

Abstract
Awareness of the need for prevention of glucocorticoid- induced fractures is growing, but glucocorticoid administration is often overlooked as the most common cause of nontraumatic osteonecrosis. Glucocorticoid- induced osteonecrosis develops in 9–40% of patients receiving long-term therapy although it may also occur with short-term exposure to high doses, after intra-articular injection, and without glucocorticoid-induced osteoporosis. The name, osteonecrosis, is misleading because the primary histopathological lesion is osteocyte apoptosis. Apoptotic osteocytes persist because they are anatomically unavailable for phagocytosis and, with glucocorticoid excess, decreased bone remodeling retards their replacement. Glucocorticoid-induced osteocyte apoptosis, a cumulative and unrepairable defect, uniquely disrupts the mechanosensory function of the osteocyte–lacunar–canalicular system and thus starts the inexorable sequence of events leading to collapse of the femoral head. Current evidence indicates that bisphosphonates may rapidly reduce pain, increase ambulation, and delay joint collapse in patients with osteonecrosis.
Keywords: Glucocorticoids, Osteonecrosis, Avascular necrosis, Apoptosis, Osteoblasts, Osteocytes, Bisphosphonates
Introduction
Less than 1 year after the introduction of cortisone for the treatment of rheumatoid arthritis in 1950, investigators became aware of injurious effects that were considered uncommon in Cushing’s syndrome [1]. These effects included insomnia, gastrointestinal bleeding, a post-cortisone- withdrawal syndrome, and hip fracture [2, 3]. As vertebral fractures and radiographic osteoporosis had not been observed, the investigators were uncertain whether the hip fractures were the result of falls due to steroid myopathy or merely coincidental with cortisone therapy. Nonetheless, efforts were made to reduce the glucocorticoid dosage [1]. Just a few years later, osteoporosis and fractures were clearly recognized as skeletal complications of prolonged treatment with cortisone, prednisolone, and prednisone. Collapse of the femoral and humeral heads after high-dose therapy was described shortly thereafter [4, 5], sometimes for treatment that was hard to justify [6]. Femoral osteonecrosis has even been noted with occlusive topical glucocorticoid ointment [7, 8]. Today, osteonecrosis develops in 9–40% of patients receiving long-term therapy and may occur without glucocorticoid-induced osteoporosis [9].
The first large series of patients with glucocorticoidinduced osteonecrosis was reported in 1971 [10]. From 482 patients with osteonecrosis seen at the Mayo Clinic from 1961 to 1968, 77 had received glucocorticoids. The report caused some confusion because “hematologic conditions,” pancreatitis, pregnancy, rheumatoid arthritis, glomerulonephritis, colitis, and gout were all listed as causes of “avascular necrosis,” but these patients had been treated with glucocorticoids and there was no evidence that the conditions listed were independent causes of osteonecrosis, although that was assumed for more than 40 years [11]. The average interval between initiation of treatment and the first symptoms of hip pain was 33 months, but several cases of osteonecrosis were noted after only 3 months. The earliest recognition was just 36 days after 16 mg/day of oral methylprednisolone (cumulative dose was 576 mg). A single “dose-pack” of methyl prednisolone may begin with more than 32 mg/day and contain a total dose of as much as 800 mg, so just one dose pack is arguably already a risk for glucocorticoid-induced osteonecrosis. Sadly, patients are seldom warned about this complication or the need to see their doctor if they experience persistent joint pain and radiological surveillance for osteonecrosis in patients receiving high-dose glucocorticoids is restricted to research studies [12]. As a result, osteonecrosis is the most common glucocorticoid-related complication associated with successful litigation [13].
Diagnosis
Persistent hip, knee or shoulder pain, especially with joint movement, tenderness or reduced range of motion, warrants magnetic resonance imaging (MRI) [9, 11, 14]. The subchondral crescent sign on radiographic examination is pathogenomic for osteonecrosis but this sign is not early and MRI can clearly show extensive osteonecrosis before any change in the shape of the femoral head or appearance of a fracture crescent (Fig. 1). A wide variety of radiographs showing glucocorticoid-induced osteonecrosis can be found at images on www.google.com.
Fig. 1.
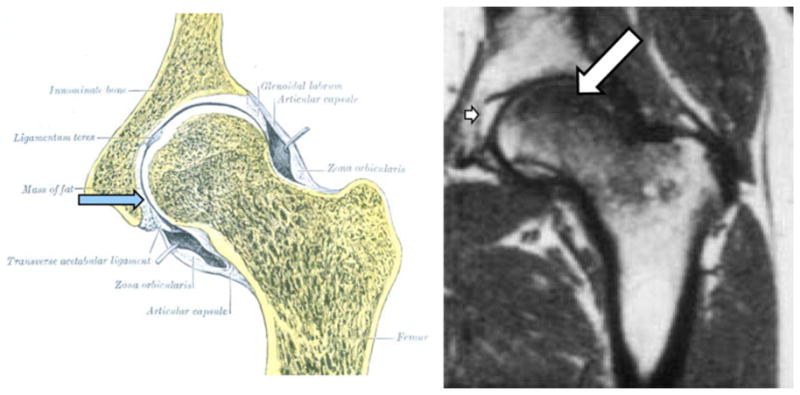
MRI of osteonecrosis. The acetabular fat pad (blue arrow) shown in the left panel gives an intense white signal with MRI as shown in the right panel (small arrow), while the loss of marrow fat (large arrow) gives a dark signal typical of edema with T1 MRI imaging
Epidemiology
An investigation of the epidemiology of osteonecrosis requires a search using several different aliases as the disorder is also known as aseptic, avascular or ischemic necrosis or bone infarcts [15]. The annual incidence of all types of osteonecrosis doubled from 1989 to 2003 (from 1.4 to 3/100,000), perhaps due to the more frequent use of MRI [16]. Most commonly, the hip is involved but almost any bone can develop osteonecrosis. The most frequent etiological factors are trauma, alcoholism, and glucocorticoids, the later causing the most devastating form of osteonecrosis [11]. Other causes include sickle cell disease, radiation, Gaucher’s disease, and Caisson disease (decompression sickness) (Table 1) [14].
Table 1.
Etiologic factors in adult osteonecrosis
| Etiologic factors |
|---|
| Trauma |
| Glucocorticoids |
| Alcohol |
| Idiopathic |
| Sickle cell disease |
| Radiation |
| Gaucher’s disease |
| Caisson disease (decompression sickness) |
In glucocorticoid-induced osteonecrosis, the risk increases with higher doses and prolonged treatment [14, 17, 18], although it may occur after short-term exposure to high doses, by intra-articular injection, and without osteoporosis (Table 2). Intra-articular glucocorticoids may be particularly dangerous because the injection may accelerate joint damage by alleviating pain, thus increasing weight bearing—a kind of Charcot’s arthropathy, in addition to the direct adverse effects of the steroids on bone [19, 20]. Osteonecrosis has been noted in just weeks to months after intra-articular injection of cumulative doses of 80–160 mg of methylprednisolone [21]. Increased susceptibility to glucocorticoid-induced osteonecrosis may occur with polymorphisms of vascular endothelial growth factor (VEGF), the glucocorticoid receptor (GR), 11β-hydroxysteroid dehydrogenase type2 (11β-HSD2), collagen type II (COL2A1), plasminogen activator inhibitor 1 (PAI1), and P-glycoprotein as well as with some underlying disorders such as renal insufficiency, transplantation, graft versus host disease, inflammatory bowel disease, HIV, and acute lymphoblastic leukemia [9, 14, 22–29].
Table 2.
Risk factors for glucocorticoid-induced osteonecrosis
| Risk factors |
|---|
| Dose and duration of therapy |
| Intra-articular administration |
| Polymorphisms in VEGF, GR, 11β-HSD2, COL2A1, PAI1, P-glycoprotein |
| Underlying disorders: renal insufficiency, transplantation, graft vs. host disease, inflammatory bowel disease, HIV, acute lymphoblastic leukemia |
| Dexamethasone causes greater skeletal complications than prednisone |
VEGF vascular endothelial growth factor, GR glucocorticoid receptor, 11β-HSD2 11β-hydroxysteroid dehydrogenase type2, COL2A1 collagen type II, PAI1 plasminogen activator inhibitor 1, HIV human immunodeficiency virus
Dexamethasone causes more skeletal complications than prednisone [27]. Natural and synthetic glucocorticoids differ in their vulnerability to 11β-HSD2, which protects target cells by inactivating glucocorticoids through conversion of the 11-hydroxyl required for hormone activity to a ketone thus preventing interaction with the GR [9, 15, 16]. In dexamethasone, the 11-hydroxyl is already present as it is in prednisolone, but the fluorine atom at the 9α position of the B ring both extends the potency and occludes the 11β location. Dexamethasone could be invulnerable to inactivation by 11β-HSD2.
Surprisingly, Cushing’s disease does not appear to be a big risk factor for osteonecrosis. Osteonecrosis in Cushing’s disease was first noted by Frost in 1961 [30], but the situation remains exceedingly rare. Only 13 cases were in the literature by 2011 [31]. However, Cushing’s disease differs in several ways from pharmacologic glucocorticoid administration. In Cushing’s disease, adrenocortical trophic hormone (ACTH) and the secretion of adrenal androgens are increased and the circulating glucocorticoid levels are generally lower than those found in patients treated with exogenous steroids. Some investigators found that ACTH could stimulate osteoblastic VEGF expression [32], but endogenous ACTH is unlikely to be the reason for the rarity of osteonecrosis in Cushing’s disease because osteonecrosis has been reported with ACTH injections alone [11]. Part of the explanation for the rarity of osteonecrosis in Cushing’s disease could be under-reporting, but the complete explanation remains a mystery.
Pathogenesis
The adverse effects of glucocorticoids on the skeleton are primarily due to direct actions on osteoblasts and osteoclasts, decreasing the production of both osteoblasts and osteoclasts and increasing the apoptosis of osteoblasts while prolonging the lifespan of osteoclasts [33–36]. Increased osteocyte apoptosis also occurs and is associated with decreases in VEGF, skeletal angiogenesis, bone interstitial fluid, and bone strength [37]. Osteocytes and the lacunar–canalicular network are the strain sensing system of bone and signal the need for remodeling to accommodate prevailing loads or repair damage. Glucocorticoidinduced osteocytes apoptosis with the resultant disruption of bone vascularity and diminution of bone hydraulic support could be the mechanisms behind the osteonecrosis and greater decline in bone strength than in loss of bone mass that occurs with glucocorticoid excess. A link between the systemic vascular system, canalicular processes, and osteocytes is suggested by the evidence that canalicular fluid transport is directly connected to the vascular space as revealed by low molecular weight tracers that traverse the venous system into the lacunocanalicular fluid within minutes [37]. Glucocorticoids rapidly disrupt this vascular connection as demonstrated by a remarkable decrease in the interstitial fluid of murine cancellous bone, revealed by fluorescent imaging of the osteocyte–lacunar– canalicular system using tail vein injections of Procion Red (molecular weight, 615 Da). Further evidence of the glucocorticoid effect on bone blood vessels in mice was obtained by microCT imaging of decalcified bones following intravenous perfusion of silicon slurry of lead chromate. Prednisolone administration dramatically decreased the vertebral and femoral vessel volume and surface area.
The direct adverse effects of administered glucocorticoids on bone cells are evident from a series of experiments in transgenic mice overexpressing the inactivating enzyme 11β-HSD2 in osteoblasts and osteocytes [34, 37]. These animals are protected from prednisolone-induced apoptosis and the resultant decrease in osteoblast number and bone formation, but still lose BMD because the osteoclasts remain exposed to the prednisolone. However, bone strength is preserved in spite of the loss of BMD, suggesting that osteocyte viability independently contributes to bone strength. Moreover, aging decreased the volume of the bone vasculature and solute transport from the peripheral circulation to the lacunar–canalicular system in wild-type mice, but similarly aged 11β-HSD2 transgenic mice were protected from the adverse effects of aging on osteoblast and osteocyte apoptosis, bone formation rate and microarchitecture, crystallinity, vasculature volume, interstitial fluid, and strength. These results suggest that endogenous glucocorticoids increase skeletal fragility in old age because of cell autonomous effects on osteoblasts and osteocytes leading to interconnected decrements in bone angiogenesis, vasculature volume, and osteocyte– lacunar–canalicular fluid. Osteonecrosis of the femoral condyles, an idiopathic lesion of elderly patients, may be due to the increased endogenous glucocorticoids that occur with aging [37, 38].
Using the same transgenic approach as described in osteoblasts and osteocytes, we found that overexpression of 11β-HSD2 in osteoclasts preserves BMD, but does not prevent the prednisolone-induced decrease in osteoblast lifespan, osteoblast number, and bone formation [36].
Osteonecrosis of the hip was once ascribed to rupture of the ligamentum teres and the resulting tearing of the arterial supply to the femoral head or to thrombosis of these vessels. However, today the ligamentum teres is considered a vestigial structure and perfusion of the femoral head through the ligament is negligible in the adult [39]. Moreover, the arteries in the ligament are closed in up to one-third of adults.
The mechanism underlying glucocorticoid-induced osteonecrosis has been postulated to be increased marrow fat resulting in an increase in intra-osseous pressure and a decrease in bone perfusion, fat embolism and hypercoagulability reducing blood flow to the femoral head, or accumulation of fatigue fractures [11, 14]. However, recent attention has focused on the role of osteocyte apoptosis [40– 45]. Glucocorticoid-induced osteocyte apoptosis, a cumulative and irreparable defect, disrupts the mechanosensory function of the osteocyte–lacunar–canalicular system and thus starts the inexorable sequence of events leading to collapse of the joint [40].
We identified abundant apoptotic osteocytes in sections of whole femoral heads obtained during total hip replacement for glucocorticoid-induced osteonecrosis, whereas apoptotic bone cells were uncommon with alcohol-induced osteonecrosis and absent in post-traumatic osteonecrosis (Figs. 2a, b; 3d, e) [40]. In addition, cell swelling and inflammation typical of necrosis did not occur with glucocorticoids. A prevalence map of osteocyte apoptosis made from sections of the femoral head showed that osteocyte apoptosis was most prevalent adjacent to the subchondral crescent and fracture cleft and decreased as the examination progressed more distally (Fig. 2c). Osteocyte apoptosis was anatomically juxtaposed to the osteonecrotic fracture. Glucocorticoid-induced osteonecrosis is a misnomer: the so-called glucocorticoid-induced “osteonecrosis” is actually osteocyte apoptosis. Apoptotic osteocytes with pyknotic nuclei have also been identified in femora and humeri of glucocorticoid-treated rabbits [41, 42].
Fig. 2.
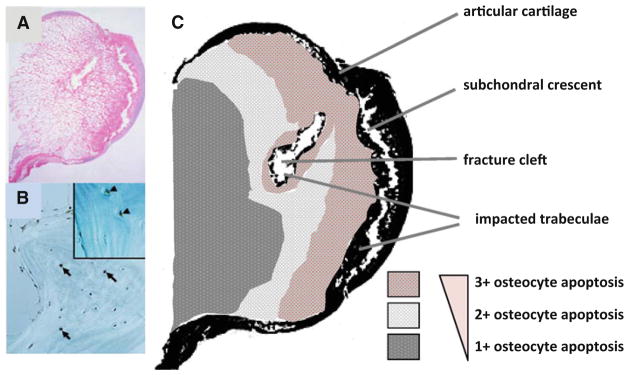
Glucocorticoid-induced osteonecrosis is osteocyte apoptosis. Abundant apoptotic osteocytes are present in sections of whole femoral heads obtained during total hip replacement for glucocorticoid- induced osteonecrosis (a, b). In c is a prevalence map of osteocyte apoptosis made from the section of the femoral head shown in (a). Osteocyte apoptosis was most prevalent (3?) adjacent to the subchondral crescent and fracture cleft and decreased (1?) as the examination progressed more distally. Osteocyte apoptosis was anatomically juxtaposed to the osteonecrotic fracture (adapted from Weinstein et al. [40]. Copyright 2000. The Endocrine Society)
Fig. 3.
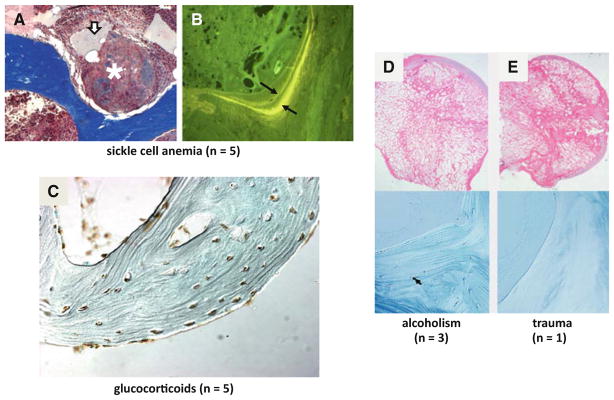
A major problem in the field of osteonecrosis is the lumping together of multiple etiologies. Femoral head core samples taken from patients with sickle cell disease show a empty osteocytic lacunae, oil cysts (arrow), and necrosis (asterisk), and b widely separated double tetracycline labels (arrows). Apoptotic cells were absent. In glucocorticoidinduced osteonecrosis (c), virtually all the osteocytes are apoptotic and remain present in their lacunae. Apoptotic osteocytes were uncommon in alcohol-induced (d) and absent in post-traumatic osteonecrosis (e) (d and e were adapted from Weinstein et al. [40]. Copyright 2000. The Endocrine Society)
A major problem in the field of osteonecrosis is the lumping together of multiple etiologies. For example, femoral head core samples taken from patients with sickle cell disease show empty osteocytic lacunae, necrosis, and oil cysts (Fig. 3a). Inflammatory cells are abundant and marrow fibrosis is occasionally noted. Four of the five specimens we examined had widely separated double tetracycline labels (Fig. 3b) (indicating bone formation and repair) and apoptotic osteocytes were absent [40]. These findings are distinctly different than those in glucocorticoid- induced osteonecrosis, where virtually all the osteocytes are apoptotic and remain resident in their lacunae even years after the initial symptoms of osteonecrosis (Fig. 3c). Other workers have also found increased osteocyte apoptosis in patients with glucocorticoid-induced osteonecrosis [44, 45]. These workers also found osteocyte apoptosis in patients with alcohol-induced osteonecrosis, which can be a high endogenous glucocorticoid condition especially with ethanol-induced episodes of hypoglycemia.
Osteocytic lacunae are rarely empty in glucocorticoidinduced osteonecrosis. After staining with a nuclear dye, condensed chromatin in apoptotic osteocytes from a femoral head removed from a patient with glucocorticoidinduced osteonecrosis show intense fluorescence (Fig. 4a), while osteocytes in the same section viewed with visible light are barely visible and their lacunae could be incorrectly thought to be empty (Fig. 4b). The devastating skeletal impact of glucocorticoids is shown by the intensely luminescent osteocytes recently buried in a new packet of bone and already apoptotic (Fig. 4c). After chronic glucocorticoid therapy, the osteocyte–canaliculi–lining cell network links dead cells (Fig. 4d). Glucocorticoid-induced osteocyte apoptosis could also account for the correlation with total steroid dose and the occurrence of osteonecrosis after glucocorticoid administration has ceased.
Fig. 4.

Osteocytic lacunae are rarely empty in glucocorticoid-induced osteonecrosis. Condensed chromatin in apoptotic osteocytes shows intense blue fluorescence after staining with Hoechst nuclear dye 33258 (a). Osteocytes in the same section viewed with visible light (b) are barely visible and their lacunae could be incorrectly thought to be empty. The osteocytes (arrows) recently buried in a new packet of bone are already apoptotic as shown by the nuclear dye (c). As a result, after chronic glucocorticoid therapy, the osteocyte–canaliculi– lining cell network links dead cells (brown staining indicates apoptotic cells; toluidine blue counterstain) (d) (adapted from Weinstein et al. [40]. Copyright 2000. The Endocrine Society)
The role of vascular factors in the glucocorticoidinduced skeletal damage is supported by evidence that glucocorticoids interfere with endothelial angiogenesis and both VEGF production and action [14, 37]. In OB-6 osteoblastic cells, dexamethasone decreased VEGF mRNA levels when the cells were cultured with vehicle or stimulated with desferrioxamine (DFO), an inhibitor of prolyl hydroxylase that mimics hypoxia. Dexamethasone also decreased DFO-stimulated hypoxia-inducible factor 1 (HIF-1) expression. The same results were found in MLOY4 osteocytic cells. These findings strongly suggest that at least some of the adverse effects of glucocorticoids on bone are due to decreased angiogenesis, caused by suppression of HIF-1 transcription and VEGF production by osteoblasts and osteocytes as well as by interference with VEGF action. Avascular necrosis was not such a bad name after all.
Treatment
A wide variety of joint-preserving treatment methods have been utilized if the osteonecrosis is recognized before reaching end-stage. The utility of these treatments varies depending on the staging of the osteonecrosis (Table 3) [11, 46]. Osteonecrosis occurs predominantly in younger patients and, therefore, efforts should be made to preserve the joint and delay hip replacement. The first approach is usually to reduce weight bearing using canes, crutches, or a walker for about 6 weeks. Another approach is core decompression with or without marrow cell transplantation, which also requires 6 weeks of reduced weight bearing. In general, core decompression is disappointing except in early disease in which the progression of the osteonecrosis is uncertain. As many as 40% of early lesions may not progress, especially if they are small (<25% of the femoral head) and medially located (away from the weight-bearing portion) [46]. For advanced disease with obliteration of the acetabular articular space and osteophyte formation, femoral head or total hip replacement is usually required. Osteonecrosis is responsible for 10% of all total hip replacement procedures and is the most common cause of total hip replacement in young adults; but problems with infection, osteolysis, dislocation and revisions are worse with total hip replacement for glucocorticoid-induced osteonecrosis than for osteoarthritis [11]. Since these replacements have about a 10-year lifespan, any delay in the need for surgery would be welcome. For these reasons, recent evidence of the utility of bisphosphonates in the treatment of osteonecrosis is quite promising.
Table 3.
Staging of osteonecrosis
| Modified classification | |
|---|---|
| Stage 0 | Known involvement of one hip and suspected in the other |
| Stage 1 | Pain and limited movement with normal X-ray and positive MRI |
| Stage 2 | Sclerosis of the femoral head |
| Stage 3 | Subchondral fracture and flattening of the femoral head |
| Stage 4 | End-stage osteonecrosis with loss of the articular space and osteophyte formation |
In a randomized, controlled, open-label trial, adults with osteonecrosis treated with alendronate had significant retardation of femoral head collapse and reduced need for total hip replacement compared to the control group over 24 months [47]. To qualify, X-rays had to show at least a 30% necrotic area. The mean rate of survival of the hips in the alendronate group at 26 months was 93% (only 2/24 collapsed) and in the control group, it was 36% (19/25 collapsed). Importantly, sustained improvement in pain and ambulation was reported within months. In another study, a 10-year follow-up of patients with osteonecrosis treated with alendronate for only the first 3 years showed decreased use of analgesics, increased ability to function as desired, and improvement in standing and ambulation time within months that lasted up to 10 years [48]. Alendronate may be a worthwhile option to postpone the need for hip surgery in young patients. The efficacy of alendronate to delay collapse of the femoral head in osteonecrosis may be due to decreased osteoclast resorption, but serial radiographic examinations of the hips in the above studies were either unchanged or progressed, suggesting that the efficacy of alendronate in osteonecrosis may involve more than just inhibition of bone resorption. Nitrogen-containing bisphosphonates also decrease glucocorticoid-induced osteocyte apoptosis [49] and preservation of osteocyte viability may play a role in the preservation of bone strength [34, 37]. Studies with molecular tracers have shown that aldolase (158,000 Da), transferrin (76,000–88,000 Da), and osteoprotegerin (100,000 Da) can penetrate the canalicular network (500–600 nm diameter) in rodents [50]. Therefore, bisphosphonates with an average molecular mass of 250–320 Da could easily reach the lacunar–canalicular system and protect osteocytes from glucocorticoid-induced apoptosis.
A thorough search of the literature failed to uncover trials of anabolic agents such as teriparatide in the treatment of glucocorticoid-induced osteonecrosis of the hip, shoulder, or knee.
Conclusions
Glucocorticoids have direct adverse effects on osteoblasts, osteoclasts, and osteocytes. These drugs decrease lacunar– canalicular fluid, bone vascularity, and bone strength via their effects on osteocytes. Glucocorticoid-induced osteocyte apoptosis accumulates and can lead to osteonecrosis, even without osteoporosis. The risk of osteonecrosis increases with increasing dose and duration of treatment and dexamethasone may be the worst culprit. Alendronate may rapidly reduce pain, increase ambulation, and delay joint collapse in patients with osteonecrosis.
Acknowledgments
I am grateful to Drs. Stavros C. Manolagas, Robert L. Jilka, Maria Almeida, and Charles A. O’Brien for their advice and helpful discussions about the adverse skeletal effects of glucocorticoids and for review of the manuscript. This study was supported by VA Merit Review Grants from the Office of Research and Development, Department of Veterans Affairs and the National Institutes of Health (P01-AG13918).
References
- 1.Boland EW, Headley NE. Management of rheumatoid arthritis with smaller (maintenance) doses of cortisone acetate. JAMA. 1950;144:365–372. doi: 10.1001/jama.1950.02920050005002. [DOI] [PubMed] [Google Scholar]
- 2.Freyberg RH, Traeger CH, Patterson M, Squires W, Adams CH, Stevenson C. Problems of prolonged cortisone treatment for rheumatoid arthritis. JAMA. 1951;147:1538–1543. doi: 10.1001/jama.1951.03670330030008. [DOI] [PubMed] [Google Scholar]
- 3.Bollet AJ, Black R, Bumin JJ. Major undesirable side-effects resulting from prednisolone and prednisone. JAMA. 1955;157:459–463. doi: 10.1001/jama.1955.02960060017005. [DOI] [PubMed] [Google Scholar]
- 4.Heiman WG, Freiberger RH. Avascular necrosis of the femoral and humeral heads after high-dosage corticosteroid therapy. N Engl J Med. 1960;263:672–675. doi: 10.1056/NEJM196010062631404. [DOI] [PubMed] [Google Scholar]
- 5.Pietrogrande V, Mastromarino R. Osteopathia da prolungato trattmento cortisonico. Ortop Tramatol. 1957;25:791–810. [Google Scholar]
- 6.Nasser SMS, Ewan PW. Depot corticosteroid treatment for hay fever causing avascular necrosis of both hips. Br Med J. 2010;322:1589–1591. doi: 10.1136/bmj.322.7302.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McClean CJ, Lobo RFJ, Brazier DJ. Cataracts, glaucoma, and femoral avascular necrosis caused by topical corticosteroid ointment. Lancet. 1995;345:330. doi: 10.1016/s0140-6736(95)90324-0. [DOI] [PubMed] [Google Scholar]
- 8.Kubo T, Kojima A, Yamazoe S, Ueshima K, Yamamoto T, Hirasawa Y. Osteonecrosis of the femoral head that developed after long-term topical steroid application. J Orthop Sci. 2001;6:92–94. doi: 10.1007/s007760170031. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein RS. Clinical practice: glucocorticoid-induced bone disease. N Engl J Med. 2011;365:62–70. doi: 10.1056/NEJMcp1012926. [DOI] [PubMed] [Google Scholar]
- 10.Fisher DE, Bickel WH. Corticosteroid-induced avascular necrosis. J Bone Jt Surg Am. 1971;53:859–873. [PubMed] [Google Scholar]
- 11.Mankin HF. Nontraumatic necrosis of bone (osteonecrosis) N Engl J Med. 1992;326:1473–1479. doi: 10.1056/NEJM199205283262206. [DOI] [PubMed] [Google Scholar]
- 12.Kawedia JD, Kaste SC, Pei D, Panetta JC, Cai X, Cheng C, Neale G, Howard SC, Evans WE, Pui CH, Rellinh MV. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2011;117:2340–2347. doi: 10.1182/blood-2010-10-311969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nash JJ, Nash AG, Leach ME, Poetker DM. Medical malpractice and corticosteroid use. Otolaryngol Head Neck Surg. 2011;144:10–15. doi: 10.1177/0194599810390470. [DOI] [PubMed] [Google Scholar]
- 14.Drescher W, Schlieper G, Floege J, Eitner F. Steroid-related osteonecrosis— an update. Nephrol Dial Transplant. 2010;26:1728–2731. doi: 10.1093/ndt/gfr212. [DOI] [PubMed] [Google Scholar]
- 15.Adler RA, Curtis JR, Saag K, Weinstein RS. Glucocorticoidinduced osteoporosis. In: Marcus R, Feldman D, Nelsen DA, Rosen CJ, editors. Osteoporosis. 3. Elsevier-Academic Press; San Diego: 2008. pp. 1135–1166. [Google Scholar]
- 16.Cooper CC, Steinbuch M, Stevenson R, Miday R, Watts NB. The epidemiology of osteonecrosis: findings from the GPRD and THIN databases in the UK. Osteoporos Int. 2010;21:569–577. doi: 10.1007/s00198-009-1003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Felson DT, Anderson JJ. Across-study evaluation of association between steroid dose and bolus steroids and avascular necrosis of bone. Lancet i. 1987:902–905. doi: 10.1016/s0140-6736(87)92870-4. [DOI] [PubMed] [Google Scholar]
- 18.McAvoy S, Baker KS, Mulrooney D, Blaes A, Arora M, Burns LJ, Majhail NS. Corticosteroid dose as a risk factor for avascular necrosis of the bone after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2010;16:1231–1236. doi: 10.1016/j.bbmt.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 19.Chandler CN, Wright V. Deleterious effect of intra-articular hydrocortisone. Lancet. 1958;ii:661–663. doi: 10.1016/s0140-6736(58)92262-1. [DOI] [PubMed] [Google Scholar]
- 20.Habib GS, Saliba W, Nashashibi M. Local effects of intraarticular corticosteroids. Clin Rheumatol. 2010;29:347–356. doi: 10.1007/s10067-009-1357-y. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto T, Schneider R, Iwamoto Y, Bullough PG. Rapid destruction of the femoral head after a single intraarticular injection of corticosteroid into the hip joint. J Rheumatol. 2006;33:1701–1704. [PubMed] [Google Scholar]
- 22.Lee YJ, Lee JS, Kang EH, Lee Y-K, Kim S-Y, Song YW, Koo K-H. Vascular endothelial growth factor polymorphisms in patients with steroid-induced femoral head osteonecrosis. J Orthop Res. 2011 doi: 10.1002/jor.21492. [DOI] [PubMed] [Google Scholar]
- 23.Russcher H, Smit P, van den Akker ELT, van Rossum EFC, Brinkman AO, de Jong FH, Lamberts SWJ, Koper JW. Two polymorphisms in the glucocorticoid receptor gene directly affect glucocorticoid-regulated gene expression. J Clin Endocrinol Metab. 2005;90:5804–5810. doi: 10.1210/jc.2005-0646. [DOI] [PubMed] [Google Scholar]
- 24.Cooper MS, Rabbitt EH, Goddard PE, Bartlett WA, Hewison M, Stewart PM. Osteoblastic 11b-hydroxysteroid dehydrogenase type 1 activity increases with age and glucocorticoid exposure. J Bone Miner Res. 2002;17:979–986. doi: 10.1359/jbmr.2002.17.6.979. [DOI] [PubMed] [Google Scholar]
- 25.Prockop DJ. Type II collagen and avascular necrosis of the femoral head. N Engl J Med. 2005;352:2268–2270. doi: 10.1056/NEJMp058072. [DOI] [PubMed] [Google Scholar]
- 26.Han N, Yan Z, Guo C-a, Shen F, Liu J, Ski Y, Zhang Z. Effects of P-glycoprotein on steroid-induced osteonecrosis of the femoral head. Calcif Tissue Int. 2010;87:246–253. doi: 10.1007/s00223-010-9385-9. [DOI] [PubMed] [Google Scholar]
- 27.Elmantaser M, Stewart G, Young D, Duncan R, Gibson B, Ahmed SF. Skeletal morbidity in children receiving chemotherapy for acute lymphoblastic leukemia. Arch Dis Child. 2010;95:805–809. doi: 10.1136/adc.2009.172528. [DOI] [PubMed] [Google Scholar]
- 28.Hautmann AH, Elad S, Lawitschka A, Greinix H, Bertz H, Halter J, Faraci M, Hofbauer LC, Lee S, Wolff D, Holler E. Metabolic bone diseases in patients after allogeneic hematopoietic stem cell transplantation: report from the consensus conference on clinical practice in chronic graft-versus-host disease. Transpl Int. 2011 doi: 10.1111/j.1432-2277.2011.01264.x. [DOI] [PubMed] [Google Scholar]
- 29.Mehta P, Nelson M, Brand A, Boag F. Avascular necrosis in HIV. Rheumatol Int. 2011 doi: 10.1007/s00296-011-2114-5. [DOI] [PubMed] [Google Scholar]
- 30.Frost HM, Villananueva AR. Human osteoblast activity: III. Effect of cortisone on lamellar osteoblast activity. Henry Ford Hosp Med Bull. 1961;9:97–99. [PubMed] [Google Scholar]
- 31.Güven M, Ünay K, Bes C, Poyanli O, Akman B. Hip osteonecrosis in Cushing’s disease treated with bone-preserving procedures. J Orthop Sci. 2009 doi: 10.1007/s00776-009-1360-1369. [DOI] [PubMed] [Google Scholar]
- 32.Zaidi M, Sun L, Robinson J, Tourkova IL, Liu L, Wang Y, Zhu L-L, Liu X, Li J, Peng Y, Yang G, Shi X, Levine A, Iqbal J, Yaroslavskly BB, Isales C, Blair HC. ACTH protects against glucocorticoid-induced osteonecrosis of bone. Proc Natl Acad Sci. 2010;107:8782–8787. doi: 10.1073/pnas.0912176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids: potential mechanisms of the deleterious effects on bone. J Clin Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Brien CA, Jia D, Plotkin LI, Bellido T, Powers CC, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoblasts and osteocytes to induce their apoptosis and reduce bone formation and strength. Endocrinology. 2004;145:1835–1841. doi: 10.1210/en.2003-0990. [DOI] [PubMed] [Google Scholar]
- 35.Weinstein RS, Chen JR, Powers CC, Stewart SA, Landes RD, Bellido T, Jilka RL, Parfitt AM, Manolagas SC. Promotion of osteoclast survival and antagonism of bisphosphonate- induced osteoclast apoptosis by glucocorticoids. J Clin Invest. 2002;109:1041–1048. doi: 10.1172/JCI14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia D, O’Brien CA, Stewart SA, Manolagas SC, Weinstein RS. Glucocorticoids act directly on osteoclasts to increase their lifespan and reduce bone density. Endocrinology. 2006;147:5592–5599. doi: 10.1210/en.2006-0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weinstein RS, Wan C, Liu Q, Wang Y, Almeida M, O’Brien CA, Thostenson J, Roberson PK, Boskey AL, Clemens TL, Manolagas SC. Endogenous glucocorticoids decrease angiogenesis, vascularity, hydration, and strength in aged mice. Aging Cell. 2010;9:147–161. doi: 10.1111/j.1474-9726.2009.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lotke PA, Ecker ML. Osteonecrosis of the knee. J Bone Jt Surg Am. 1988;70:470–473. [PubMed] [Google Scholar]
- 39.Bardakos NV, Villar RN. The ligamentum teres of the adult hip. J Bone Joint Surg Br. 2009;91:8–15. doi: 10.1302/0301-620X.91B1.21421. [DOI] [PubMed] [Google Scholar]
- 40.Weinstein RS, Nicholas RW, Manolagas SC. Apoptosis of osteocytes in glucocorticoid-induced osteonecrosis of the hip. Endocrinology. 2000;85:2907–2912. doi: 10.1210/jcem.85.8.6714. [DOI] [PubMed] [Google Scholar]
- 41.Kabata T, Kubo T, Matsumoto T, Nishino M, Tomita K, Katsuda S, Horii T, Uto N, Kitajima I. Apoptotic cell death in steroid induced osteonecrosis: an experimental study in rabbits. J Rheumatol. 2000;27:2166–2177. [PubMed] [Google Scholar]
- 42.Eberhardt AW, Yeager-Jones A, Blair HC. Regional trabecular bone matrix degeneration and osteocyte death in femora of glucocorticoid-treated rabbits. Endocrinology. 2001;142:1333–1340. doi: 10.1210/endo.142.3.8048. [DOI] [PubMed] [Google Scholar]
- 43.Calder JDF, Buttery L, Revell PA, Pearse M, Polak JM. Apoptosis—a significant cause of bone cell death in osteonecrosis of the femoral head. J Bone Joint Surg Br. 2004;86:1209–1213. doi: 10.1302/0301-620x.86b8.14834. [DOI] [PubMed] [Google Scholar]
- 44.Ko JY, Wang FS, Wang CJ, Wong T, Chou WY, Tseng SL. Increased Dickkopf-1 expression accelerates bone cell apoptosis in femoral head osteonecrosis. Bone. 2010;46:584–591. doi: 10.1016/j.bone.2009.10.030. [DOI] [PubMed] [Google Scholar]
- 45.Youm YS, Lee SY, Lee SH. Apoptosis in the osteonecrosis of the femoral head. Clin Orthop Surg. 2010;2:250–255. doi: 10.4055/cios.2010.2.4.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mont MA, Zywiel MG, Marker DR, McGrath MS, Delanois RE. The natural history of asymptomatic osteonecrosis of the femoral head. J Bone Jt Surg Am. 2010;92:2165–2170. doi: 10.2106/JBJS.I.00575. [DOI] [PubMed] [Google Scholar]
- 47.Lai KA, Shen WJ, Yang CY, Shao CJ, Hsu JT, Lin RM. The use of alendronate to prevent early collapse of the femoral head in patients with nontraumatic osteonecrosis. J Bone Jt Surg Am. 2005;87:2155–2159. doi: 10.2106/JBJS.D.02959. [DOI] [PubMed] [Google Scholar]
- 48.Agarwala S, Shah SB. Ten-year follow-up of avascular necrosis of femoral head treated with alendronate for 3 years. J Arthroplast. 2011 doi: 10.1016/j.arth.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 49.Plotkin LI, Weinstein RS, Parfitt AM, Manolagas SC, Bellido T. Prevention of osteocyte and osteoblast apoptosis by bisphosphonates and calcitonin. J Clin Invest. 1999;104:1363–1374. doi: 10.1172/JCI6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weinstein RS, O’Brien CA, Zhao H, Roberson PK, Manolagas SC. Osteoprotegerin prevents glucocorticoid-induced osteocyte apoptosis in mice. Endocrinology. 2011;152:3323–3331. doi: 10.1210/en.2011-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]