

Abstract
We previously generated separate lines of transgenic mice that specifically overexpress either the Fibroblast growth factor (FGF)-2 low-molecular-mass isoform (TgLMW) or the high-mass isoforms (TgHMW) in the osteoblast lineage. Vector/control (TgVector) mice were also made. Here we report the use of isolated calvarial osteoblasts (COBs) from those mice to investigate whether the FGF-2 protein isoforms differentially modulate bone formation in vitro. Our hypothesis states that FGF-2 isoforms specifically modulate bone morphogenetic protein 2 (BMP-2) function and subsequently bone differentiation genes and their related signaling pathways. We found a significant increase in alkaline phosphatase-positive colonies in TgLMW COBs compared with TgVector controls. BMP-2 treatment significantly increased mineralized colonies in TgVector and TgLMW COBs. BMP-2 caused a further significant increase in mineralized colonies in TgLMW COBs compared with TgVector COBs but did not increase alkaline phosphatase-positive colonies in TgHMW COBs. Time-course studies showed that BMP-2 caused a sustained increase in phosphorylated mothers against decapentaplegic-1/5/8 (Smad/1/5/8), runt-related transcription factor-2 (Runx-2), and osterix protein in TgLMW COBs. BMP-2 caused a sustained increase in phospho-p38 MAPK in TgVector but only a transient increase in TgLMW and TgHMW COBs. BMP-2 caused a transient increase in phospho-p44/42 MAPK in TgVector COBs and no increase in TgLMW COBs, but a sustained increase was found in TgHMW COBs. Basal expression of FGF receptor 1 protein was significantly increased in TgLMW COBs relative to TgVector COBs, and although BMP-2 caused a transient increase in FGF receptor 1 expression in TgVector COBs and TgHMW COBs, there was no further increase TgLMW COBs. Interestingly, although basal expression of FGF receptor 2 was similar in COBs from all genotypes, BMP-2 treatment caused a sustained increase in TgLMW COBs but decreased FGF receptor 2 in TgVector COBs and TgHMW COBs.
Osteoblast differentiation and bone formation are regulated by many local factors, and, among those, bone morphogenetic proteins (BMPs) are the most potent. BMPs, synthesized and released by osteoblasts in the bone matrix (1, 2), signal through heterotetrameric complexes formed by two receptor type II and 2 receptor type I subunits (3). BMP-2 specific mothers against decapentaplegic (R-Smads; Smad 1/5/8) are the major transducer for BMP receptors, and, once activated, they form heteromeric complexes with Smad 4 for nuclear translocation to regulate target genes by interacting with various transcription factors (3). Disorders in BMP signaling have been implicated in multiple bone diseases including tumor metastasis, brachydactyly type A2, and osteoarthritis (3, 4). It is known that BMP-2 controls the expression and function of runt-related transcription factor-2 (Runx-2; Cbfa1) through Smad signaling (5, 6) in which Runx-2 is an essential protein for osteoblast maturation (7). Indeed, in humans, heterozygous loss of Runx-2 function segregates with abnormal skeletal phenotypes (8, 9), and murine Runx-2 haploinsufficiency causes similar phenotypes with a primary defect in intramembranous bone formation (7, 10). Moreover, BMP-2 strongly increases osterix, the secondary regulator of osteoblastogenesis (11–13) that is specifically expressed in the osteoblasts and osteocytes of all developing bones, in which it likely acts downstream of Runx-2 (14).
Fibroblast growth factor (FGF)-2 potently stimulates proliferation of osteoblast precursor and is therefore anabolic in experimental mice (15), whereas Fgf2−/− mice show significantly decreased bone mass and bone formation (16). The FGF-2 gene encodes multiple protein isoforms from alternative translation start sites (17, 18). The human FGF-2 high-molecular-mass isoforms 22, 23, and 24 kDa contain a nuclear localization signal in the amino- terminus extension to confer an intracrine function (18–20). The low-molecular-mass 18-kDa FGF-2 protein is exported from cells, presumably conferring a different function vs the high-molecular-mass (HMW) FGF protein isoforms. In rodents, there are two nuclear isoforms of 19 kDa and 21 kDa and then a low-molecular-mass, 17.5-kDa FGF-2 protein that is exported from cells and stored in the extracellular matrix (21–23).
Previously we reported that FGF-2 null mice developed osteopenia with aging (16), demonstrating that FGF-2 is an important regulator of bone formation in vivo. Alternatively, global transgenic overexpression of the cDNA that encodes for all FGF-2 protein isoforms resulted in decreased endochondral and intramembranous bone formation (24). We have also shown that overexpression of the HMW/nuclear isoforms preferentially mediate augmented osteoblastic ROS17/2.8 cell proliferation (25), whereas the low-molecular-weight (LMW)/exported isoform is involved in the prostaglandin F2α (PGF2α) effects found in mouse primary calvarial osteoblasts (26–28). In addition, we demonstrated that the anabolic effects of PTH on bone are, at least in part, mediated by FGF-2 (29). In other studies we reported that BMP-2 increased FGF-2 mRNA and protein in osteoblasts from Fgf2+/+ mice (30). We showed that the BMP-2 induced nuclear accumulation of Runx-2 was markedly impaired in osteoblasts from Fgf2−/− mice, whereas an increased Runx-2 accumulation and nuclear localization were found after BMP-2 treatment in Fgf2−/− osteoblasts transfected with a retroviral Fgf2 construct (31). Thus, it is evident that a reciprocal regulation of FGF-2 and BMP-2 exists in osteoblasts. However, so far it is unclear whether the LMW and HMW FGF-2 isoforms selectively modulate the effect of BMP-2-induced bone formation or the regulation of the BMP-2 downstream signaling in osteoblasts.
Therefore, the aim of this in vitro study was to explore the potential role of FGF-2 isoforms for BMP-2 signaling in osteoblasts differentiation. For this purpose, we used primary calvarial osteoblasts isolated from different lines of transgenic (Tg) mice that selectively overexpress either the LMW isoforms of FGF-2 (TgLMW) or the HMW isoforms of FGF-2 (TgHMW).
Materials and Methods
Animals
Generation of mice expressing HMW and LMW isoforms of FGF-2
Transgenic mouse lines overexpressing specific FGF-2 isoforms, regulated by collagen type I 5′ elements for osteoblast specificity, were developed as previously described (32), and they are as follows: TgLMW, which specifically overexpresses low-molecular-mass human FGF-2 (18 kDa); TgHMW, which specifically overexpresses the three high-molecular-mass human FGF-2 isoforms (22, 23, and 24 kDa); and TgVector, which overexpresses the transgene cassette without FGF-2 coding sequences. All mice were bred and housed in the transgenic facility in the Center for Laboratory Animal Care at the University of Connecticut Health Center.
Primary calvarial osteoblasts (COBs)
Newborn mice were killed by CO2 narcosis and cervical dislocation. The Animal Care and Use Committee of the University of Connecticut Health Center approved all animal protocols. Because the mice were newborn, it was not possible to identify the specific sex. Cells were digested from calvariae of newborn control TgVector as well as TgLMW and TgHMW mice in a solution of 1 mL trypsin as previously described (16). Cells were pooled and then plated in 100-mm dishes and cultured to confluence in DMEM without phenol red (Sigma Chemical Co, St Louis, Missouri) containing 10% heat-inactivated-fetal calf serum (HIFCS), penicillin (100 U/mL), and streptomycin (50 μg/mL) in a humidified atmosphere of 5% CO2 at 37°C. Then cultured cells were shipped frozen in medium plus 10% dimethylsulfoxide to the University of Camerino (Camerino, Italy), where they were thawed and cultured immediately upon receipt to perform the in vitro experiments described below. No significant effects on cell morphology and viability were observed.
Alkaline phosphatase (ALP) assay and Alizarin red S histochemical staining
COBs from TgVector mice and the two FGF-2 transgenic lines (described above) were plated at 5 × 104 cells/well in 6-well culture plates and cultured for 6 days in differentiation media (DMEM, 10% HIFCS, 8 mM β-glycerophosphate, 50 μg/mL ascorbic acid, 100 nM dexamethasone) in the presence or absence of BMP-2 (50 ng/mL) or vehicle. ALP staining was per formed with a commercial kit (Sigma-Aldrich, Milano, Italy) according to the manufacturer's instructions. Using Alizarin red S, COBs cultured on 6-well culture dishes containing coverslips, were stained on day 14 for assessing the mineralized matrix. The medium was removed, and the cell layers were rinsed with PBS and fixed in 4% paraformaldehyde diluted in PBS for 25 minutes at room temperature. Then cultures were washed with PBS and stained with 2% Alizarin red S (pH 7.2; Sigma-Aldrich) for 20 minutes at 37°C. Cultures were observed at light microscope level. Then stain was desorbed and the collected solutions were distributed as 100 μL/well on 96-well plates for absorbance reading at 590 nm by spectrophotometry (Tecan Infinite reader; Tecan, Milano, Italy).
Western blotting
COBs from TgVector mice and the two FGF-2 transgenic lines (described above) were plated at 10 × 106 cells/well in 6-well culture dishes and grown in appropriate medium with 10% HIFCS, penicillin, and streptomycin until confluence. Cells were serum deprived for 24 hours and stimulated with BMP-2 (50 ng/mL) or vehicle for selected time periods. Proteins were extracted with cell lysis buffer (Cell Signaling Celbio, Milano, Italy), and the concentration was determined by the BCA protein assay reagent (Pierce, Celbio, Italy). After SDS-PAGE on 12% gels, proteins were transferred to polyvinyl difluoride (PVDF) membranes (Amersham Biosciences Europe Gmbh, Cologno Monzese, Milano, Italy). The next steps were performed by enhanced chemiluminescence Advance Western blotting detection kit (Amersham Biosciences Europe). Briefly, membranes were blocked with Advance blocking agent in PBS containing 0.1% Tween 20 (PBS-T) for 1 hour at room temperature. Then, membranes were incubated for 2 hours at room temperature with the following primary antibodies: rabbit antiphospho-Smads 1/5/8, rabbit antiphospho-p38, rabbit antiphospho-p44/42, rabbit antiphospho-Akt, rabbit anti-Akt antibodies (Cell Signaling), all diluted at 1:800; and rabbit anti-Pebp2aA (Runx-2), rabbit antiosterix, rabbit anti-C-term-FGFR1 (Flg C-15), and rabbit anti-C-term-FGFR2 (Bek C-17) antibodies (Santa Cruz Biotechnology, Tebu-Bio, Milano, Italy), all diluted 1:200. After washing with PBS-T, the blots were incubated with horseradish peroxidase-conjugated donkey antirabbit IgG antibody (Amersham Biosciences Europe) diluted 1:100 000 in blocking solution for 1 hour at room temperature. After further washing with PBS-T, the immunoreactive bands were visualized using luminol reagents and Hyperfilm-enhanced chemiluminescence film (Amersham Biosciences Europe) in accordance with the manufacturer's instructions. To normalize the bands, the filters were stripped and reprobed with a mouse anti-α-tubulin antibody (Sigma-Aldrich). Band densities were quantified densitometrically.
RNA interference
To specifically study the effects of BMP-2 treatments on FGF receptor-1 (FGFR1) and FGFR2, COBs from TgVector and the two FGF-2 transgenic lines (described above) were plated in 6-well culture dishes at a density of 10 × 106 cells/well and grown for 5 days in DMEM containing 10% HIFCS, penicillin, and streptomycin. Other cultures were transfected with FGFR1 small interfering RNA (siRNA) or with FGFR2 siRNA according to siRNA transfection protocol from the manufacturer (Santa Cruz Biotechnology). A control siRNA (nonhomologous to any known gene sequence) was used as a negative control. The levels of FGFR1 and FGFR2 were analyzed by Western blotting using a rabbit anti-C-term-FGFR1 (Flg C-15) and a rabbit anti-C-term-FGFR2 antibody (Bek C-17) antibody (Santa Cruz Biotechnology), and the specificity of the silencing was confirmed in 3 independent experiments. Cells were 24 hours serum deprived before the addition of BMP-2 (50 ng/mL) or vehicle for 2 hours. Then proteins were extracted and phospho-Smads 1/5/8 and phospho-p44/42 as well as Runx-2 and osterix levels were analyzed by Western blotting as described above. To normalize the bands, filters were stripped and reprobed with a monoclonal anti-α-tubulin (Sigma-Aldrich). Band densities were quantified using densitometry.
Statistical analysis
All experiments were repeated at least 3 times with overlapping results. An unpaired 2-tailed Student's t test was used to test for significant differences between 2 groups with a minimum threshold of P < .05.
Results
BMP-2 effects on bone mineralization marker expression in COBs from TgVector and FGF-2 transgenic mouse lines
We evaluated whether the overexpression of specific FGF-2 isoforms differentially modulated bone formation in vitro or influenced the BMP-2 regulation of bone differentiation markers that control matrix mineralization. After 6 days of treatment, vehicle-only-treated cultures showed a significantly higher level of ALP-positive colonies in TgLMW COBs compared with TgHMW COBs (Figure 1). Treatment with BMP-2 caused a further significant increase in ALP-positive colonies in both TgVector and TgLMW COBs. In contrast, no significant changes between BMP-2-treated and -untreated cultures were observed in TgHMW COBs, as shown in the graphic derived from a pool of 3 independent experiments (Figure 1, A and B). Alizarin red staining of untreated cultures after 14 days revealed a significant decrease in calcified nodules in TgHMW COBs compared with TgVector and TgLMW COBs (Figure 2). Moreover, BMP-2 treatment caused a significant increase in calcified nodules only in TgVector and TgLMW COBs (Figure 2, A and B).
Figure 1.
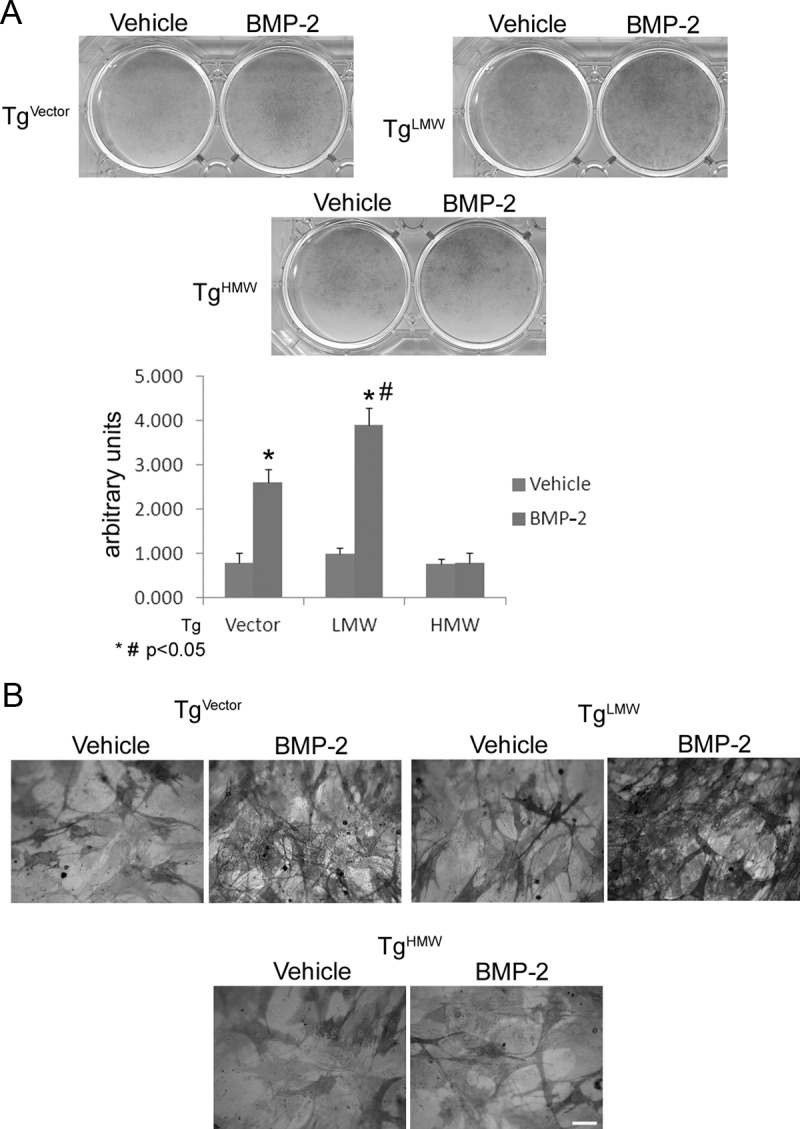
ALP staining. COBs from TgVector mice and FGF-2 transgenic mice were grown for 6 days on 6-well culture dishes in differentiation medium with BMP-2 (50 ng/mL) or vehicle. Colony area was measured by NIH Image (National Institutes of Health, Bethesda, Maryland), and values are reported as means ± SD and statistically analyzed with a t test. *, P < .05 vs the corresponding vehicle-treated COBs; #, P < .05 vs the corresponding BMP-2-treated COBs (A) were in accordance with ALP staining observed with a light microscope. Bar, 100 μm (B).
Figure 2.
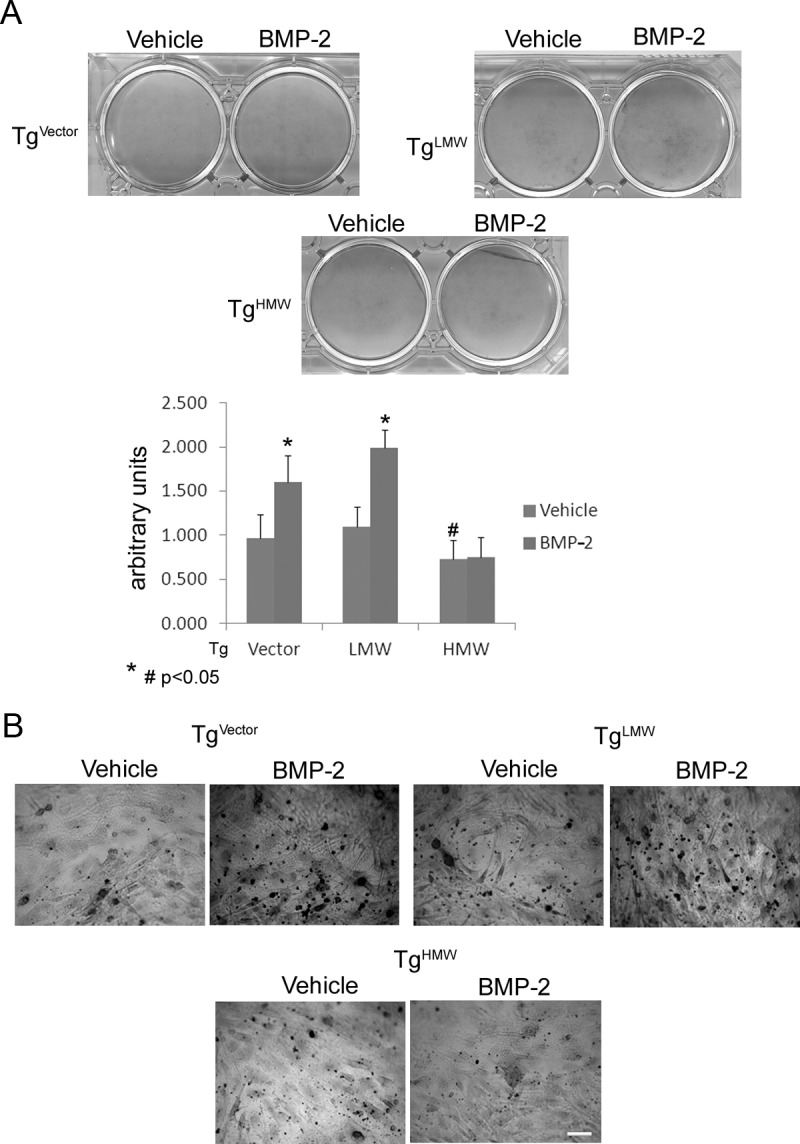
Alizarin red S staining. COBs from TgVector, TgLMW, and TgHMW mice were plated on coverslips in 6-well culture dishes and then growth for 14 days in differentiation medium in presence of BMP-2 (50 ng/mL) or vehicle. The results show a quantitative analysis of Alizarin red staining in which values are reported as means ± SD and statistically analyzed with a t test. #, P < .05 vs the corresponding vehicle-treated TgVector and TgLMW COBs; *, P < .05 vs the corresponding vehicle-treated COBs and were in accordance with the Alizarin red S staining observed with a light microscope. Bar, 100 μm (B).
Expression and BMP-2 regulation of the intracellular signaling molecules in COBS from TgVector, Tg LMW, or TgHMW mice
The results shown in Figure 1 demonstrated that overexpression of the FGF-2 isoforms differentially influenced the expression of bone formation markers in both untreated and BMP-2-treated conditions. Therefore, we determined whether the FGF-2 isoforms influenced the BMP-2-activated downstream intracellular signaling molecules. Those pathways included the classically BMP-2 activated Smad signaling pathways (2) as well as p38 MAPK and ERKs p44/42 that were reported to be significant in FGF-2 (15) and BMP-2 signal cascades (33). The basal expression of phospho-Smad 1/5/8, phospho-p38 and phospho-p44/42 in 6-day cultured and 24-hour serum-deprived COBs from TgVector and the FGF-2 transgenic mouse lines are shown in Figure 3. COBs isolated from TgHMW overexpression mice showed significantly up-regulated levels of phospho-Smad 1/5/8 level compared with TgVector COBs. As shown in Figure 4, A and B, treatment with BMP-2 increased phospho-Smad 1/5/8 levels in TgVector and in TgLMW at 30 minutes and 2 and 6 hours. Conversely, in TgHMW COBs (Figure 4C), the increase was evidenced only at 30 minutes and 2 hours. We observed no differences in basal phospho-p38 expression in COBs from any of the FGF-2 transgenic mice (Figure 3). However, as shown in Figure 4A, phospho-p38 was increased by BMP-2 in TgVector COBs at all time points tested, with the maximal up-regulation at 2 hours, although phospho-p38 was increased only at 2 hours in TgLMW (Figure 4B) and in TgHMW COBs, as shown in Figure 4C. The basal level of phospho-p44/42, observed in TgLMW COBs, was comparable with that found in TgVector COBs (Figure 3) and was unchanged by treatment with BMP-2 (Figure 4B). In contrast, BMP-2 up-regulated phospho-p44/42 levels at 2 hours in TgVector COBs (Figure 4A), and then BMP-2 also increased phospho-p44/42 levels in TgHMW COBs at all time points (Figure 4C).
Figure 3.
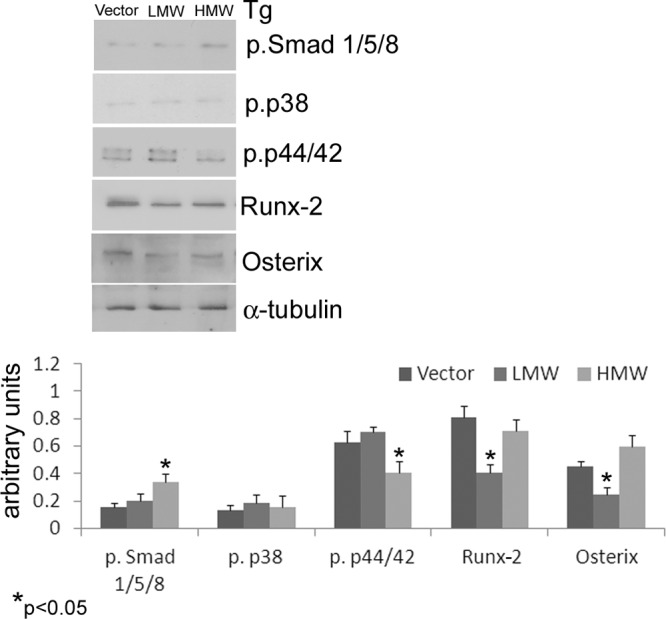
COBs from TgVector, TgLMW, and TgHMW mice were serum deprived for 24 hours. Proteins were extracted, subjected to SDS-PAGE, transferred to a PVDF membrane, and then probed with the rabbit antiphospho-Smad 1/5/8, the rabbit antiphospho-p38, the rabbit antiphospho-p44/42, the rabbit anti-Runx-2, and the rabbit anti-osterix antibodies; next, the filters were stripped and reprobed with the mouse anti-α-tubulin antibody. Results from 3 independent experiments are shown as means ± SD. *, P < .05 vs TgVector COBs.
Figure 4.

COBs isolated from TgVector (A), TgLMW (B), and TgHMW (C) mice were serum deprived for 24 hours and then treated with BMP-2 (50 ng/mL) or vehicle for 30 minutes, 2 hours, and 6 hours. Proteins were extracted, subjected to SDS-PAGE, transferred to a PVDF membrane, and probed with antibodies to either rabbit antiphospho-Smads (1, 5, 8), rabbit antiphospho-p38, rabbit antiphospho-p44/42, rabbit anti-Runx-2, or rabbit anti-osterix; then the filters were stripped and reprobed with the mouse anti-α-tubulin antibody for quantitative analysis. Results from 3 independent experiments are shown as means ± SD. *, P < .05 vs the corresponding unstimulated COBs.
We also examined the levels of the osteogenic master transcription factors Runx-2 and osterix. The levels of both proteins were decreased in TgLMW COBs compared with TgVector COBs (Figure 3). As shown in Figure 4A, BMP-2 treatment increased Runx-2 only at 6 hours; however, BMP-2 significantly increased osterix protein during the entire treatment period in TgVector COBs. In addition, Runx-2 and osterix were strongly and stably increased by BMP-2 in TgLMW COBs (Figure 4B). In contrast, in TgHMW COBs, BMP-2 down-regulated Runx-2 at 6 hours, and, in a time dependent manner, it also reduced osterix synthesis (Figure 4C).
Because recent studies demonstrated that the requirement of protein kinase B (Akt) for BMP-2 stimulated osteoblast differentiation (34), we also examined its expression. Interestingly, a consistent and stable increase of phospho-Akt was observed only in TgLMW COBs (Figure 5).
Figure 5.
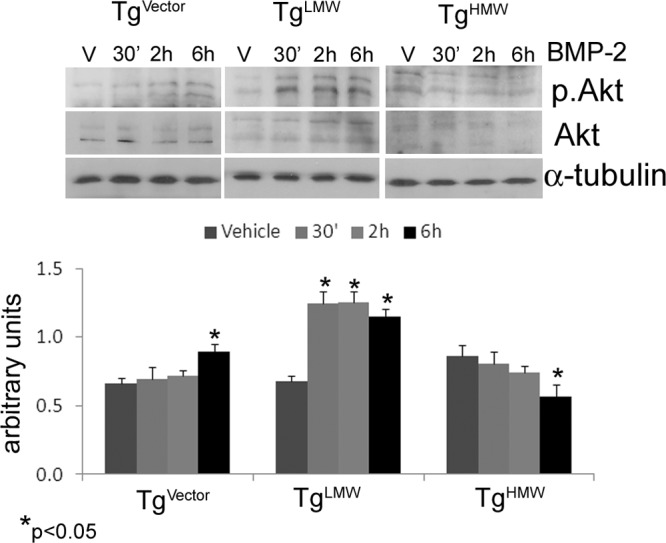
COBs from TgVector, TgLMW, and TgHMW mice were serum deprived for 24 hours and treated with BMP-2 (50 ng/mL) or vehicle for 30 minutes, 2 hours, and 6 hours. Proteins were extracted, subjected to SDS-PAGE, transferred to a PVDF membrane, and probed with the rabbit antiphospho-Akt and the rabbit anti-Akt; then the filters were stripped and reprobed with the mouse anti-α-tubulin antibody. Results from 3 independent experiments are shown as means ± SD. *, P < .05 vs the corresponding unstimulated COBs.
Expression and regulation of the FGFR1 and the FGFR2 by BMP-2 in COBs from TgVector, TgLMW, and TgHMW mice
We first evaluated whether there was FGF-2 isoform-specific regulation of the FGFR1 and FGFR2 proteins and whether BMP-2 differentially regulated FGFR1 and FGFR2 protein synthesis in FGF-2 isoform-specific COBs. As shown in Figure 6, in 24-hour serum-derived cultures, a higher level of FGFR1 was detected in TgLMW COBs compared with TgVector COBs, whereas the receptor level was similar in TgHMW COBs and in TgVector COBs. Interestingly, treatment with BMP-2 at 6 hours significantly increased the FGFR1 level in TgVector COBs but did not affect the receptor level in TgLMW COBs at any time point. However, a strong up-regulation of FGFR1 was observed in TgHMW COBs incubated for 30 minutes and 2 hours with BMP-2. In contrast to FGFR1, as shown in Figure 6, basal expression of FGFR2 was similar in TgLMW COBs and TgHMW COBs compared with TgVector COBs in which FGFR2 protein was decreased by BMP-2 at 30 minutes in TgVector COBs and at 6 hours in TgHMW COBs. Interestingly, a significant increase in FGFR2 protein was observed in TgLMW COBs for the entire time course of BMP-2 treatment.
Figure 6.
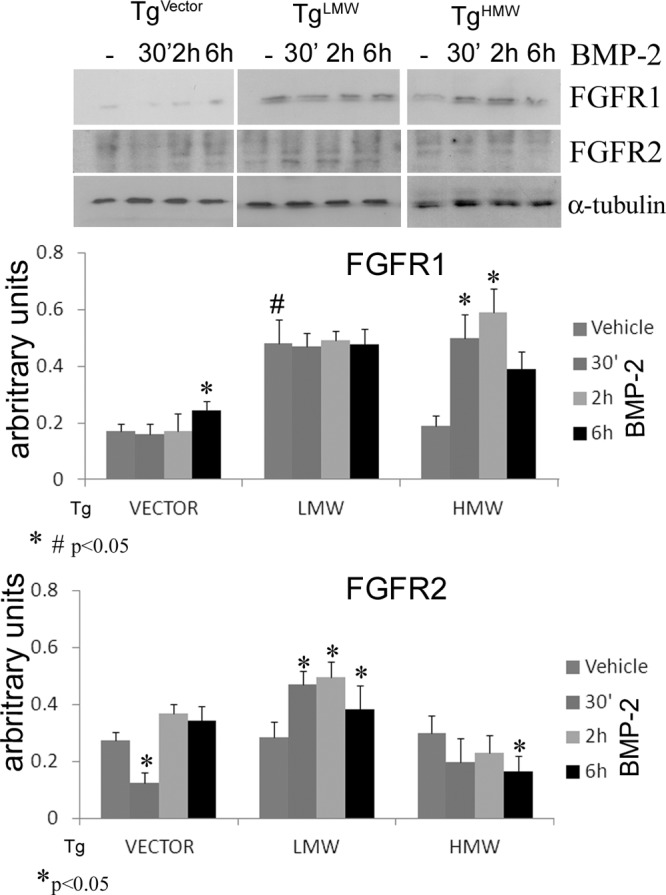
COBs from TgVector, TgLMW, and TgHMW mice were serum deprived for 24 hours and treated with BMP-2 (50 ng/mL) or vehicle for 30 minutes, 2 hours, and 6 hours. Proteins were extracted, subjected to SDS-PAGE, transferred to a PVDF membrane, and probed with the rabbit anti-C-term-FGFR1 antibody (Flg C-15) or with the rabbit anti anti-C-term-FGFR2 antibody (Bek C-17); filters were stripped and reprobed with the mouse anti-α-tubulin antibody. Results from 3 independent experiments are shown as means ± SD. *, P < .05 vs corresponding unstimulated control; #, P < .05 vs TgVector unstimulated COBs.
FGFR2 and FGFR1 knockdown in FGF-2 transgenic COBs
To analyze the potential involvement of the FGFR2 in BMP-2-modulated intracellular signaling proteins, we conducted FGFR2 knockdown experiments in BMP-2-treated, FGF-2 transgenic COBs (Figure 6). COBs isolated from TgVector, FGF-2 TgLMW, and TgHMW mice were cultured in either 2-hour vehicle-treated or BMP-2-treated conditions (Figure 7). siRNA against the FGFR2 message efficiently and specifically silenced (>80%) FGFR2 protein expression in TgVector COBs, in TgLMW COBs, and in TgHMW COBs (Supplemental Data, published on The Endocrine Society's Journals Online web site at http://endo.endojournals.org). Depletion of FGFR2 strongly decreased phospho-Smad 1/5/8 in vehicle-treated and in BMP-2-treated TgVector COBs (Figure 7A) and partially inhibited the BMP-2-induced increase of phospho-Smad in TgLMW COBs (Figure 7B); moreover, the FGFR2 siRNA totally inhibited the BMP-2-induced increase of phospho-Smads in TgHMW COBs (Figure 7C). Lack of FGFR2 also caused a decrease of phospho-p44/42 in vehicle-treated and BMP-2-treated TgVector COBs and TgLMW COBs (Figure 7, A and B). Interestingly, phospho-p44/42 was increased in vehicle-treated FGFR2 knocked-down TgHMW COBs, which was unchanged by treatment with BMP-2 (Figure 7C). In FGFR2 knocked-down TgVector COBs, treatment with BMP-2 decreased Runx-2 and osterix protein level (Figure 7A). FGFR2 knockdown also decreased both Runx-2 and osterix proteins in TgLMW COBs, in TgHMW vehicle-treated, and BMP-2-treated COBs (Figure 7, B and C).
Figure 7.

Depletion of FGFR2 and FGFR1 expression by siRNA in COBs from TgVector (A), TgLMW (B), and TgHMW (C) mice. COBs were transfected with FGFR1 or FGFR2 siRNA. After transfection, as described above, cells were serum deprived for 24 hours and treated with BMP-2 (50 ng/mL) or vehicle for 2 hours. Proteins were extracted for Western blotting analysis and probed with antibodies to either rabbit antiphospho-Smads (Smads 1, 5, 8), rabbit antiphospho-p44/42, rabbit anti-Runx-2, or rabbit anti-osterix; then the filters were stripped and reprobed with the mouse anti-α-tubulin antibody. Results from 3 independent experiments are shown as means ± SD and statistically analyzed with a t test. *, P < .05 vs unstimulated COBs expressing the receptor; §, P < .05 vs unstimulated COBs expressing the receptor; #, P < .05 vs BMP-2-treated COBs expressing the receptor.
We also conducted similar FGFR1 knockdown experiments on FGF-2 transgenic COBs (Supplemental Data). As shown in Figure 7, the use of FGFR1 siRNA in TgVector COBs and in TgLMW COBs decreased phospho-Smad in both the presence and absence of BMP-2 (Figure 7, A and B). Conversely, in TgHMW COBs, the decrease of phospho-Smad levels was observed only in vehicle-treated cells (Figure 7C). Moreover, the depletion of FGFR1 caused a decrease of phospho-p44/42 levels in TgVector COBs and in TgLMW COBs, both under vehicle-treated and BMP-2-treated conditions (Figure 7, A and B). In addition, FGFR1 depletion blocked the up-regulation of phospho-p44/42 induced by BMP-2 in TgHMW COBs (Figure 7C). The levels of Runx-2 and osterix in the absence and the presence of BMP-2 in TgVector COBs, lacking FGFR1, were similar to that found in Tg COBs expressing FGFR1 (Figure 7A). Finally, in TgLMW COBs, Runx-2 and osterix proteins were increased only in vehicle-treated conditions (Figure 7B), whereas in TgHMW COBs, decreased Runx-2 and osterix was observed in the presence and absence of BMP-2 (Figure 7C).
Discussion
In this study we evaluated whether the FGF-2 isoform-specific regulation of bone formation was regulated by interaction with BMP-2 that, in turn, regulates bone differentiation markers to control matrix mineralization. Our results clearly demonstrate a differential effect from the specific expression of the FGF-2 isoforms on bone formation, in which the exported LMW isoform enhanced matrix deposition, whereas the HMW isoforms had an inhibitory effect. These results are consistent with our previous observations of increased bone formation in vivo in TgLMW (32) and decreased bone formation in vivo in TgHMW mice (35).
The results also show a differential response for BMP-2 in FGF-2 isoform COBs in which BMP-2 enhances the anabolic effects of the LMW FGF-2 isoform but could not rescue the inhibitory effects of the HMW FGF-2 isoform on osteoblast differentiation and mineralization. The major signaling pathway for BMP-2 action occurs via the activation of phospho-Smad, but BMPs also activate the MAPK signaling pathway (2). In the present study, BMP-2 activated phospho-Smad 1/5/8 in TgVector COBs as well as in TgLMW and in TgHMW COBs. Therefore, regulation of Smads does not mediate the differential effects of BMP-2 on bone formation in FGF-2 isoform mice, suggesting modulation through other signaling molecules. Indeed, we previously demonstrated that BMP-2 caused a similar induction of phospho-Smad in COBs from Fgf2+/+ and Fgf2−/− mice (30). These data extend those results in a very significant manner.
Both the p38 MAPK and ERKs p44/42 were reported to be important in BMP-2-induced osteoblast differentiation (33). In particular, the p38 MAPK pathway plays a positive role in BMP-2-induced osterix expression (36). The fact that BMP-2 increased phospho-p38 at all time points in TgVector COBs but not in COBs from TgLMW or TgHMW mice suggests that p38 does not play a significant role in BMP-2 effects on bone formation in FGF-2 isoform-specific transgenic mice. Indeed, we found that phospho-p38 proteins were comparably regulated by BMP-2 in COBs from Fgf2+/+ and Fgf2−/− mice (30).
The function of MAPK in osteoblasts is still controversial. Indeed, it was reported that ERK activation stimulates Runx-2 activity and osteoblast differentiation (37, 38). Likewise, constitutively active MAPK kinase-1, a MAPK activator, induced Runx-2 stimulation (39), whereas FGF-2 also increased the stabilization, acetylation, and activation of Runx-2. Previously this function was restricted to ERK (40). In this context, we previously reported that PTH increased Runx-2 only in Fgf2+/+ COBs (29). Other reports demonstrated that persistent ERK signaling is associated with inhibition of bone formation (14, 41, 42). Moreover, it was proposed that the pathogenesis of achrondroplasia may be mediated by inhibition of Smad 1/5/8 activity through increased MAPK phosphorylation (43). The new results presented here suggest that the marked and stable increase of phospho-p44/42 by BMP-2, observed only in TgHMW COBs, negatively affected the osteogenic master transcription factors, Runx-2 and osterix, which were found to be strongly decreased by these effectors. However, the robust and stable up-regulation of Runx-2 and osterix by BMP-2 that we found in TgLMW COBs, consistent with other reports (34, 44), could be partly correlated with the increase of phospho-Akt that occurred only in TgLMW COBs.
In support of this hypothesis, our recent demonstration that BMP-2 increased phospho-Akt in COBs from Fgf2+/+ mice but decreased these phospho-proteins in COBs from Fgf2−/− mice emphasizes the potential role of Akt in the BMP-2/FGF-2 signal (45). In COBs from vector mice, although Runx-2 was slightly increased only after 6 hours of treatment, osterix was up-regulated during the entire time period. The former result is consistent with previous studies, indicating that BMP-2-induced osterix mRNA expression is not dependent on Runx-2 (46). Thus, the results in toto add new insight to our previous data about the importance of the LMW FGF-2 isoform in mediating bone remodeling.
Current models hold that most of the biological functions for FGF-2 are mediated by binding to and activation of its specific tyrosine kinase receptors in the cell membrane, whereas other effects involve the translocation of the receptors into the nucleus (15, 26–28). Our data show, for the first time, differential basal expression of FGFR1, but not FGFR2, in COBs from FGF-2 isoform genotype mice. We also demonstrate, for the first time, that BMP-2 differentially regulates FGFR1 and FGFR2 synthesis in COBs. Moreover, the specific receptor regulation by BMP-2 is dependent on the overexpression of the specific FGF-2 isoforms. BMP-2 did not increase FGFR1 but did increase FGFR2 protein in TgLMW COBs, suggesting that FGFR2 but not FGFR1 may be important in the enhanced anabolic effect of BMP-2 on bone formation in TgLMW COBs. A role for FGFR2 in BMP-2-modulated intracellular signaling proteins and bone formation is further supported by the results from siRNA depletion of FGFR2, which showed a marked decreased of Runx-2 and osterix protein levels in vehicle-treated and BMP-2-treated TgLMW COBs. These results are consistent with previous studies that identified FGFR2 as an inducer of osteogenic differentiation of mouse calvarial osteoblasts (47, 48). Similarly, we observed a decrease of Runx-2 and osterix in the TgHMW COBs that were FGFR2 depleted. The notable increase of phospho-p44/42, which was found in the FGFR2-silenced TgHMW COBs that were either vehicle or BMP-2 treated, seems to confirm our previous observation that their activation is not involved in the regulation of Runx-2 and osterix; indeed, both proteins were down-regulated in these Tg COBs, with or without BMP-2 treatment.
Based on our data, FGFR1 may participate in the osteoblast differentiation process by playing a negative role in the untreated condition and a positive function in BMP-2-induced osteoblast maturation. Indeed, although we found that FGFR1 ablation in TgLMW COBs increases the basal level of Runx-2 and osterix proteins, the results also showed that the lack of FGFR1 blocks the increase of Runx-2 and osterix induced by BMP-2. In summary, this study, consistent with our previous in vivo studies showing the importance of the exported 18-kDa FGF-2 isoform in osteoblast differentiation (32), provides new knowledge regarding the influence of FGF-2 isoforms on signaling pathways implicated in bone remodeling processes. We previously reported that low doses of FGF-2 in combination with BMP-2 increases mineralization more than BMP-2 alone (49). The actual intriguing evidence, in which differential osteoblast responses to BMP-2 are clearly dependent on both the FGF-2 isoform species and the specific expression of FGFRs, will prove to be very useful in bone remodeling research, and, more importantly, it will provide additional insights into potential therapeutic applications of BMP-2.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant AG21189 (to M.M.H.) and Camerino University Grant FAR 2010.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ALP
- alkaline phosphatase
- BMP
- bone morphogenetic protein
- COB
- calvarial osteoblast
- FGF
- fibroblast growth factor
- FGFR
- FGF receptor
- HIFCS
- heat-inactivated-fetal calf serum
- HMW
- high molecular mass
- LMW
- low molecular weight
- PBS-T
- PBS containing Tween 20
- PVDF
- polyvinyl difluoride
- Runx
- runt-related transcription factor
- siRNA
- small interfering RNA
- Smad
- mothers against decapentaplegic
- Tg
- transgenic.
References
- 1. Yamaguchi A. Regulation of differentiation pathway of skeletal mesenchymal cells in cell lines by transforming growth factor-β superfamily. Semin Cell Biol. 1995;6:165–173 [DOI] [PubMed] [Google Scholar]
- 2. Rosen V, Wozney JM. Bone morphogenetic proteins. In: Belizikian J, Raisz LG, Rodan G, eds. Principles of Bone Biology. 2nd ed San Diego: Academic Press; 2002:919–928 [Google Scholar]
- 3. Miyazono K, Maeda S, Imamura T. BMP receptor signaling: transcriptional targets, regulation of signals, and signaling cross-talk. Cytokine Growth Factor Rev. 2005;16:251–263 [DOI] [PubMed] [Google Scholar]
- 4. Papachroni KK, Karatzas DN, Papavassiliou KA, Basdra EK, Papavassiliou AG. Mechanotransduction in osteoblast regulation and bone disease. Trends Mol Med. 2009;15:208–216 [DOI] [PubMed] [Google Scholar]
- 5. Hanai J, Chen LF, Kanno T, et al. Interaction and functional cooperation of PEBP2/CBF with Smads. Synergistic induction of the immunoglobulin germline Cα promoter. Biol Chem. 1999;274:31577–31582 [DOI] [PubMed] [Google Scholar]
- 6. Javed A, Barnes GL, Jasanya BO, et al. Runt homology domain transcription factors (Runx, Cbfa, and AML) mediate repression of the bone sialoprotein promoter: evidence for promoter context dependent activity of Cbfa proteins. Mol Cell Biol. 2001;21:2891–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771 [DOI] [PubMed] [Google Scholar]
- 8. Otto F, Kanegane H, Mundlos S. Mutations in the RUNX2 gene in patients with cleidocranial dysplasia. Hum Mutat. 2002;19:209–216 [DOI] [PubMed] [Google Scholar]
- 9. Yoshida T, Kanegane H, Yoshida T, et al. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet. 2002;71:724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764 [DOI] [PubMed] [Google Scholar]
- 11. Karsenty G. Minireview: transcriptional control of osteoblast differentiation. Endocrinology. 2001;142:2731–2733 [DOI] [PubMed] [Google Scholar]
- 12. Stein GS, Lian JB, Stein J, Van Wijnen AJ, Montecino M. Transcriptional control of osteoblast growth and differentiation. Physiol Rev. 1996;76:593–629 [DOI] [PubMed] [Google Scholar]
- 13. Yang X, Karsenty G. Transcription factors in bone: developmental and pathological aspects. Trends Mol Med. 2002;8:340–345 [DOI] [PubMed] [Google Scholar]
- 14. Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29 [DOI] [PubMed] [Google Scholar]
- 15. Hurley MM, Marie P, Forkiewicz R. Fibroblasts growth factor and fibroblasts growth factor receptor families. In: Bilezikian JP, Raisz LG, Rodan G, eds. Principles of Bone Biology. San Diego: Academic Press; 2002:825–852 [Google Scholar]
- 16. Montero A, Okada Y, Tomita M, et al. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J Clin Invest. 2000;105:1085–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abraham JA, Whang JL, Tumolo A, et al. Human basic fibroblast growth factor: nucleotide sequence and genomic organization. EMBO J. 1986;5:2523–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Florkiewicz RZ, Sommer A. Human basic fibroblast growth factor gene encodes four polypeptides: three initiate translation from non-AUG codons. Proc Natl Acad Sci USA. 1989;86:3981–3987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prats H, Kaghad M, Prats AC, et al. High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons. Proc Natl Acad Sci USA. 1989;86:1836–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Powell PP, Klagsbrun M. Three forms of rat basic fibroblast growth factor are made from a single mRNA and localize to the nucleus. J Cell Physiol. 1991;148:202–210 [DOI] [PubMed] [Google Scholar]
- 21. Stachowiak M, Moffett J, Joy A, Puchacz E, Florkiewicz R, Stachowiak E. Regulation of bFGF gene expression and subcellular distribution of bFGF protein in adrenal medullary cells. J Cell Biol. 1994;127:203–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol Cell. 2003;95:169–178 [DOI] [PubMed] [Google Scholar]
- 23. Sabbieti MG, Marchetti L, Abreu C, et al. Prostaglandins regulate the expression of fibroblast growth factor-2 in bone. Endocrinology. 1999;140:434–444 [DOI] [PubMed] [Google Scholar]
- 24. Sobue T, Naganawa T, Xiao L, et al. Over-expression of fibroblast growth factor-2 causes defective bone mineralization and osteopenia in transgenic mice. J Cell Biochem. 2005;95:83–94 [DOI] [PubMed] [Google Scholar]
- 25. Xiao L, Liu P, Sobue T, Lichtler A, Coffin JD, Hurley MM. Effect of overexpressing fibroblast growth factor 2 protein isoforms in osteoblastic ROS 17/2.8 cells. J Cell Biochem 2003;89:1291–1301 [DOI] [PubMed] [Google Scholar]
- 26. Sabbieti MG, Agas D, Materazzi S, et al. Prostaglandin F2α involves heparin sulphate sugar chains and FGFRs to modulate osteoblast growth and differentiation. J Cell Physiol. 2008;217:48–59 [DOI] [PubMed] [Google Scholar]
- 27. Agas D, Marchetti L, Menghi G, et al. Anti-apoptotic Bcl-2 enhancing requires FGF-2/FGF receptor 1 binding in mouse osteoblasts. J Cell Physiol. 2008;214:145–152 [DOI] [PubMed] [Google Scholar]
- 28. Sabbieti MG, Agas D, Marchetti L, et al. Signaling pathways implicated in PGF2α effects on Fgf2+/+ and Fgf2−/− osteoblasts. J Cell Physiol. 2010;224:465–474 [DOI] [PubMed] [Google Scholar]
- 29. Sabbieti MG, Agas D, Xiao L, et al. Endogenous FGF-2 is critically important in PTH anabolic effects on bone. J Cell Physiol. 2009;219:143–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Naganawa T, Xiao L, Coffin JD, et al. Reduced expression and function of bone morphogenetic protein-2 in bones of Fgf2 null mice. J Cell Biochem. 2008;103:1975–1988 [DOI] [PubMed] [Google Scholar]
- 31. Naganawa T, Xiao L, Abogunde E, et al. In vivo and in vitro comparison of the effects of FGF-2 null and haplo-insufficiency on bone formation in mice. Biochem Biophys Res Commun. 2006;339:490–498 [DOI] [PubMed] [Google Scholar]
- 32. Xiao L, Liu P, Li X, et al. Exported 18-kDa isoform of fibroblast growth factor-2 is a critical determinant of bone mass in mice. J Biol Chem. 2009;284:3170–3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuo P, Hsu Y, Chang CH, Chang JK. Ostholemediated cell differentiation through bone morphogenetic protein-2/p38 and extracellular signal-regulated kinase pathway in human osteoblast cells. J Pharm Exp Ther. 2005;314:1290–1299 [DOI] [PubMed] [Google Scholar]
- 34. Mukherjee A., Rotwein P. Akt promotes BMP-2-mediated osteoblast differentiation and bone development. J Cell Sci. 2009;122:716–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao L, Naganawa T, Lorenzo J, Carpenter TO, Coffin JD, Hurley MM. Nuclear isoforms of fibroblast growth factor 2 are novel inducers of hypophosphatemia via modulation of FGF23 and KLOTHO. J Biol Chem. 2010;285:2834–2846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang X, Goh CH, Li B. p38 mitogen-activated protein kinase regulates osteoblast differentiation through osterix. Endocrinology. 2007;148:1629–1637 [DOI] [PubMed] [Google Scholar]
- 37. Franceschi RT, Xiao G, Jiang D, Gopalakrishnan R, Yang S, Reith E. Multiple signaling pathways converge on the Cbfa1/Runx2 transcription factor to regulate osteoblast differentiation. Connect Tissue Res 2003;44(suppl 1):109–116 [PMC free article] [PubMed] [Google Scholar]
- 38. Kanno T, Takahashi T, Tsujisawa T, Ariyoshi W, Nishihara T. Mechanical stress-mediated Runx2 activation is dependent on Ras/ERK1/2 MAPK signaling in osteoblasts. J Cell Biochem. 2007;101:1266–1277 [DOI] [PubMed] [Google Scholar]
- 39. Xiao G, Gopalakrishnan R, Jiang D, Reith E, Benson MD, Franceschi RT. Bone morphogenetic proteins, extracellular matrix, and mitogen-activated protein kinase signaling pathways are required for osteoblast-specific gene expression and differentiation in MC3T3–E1 cells. J Bone Miner Res. 2002;17:101–110 [DOI] [PubMed] [Google Scholar]
- 40. Park OJ, Kim HJ, Woo KM, Baek JH, Ryoo HM. FGF2-activated ERK mitogen-activated protein kinase enhances Runx2 acetylation and stabilization. J Biol Chem. 2010;285:3568–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Higuchi C, Myoui A, Hashimoto N, et al. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res 2002;17:1785–1794 [DOI] [PubMed] [Google Scholar]
- 42. Chow JY, Quach KT, Cabrera BL, Cabral JA, Beck SE, Carethers JM. RAS/ERK modulates TGFβ-regulated PTEN expression in human pancreatic adenocarcinoma cells. Carcinogenesis. 2007; 28:2321–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eivers E, Fuentealba LC, De Robertis EM. Integrating positional information at the levels of Smad 1/5/8. Curr Opin Genet Dev. 2008;18:304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raucci A, Bellosta P, Grassi R, Basilico C, Mansukhani A. Osteoblast proliferation or differentiation is regulated by relative strengths of opposing signaling pathways. J Cell Physiol. 2008;215:442–451 [DOI] [PubMed] [Google Scholar]
- 45. Agas D, Sabbieti MG, Marchetti L, Xiao L, Hurley MM. FGF-2 enhances Runx-2/Smads nuclear localization in BMP-2 canonical signaling in osteoblasts. [published online April 5, 2013] J Cell Physiol. doi: 10.1002/jcp. 24382 [DOI] [PubMed] [Google Scholar]
- 46. Lee MH, Kwon TG, Park HS, Wozney JM, Ryoo HM. BMP-2-induced Osterix expression is mediated by Dlx5 but is independent of Runx2. Biochem Biophys Res Commun. 2003;309:689–694 [DOI] [PubMed] [Google Scholar]
- 47. Tanimoto Y, Yokozeki M, Hiura K, et al. A soluble form of fibroblast growth factor receptor 2 (FGFR2) with S252W mutation acts as an efficient inhibitor for the enhanced osteoblastic differentiation caused by FGFR2 activation in Apert syndrome. J Biol Chem. 2004;279:45926–45934 [DOI] [PubMed] [Google Scholar]
- 48. Miraoui H, Oudina K, Petite H, Tanimoto Y, Moriyama K, Marie PJ. Fibroblast growth factor receptor 2 promotes osteogenic differentiation in mesenchymal cells via ERK1/2 and protein kinase C signaling. J Biol Chem. 2009;284:4897–4904 [DOI] [PubMed] [Google Scholar]
- 49. Kuhn LT, Ou G, Charles L, Hurley MM, Rodner CM, Gronowicz G. Fibroblast growth factor-2 and bone morphogenetic protein-2 have a synergistic stimulatory effect on bone formation in cell cultures from elderly mouse and human bone. [published online March 26, 2013] J Gerontol A Biol Sci Med Sci. doi:10.1093/gerona/glt018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.