PubMed] [Google Scholar]
- Berks BC. A common export pathway for proteins binding complex redox cofactors? Mol Microbiol. 1996;22:393–404. doi: 10.1046/j.1365-2958.1996.00114.x. [DOI] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaudeck N, Kreutzenbeck P, Muller M, Sprenger GA, Freudl R. Isolation and characterization of bifunctional Escherichia coli TatA mutant proteins that allow efficient tat-dependent protein translocation in the absence of TatB. J Biol Chem. 2005;280:3426–3432. doi: 10.1074/jbc.M411210200. [DOI] [PubMed] [Google Scholar]
- Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273:18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- Bolhuis A, Mathers JE, Thomas JD, Barrett CM, Robinson C. TatB and TatC form a functional and structural unit of the twin-arginine translocase from Escherichia coli. J Biol Chem. 2001;276:20213–20219. doi: 10.1074/jbc.M100682200. [DOI] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Cline K, Mori H. Thylakoid DeltapH-dependent precursor proteins bind to a cpTatC–Hcf106 complex before Tha4-dependent transport. J Cell Biol. 2001;154:719–729. doi: 10.1083/jcb.200105149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline K, Theg SM. The Sec and Tat protein translocation pathways in chloroplasts. In: Dalbey RE, Koehler CM, Tamanoi F, editors. Molecular Machines Involved in Protein Transport across Cellular Membranes. London: Elsevier; 2007. pp. 463–492. [Google Scholar]
- Dabney-Smith C, Cline K. Clustering of C-terminal stromal domains of Tha4 homo-oligomers during translocation by the Tat protein transport system. Mol Biol Cell. 2009;20:2060–2069. doi: 10.1091/mbc.E08-12-1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabney-Smith C, Mori H, Cline K. Oligomers of Tha4 organize at the thylakoid Tat translocase during protein transport. J Biol Chem. 2006;281:5476–5483. doi: 10.1074/jbc.M512453200. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leeuw E, Porcelli I, Sargent F, Palmer T, Berks BC. Membrane interactions and self-association of the TatA and TatB components of the twin-arginine translocation pathway. FEBS Lett. 2001;506:143–148. doi: 10.1016/s0014-5793(01)02904-0. [DOI] [PubMed] [Google Scholar]
- Dilks K, Gimenez MI, Pohlschroder M. Genetic and biochemical analysis of the twin-arginine translocation pathway in halophilic archaea. J Bacteriol. 2005;187:8104–8113. doi: 10.1128/JB.187.23.8104-8113.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driessen AJ, Nouwen N. Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem. 2008;77:643–667. doi: 10.1146/annurev.biochem.77.061606.160747. [DOI] [PubMed] [Google Scholar]
- Frobel J, Rose P, Muller M. Early contacts between substrate proteins and TatA translocase component in Twin-arginine translocation. J Biol Chem. 2011;286:43679–43689. doi: 10.1074/jbc.M111.292565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohlke U, Pullan L, McDevitt CA, Porcelli I, de Leeuw E, Palmer T, et al. The TatA component of the twin-arginine protein transport system forms channel complexes of variable diameter. Proc Natl Acad Sci USA. 2005;102:10482–10486. doi: 10.1073/pnas.0503558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene NP, Porcelli I, Buchanan G, Hicks MG, Schermann SM, Palmer T, Berks BC. Cysteine scanning mutagenesis and disulfide mapping studies of the TatA component of the bacterial twin arginine translocase. J Biol Chem. 2007;282:23937–23945. doi: 10.1074/jbc.M702972200. [DOI] [PubMed] [Google Scholar]
- Gust B, Challis GL, Fowler K, Kieser T, Chater KF. PCR-targeted Streptomyces gene replacement identifies a protein domain needed for biosynthesis of the sesquiterpene soil odor geosmin. Proc Natl Acad Sci USA. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ize B, Stanley NR, Buchanan G, Palmer T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol. 2003;48:1183–1193. doi: 10.1046/j.1365-2958.2003.03504.x. [DOI] [PubMed] [Google Scholar]
- Jack RL, Sargent F, Berks BC, Sawers G, Palmer T. Constitutive expression of Escherichia coli tat genes indicates an important role for the twin-arginine translocase during aerobic and anaerobic growth. J Bacteriol. 2001;183:1801–1804. doi: 10.1128/JB.183.5.1801-1804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jongbloed JD, Grieger U, Antelmann H, Hecker M, Nijland R, Bron S, van Dijl JM. Two minimal Tat translocases in Bacillus. Mol Microbiol. 2004;54:1319–1325. doi: 10.1111/j.1365-2958.2004.04341.x. [DOI] [PubMed] [Google Scholar]
- Jongbloed JD, van der Ploeg R, van Dijl JM. Bifunctional TatA subunits in minimal Tat protein translocases. Trends Microbiol. 2006;14:2–4. doi: 10.1016/j.tim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Koch S, Fritsch MJ, Buchanan G, Palmer T. The Escherichia coli TatA and TatB proteins have an N-out C-in topology in intact cells. J Biol Chem. 2012;287:14420–14431. doi: 10.1074/jbc.M112.354555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostecki JS, Li H, Turner RJ, DeLisa MP. Visualizing interactions along the Escherichia coli twin-arginine translocation pathway using protein fragment complementation. PLoS ONE. 2010;5:e9225. doi: 10.1371/journal.pone.0009225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Leake MC, Greene NP, Godun RM, Granjon T, Buchanan G, Chen S, et al. Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci USA. 2008;105:15376–15381. doi: 10.1073/pnas.0806338105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Buchanan G, Stanley NR, Berks BC, Palmer T. Truncation analysis of TatA and TatB defines the minimal functional units required for protein translocation. J Bacteriol. 2002;184:5871–5879. doi: 10.1128/JB.184.21.5871-5879.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PA, Orriss GL, Buchanan G, Greene NP, Bond PJ, Punginelli C, et al. Cysteine-scanning mutagenesis and disulfide mapping studies of the conserved domain of the twin-arginine translocase TatB component. J Biol Chem. 2006;281:34072–34085. doi: 10.1074/jbc.M607295200. [DOI] [PubMed] [Google Scholar]
- de Leeuw E, Granjon T, Porcelli I, Alami M, Carr SB, Muller M, et al. Oligomeric properties and signal peptide binding by Escherichia coli Tat protein transport complexes. J Mol Biol. 2002;322:1135–1146. doi: 10.1016/s0022-2836(02)00820-3. [DOI] [PubMed] [Google Scholar]
- Lindenstrauss U, Matos CF, Graubner W, Robinson C, Bruser T. Malfolded recombinant Tat substrates are Tat-independently degraded in Escherichia coli. FEBS Lett. 2010;584:3644–3648. doi: 10.1016/j.febslet.2010.07.039. [DOI] [PubMed] [Google Scholar]
- McDevitt CA, Hicks MG, Palmer T, Berks BC. Characterisation of Tat protein transport complexes carrying inactivating mutations. Biochem Biophys Res Commun. 2005;329:693–698. doi: 10.1016/j.bbrc.2005.02.038. [DOI] [PubMed] [Google Scholar]
- McDevitt CA, Buchanan G, Sargent F, Palmer T, Berks BC. Subunit composition and in vivo substrate-binding characteristics of Escherichia coli Tat protein complexes expressed at native levels. FEBS J. 2006;273:5656–5668. doi: 10.1111/j.1742-4658.2006.05554.x. [DOI] [PubMed] [Google Scholar]
- Maegawa S, Koide K, Ito K, Akiyama Y. The intramembrane active site of GlpG, an E. coli rhomboid protease, is accessible to water and hydrolyses an extramembrane peptide bond of substrates. Mol Microbiol. 2007;64:435–447. doi: 10.1111/j.1365-2958.2007.05679.x. [DOI] [PubMed] [Google Scholar]
- Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- Mori H, Cline K. A twin arginine signal peptide and the pH gradient trigger reversible assembly of the thylakoid [Delta]pH/Tat translocase. J Cell Biol. 2002;157:205–210. doi: 10.1083/jcb.200202048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oates J, Barrett CM, Barnett JP, Byrne KG, Bolhuis A, Robinson C. The Escherichia coli twin-arginine translocation apparatus incorporates a distinct form of TatABC complex, spectrum of modular TatA complexes and minor TatAB complex. J Mol Biol. 2005;346:295–305. doi: 10.1016/j.jmb.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Orriss GL, Tarry MJ, Ize B, Sargent F, Lea SM, Palmer T, Berks BC. TatBC, TatB, and TatC form structurally autonomous units within the twin arginine protein transport system of Escherichia coli. FEBS Lett. 2007;581:4091–4097. doi: 10.1016/j.febslet.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer T, Berks BC, Sargent F. Analysis of Tat targeting function and twin-arginine signal peptide activity in Escherichia coli. Methods Mol Biol. 2010a;619:191–216. doi: 10.1007/978-1-60327-412-8_12. [DOI] [PubMed] [Google Scholar]
- Palmer T, Sargent F, Berks BC. The Tat protein export pathway. In: Böck A, Curtiss R III, Kaper JB, Karp PD, Neidhardt FC, Nyström T, et al., editors. EcoSal –Escherichia coli and Salmonella: Cellular and Molecular Biology. Washington, DC: ASM Press; 2010b. [WWW document]. URL http://www.ecosal.org. [Google Scholar]
- Porcelli I, de Leeuw E, Wallis R, van den Brink-van der Laan E, de Kruijff B, Wallace BA, et al. Characterization and membrane assembly of the TatA component of the Escherichia coli twin-arginine protein transport system. Biochemistry. 2002;41:13690–13697. doi: 10.1021/bi026142i. [DOI] [PubMed] [Google Scholar]
- Richter S, Bruser T. Targeting of unfolded PhoA to the TAT translocon of Escherichia coli. J Biol Chem. 2005;280:42723–42730. doi: 10.1074/jbc.M509570200. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sargent F, Bogsch EG, Stanley NR, Wexler M, Robinson C, Berks BC, Palmer T. Overlapping functions of components of a bacterial Sec-independent protein export pathway. EMBO J. 1998;17:3640–3650. doi: 10.1093/emboj/17.13.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sargent F, Stanley NR, Berks BC, Palmer T. Sec-independent protein translocation in Escherichia coli. A distinct and pivotal role for the TatB protein. J Biol Chem. 1999;274:36073–36082. doi: 10.1074/jbc.274.51.36073. [DOI] [PubMed] [Google Scholar]
- Sargent F, Gohlke U, Leeuw ED, Stanley NR, Palmer T, Saibil HR, Berks BC. Purified components of the Escherichia coli Tat protein transport system form a double-layered ring structure. Eur J Biochem. 2001;268:3361–3367. doi: 10.1046/j.1432-1327.2001.02263.x. [DOI] [PubMed] [Google Scholar]
- Silvestro A, Pommier J, Giordano G. The inducible trimethylamine-N-oxide reductase of Escherichia coli K12: biochemical and immunological studies. Biochim Biophys Acta. 1988;954:1–13. doi: 10.1016/0167-4838(88)90049-0. [DOI] [PubMed] [Google Scholar]
- Simons RW, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- Stanley NR, Palmer T, Berks BC. The twin arginine consensus motif of Tat signal peptides is involved in Sec-independent protein targeting in Escherichia coli. J Biol Chem. 2000;275:11591–11596. doi: 10.1074/jbc.275.16.11591. [DOI] [PubMed] [Google Scholar]
- Stevenson LG, Strisovsky K, Clemmer KM, Bhatt S, Freeman M, Rather PN. Rhomboid protease AarA mediates quorum-sensing in Providencia stuartii by activating TatA of the twin-arginine translocase. Proc Natl Acad Sci USA. 2007;104:1003–1008. doi: 10.1073/pnas.0608140104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strisovsky K, Sharpe HJ, Freeman M. Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol Cell. 2009;36:1048–1059. doi: 10.1016/j.molcel.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarry MJ, Schafer E, Chen S, Buchanan G, Greene NP, Lea SM, et al. Structural analysis of substrate binding by the TatBC component of the twin-arginine protein transport system. Proc Natl Acad Sci USA. 2009;106:13284–13289. doi: 10.1073/pnas.0901566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang Y, Ha Y. Crystal structure of a rhomboid family intramembrane protease. Nature. 2006;444:179–180. doi: 10.1038/nature05255. [DOI] [PubMed] [Google Scholar]
- Weiner JH, Bilous PT, Shaw GM, Lubitz SP, Frost L, Thomas GH, et al. A novel and ubiquitous system for membrane targeting and secretion of cofactor-containing proteins. Cell. 1998;93:93–101. doi: 10.1016/s0092-8674(00)81149-6. [DOI] [PubMed] [Google Scholar]
- Westermann M, Pop OI, Gerlach R, Appel TR, Schlormann W, Schreiber S, Muller JP. The TatAd component of the Bacillus subtilis twin-arginine protein transport system forms homo-multimeric complexes in its cytosolic and membrane embedded localisation. Biochim Biophys Acta. 2006;1758:443–451. doi: 10.1016/j.bbamem.2006.03.018. [DOI] [PubMed] [Google Scholar]
- Wexler M, Sargent F, Jack RL, Stanley NR, Bogsch EG, Robinson C, et al. TatD is a cytoplasmic protein with DNase activity. No requirement for TatD family proteins in sec-independent protein export. J Biol Chem. 2000;275:16717–16722. doi: 10.1074/jbc.M000800200. [DOI] [PubMed] [Google Scholar]
- Zamenhof PJ, Villarejo M. Construction and properties of Escherichia coli strains exhibiting – complementation of – galactosidase fragments in vivo. J Bacteriol. 1972;110:171–178. doi: 10.1128/jb.110.1.171-178.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.