

Abstract
The β-isoform of group VIA calcium-independent phospholipase A2 (iPLA2β) does not require calcium for activation, is stimulated by ATP, and is sensitive to inhibition by a bromoenol lactone suicide substrate. Several potential functions have been proposed for iPLA2β. Our studies indicate that iPLA2β is expressed in β-cells and participates in glucose-stimulated insulin secretion but is not involved in membrane phospholipid remodeling. If iPLA2β plays a signaling role in glucose-stimulated insulin secretion, then conditions that impair iPLA2β functions might contribute to the diminished capacity of β-cells to secrete insulin in response to glucose, which is a prominent characteristic of type 2 diabetes. Our recent studies suggest that iPLA2β might also participate in β-cell proliferation and apoptosis and that various phospholipid-derived mediators are involved in these processes. Detailed characterization of the iPLA2β protein level reveals that β-cells express multiple isoforms of the enzyme, and our studies involve the hypothesis that different isoforms have different functions.
CLASSIFICATION OF PHOSPHOLIPASE A2 AND FEATURES OF A GROUP VIA PHOSPHOLIPASE A2 (iPLA2β)
Phospholipase A2 (PLA2) (1) is a diverse group of enzymes that catalyze hydrolysis of the sn-2 substituent from glycerophospholipid substrates to yield a free fatty acid and a 2-lysophospholipid (1). At present, the recognized PLA2s are classified into 14 different groups, based on their calcium requirement for activation and sequence homology (2). These include the low–molecular weight secretory PLA2s (groups IB, IIA, IID, IIE, IIF, III, V, X, and XII) and the higher–molecular weight Ca2+-dependent cytosolic PLA2s (groups IVA, IVB, and IVC) and the Ca2+-independent PLA2s (groups VIA and VIB).
Among the PLA2s is an 84-kDa (752 amino acid residues) cytosolic PLA2 that does not require Ca2+ for catalysis. This PLA2 has now been cloned from several sources (3–5), including rat and human pancreatic islet β-cells (4,6), is classified as group VIA PLA2, and is designated the β-isoform of group VIA calcium-independent phospholipase A2 (iPLA2β) (7–9). Salient features (10,11) of the iPLA2β amino acid sequence (Fig. 1) include eight NH2-terminal ankyrin repeats, a caspase-3 cleavage site, an ATP-binding domain, a serine lipase consensus sequence (GXSXG), a bipartite nuclear localization sequence, and a COOH-terminal calmodulin-binding domain(s). An 88-kDa iPLA2β isoform is also expressed in human pancreatic islets and is the product of an mRNA species that arises from alternate splicing (6). This isoform contains a 54–amino acid sequence that interrupts the eighth ankyrin repeat.
FIG. 1.

Rat pancreatic islet iPLA2β-deduced amino acid sequence. The deduced 752–amino acid sequence of rat pancreatic islet iPLA2β, updated from our earlier illustration (4), is shown to contain an underlined region of amino acid sequences homologous to a repetitive motif in ankyrin, in addition to a caspase-3 cleavage site, an ATP-binding domain, a catalytic serine lipase consensus sequence, a bipartite nuclear localization consensus sequence, and calmodulin-binding domain(s), which are underlined and numbered 1–5, respectively.
PROPOSED FUNCTIONS FOR iPLA2β
Phospholipid remodeling
A housekeeping role involving generation of lysophospholipid acceptors for incorporation of arachidonic acid into phospholipids has been proposed for iPLA2β, based on experiments involving inhibition of iPLA2β activity in P388D1 cells with the bromoenol lactone (BEL) suicide substrate or with an antisense oligonucleotide (10,11). Inhibition of iPLA2β activity in P388D1 cells suppressed (~60%) incorporation of [3H]-arachidonic acid into phospholipids while reducing (~60%) [3H]-lysophosphatidylcholine levels in [3H]-choline–labeled P388D1 cells. However, [3H]-palmitic acid incorporation was reduced only slightly. This is thought to represent the mechanism whereby iPLA2β inhibition reduces incorporation of [3H]-arachidonic acid into P388D1 cell phospholipids. Such incorporation reflects a deacylation/reacylation cycle (12) of phospholipid remodeling rather than de novo synthesis (13), and the level of lysophosphatidylcholine acceptors is thought to limit the rate of [3H]-arachidonic acid incorporation into P388D1 cell phosphatidylcholine (10,11).
A second housekeeping function for iPLA2β is suggested from studies with CTP:phosphocholine cytidyltransferase (CT)-overexpressing cells (14). CT catalyzes the rate-limiting step in phosphatidylcholine biosynthesis via the Kennedy pathway, and cells overexpressing CT exhibit increased rates of phosphatidylcholine biosynthesis and degradation and little net change in phosphatidylcholine accumulation (14). The increased phosphatidylcholine degradation observed in CT-overexpressing cells is prevented by BEL, and immunoreactive iPLA2β protein and activity increase in such cells, suggesting that iPLA2β is upregulated in response to CT overexpression (14). If general, this could represent an important role for iPLA2β in cell biology because phosphatidylcholine biosynthesis is involved in regulation of the cell cycle and apoptosis (15).
Cell proliferation
Inhibition of iPLA2β with BEL is reported to reduce arachidonic acid release, [3H]-thymidine incorporation, and rates of lymphocyte (16) and Caco-2 (17) cell proliferation. In view of findings that arachidonic acid and/or its metabolites can stimulate c-fos, c-jun, or mitogen-activated protein kinase (18), it is possible that iPLA2β might affect nuclear events involved in cell division.
Apoptosis
Involvement of iPLA2β in processes leading to apoptotic cell death is suggested by several lines of evidence. Induction of apoptosis of U927 cells by anti-fas antibody is associated with hydrolysis of arachidonic acid from membrane phospholipids that is not catalyzed by group IV cytosolic phospholipase A2, which is inactivated by caspases during apoptosis. The release of arachidonic acid is not inhibited by inhibitors of group II secretory phospholipase A2, but the release is suppressed by the iPLA2β inhibitor BEL (19). Both arachidonate 12-lipoxygenase and inducible nitric oxide synthase (iNOS) knockout mice are resistant to the diabetogenic effects of low doses of streptozotocin (20,21). Interleukin-1β–induced generation of nitric oxide in pancreatic islets causes accumulation of arachidonic acid and augments production of the arachidonic acid metabolite 12-hydroxy-(5,8,10,14)-eicosatetraenoic acid (12-HETE), and these effects are prevented by BEL (22). Induction of apoptosis in human promonocytic U937 cells is associated with activation of iPLA2β after proteolysis (23). Collectively, these observations suggest that iPLA2β is involved in releasing arachidonic acid from membrane phospholipids in apoptosis and that arachidonic acid and/or its metabolites serve mediator functions in apoptosis.
Signal transduction
Studies in various cellular systems suggest that iPLA2β might also participate in various signaling pathways. Evidence in support of this role include the observations that BEL, at concentrations that inhibit iPLA2β, suppresses 1) parathyroid-induced generation of arachidonic acid in rat proximal tubules (24), 2) stimulated superoxide generation in neutrophils (25,26), 3) interleukin-1β–stimulated increases in iNOS protein and nitric oxide generation in cardiac myocytes (27), 4) cAMP response element binding protein phosphorylation during the period of myocardial ischemia (28), and 5) virus-induced or double-stranded RNA-induced activation of iNOS expression by macrophages (29). Because pancreatic β-cells also express iPLA2β, we have examined the potential role(s) of this group VIA Ca2+-independent iPLA2β in β-cells, and our findings are discussed below.
POTENTIAL ROLES OF iPLA2β IN β-CELLS
Evidence for a role in signal transduction
Glucose and other fuel secretagogues induce hydrolysis of phospholipids in β-cell membranes, and this is reflected by accumulation of phospholipid-derived mediators including inositol 1,4,5-triphosphate, free arachidonic acid, and arachidonate metabolites (30). Metabolism of fuel secretagogues to yield ATP is an obligatory event in their induction of hydrolysis of arachidonate from β-cell membrane phospholipids (30,31) just as it is for insulin secretion (32). The fact that fuel secretagogue-induced hydrolysis of membrane phospholipids is, in part, independent of Ca2+ influx (33) suggests that a Ca2+-independent phospholipase such as a iPLA2β might be involved in this process.
Pancreatic islets, islet β-cells, and glucose-responsive insulinoma cells all express iPLA2β mRNA and iPLA2β enzymatic activity that is stimulated by ATP and inhibited by BEL (4,34–37). Inhibition of β-cell iPLA2β activity with BEL suppresses glucose-stimulated hydrolysis of arachidonic acid from membrane phospholipids, the rise in cytosolic [Ca2+]i, and insulin secretion (34–37). However, BEL does not inhibit incorporation of labeled fatty acids into either pancreatic islet or insulinoma cell membrane phospholipids (9,38). Whereas BEL, at concentrations that inhibit iPLA2β, does not inhibit group IV cytosolic PLA2 (37) or glucose oxidation (35), which requires both glycolytic metabolism and mitochondrial oxidation of glucose, in pancreatic islets, it has been recognized to decrease the ATP/ADP ratio in mouse islets (39) and also inhibit phosphatidate phosphohydrolase (40). In view of such reports, nonpharmacological approaches involving molecular biological manipulations were used to explore involvement of the iPLA2β protein in β-cell function. Attempts to suppress β-cell iPLA2β activity with antisense oligonucleotides were ineffective (38); therefore, effects of an alternate approach of overexpressing iPLA2β in insulinoma cells were examined.
INS-1 cells were stably transfected with a retroviral vector containing an iPLA2β cDNA insert (9). Two stably transfected lines were isolated that overexpressed iPLA2β activity and protein by 10-fold compared with the parental cell line. These were designated overexpressing (OE) cells. Insulin secretion and phospholipid remodeling in OE cells were then examined. Relative to the secretory responses observed with INS-1 cells transfected with empty retroviral vector that did not contain iPLA2β cDNA, INS-1 cells that overexpressed iPLA2β exhibited a much greater insulin secretory response to glucose alone and to glucose in combination with the cAMP-elevating agents forskolin or isobutylmethylxanthine (9,41). Pretreatment of INS-1 cells with BEL suppressed stimulated insulin secretion, but neither overexpression of iPLA2β nor inhibition of iPLA2β enzymatic activity in the iPLA2β-overexpressing INS-1 cells with BEL affected the rate or extent of arachidonic acid incorporation into INS-1 cell phosphatidylcholine.
Overexpression of iPLA2β as a fusion protein with enhanced green fluorescence protein in INS-1 cells permitted examination of changes in its intracellular location after stimulation (Fig. 2), and glucose plus cAMP-elevating agents were found to induce accumulation of iPLA2β in the perinuclear region. Such intracellular translocation appears to require cAMP-dependent protein kinase A–mediated phosphorylation events because inhibition of protein kinase A with H89 prevented stimulated perinuclear accumulation of iPLA2β (41). Nuclear association of iPLA2β induced by cAMP-elevating agents in INS-1 cells is of interest because glucose promotes both β-cell insulin secretion and proliferation, and glucose-induced INS-1 cell mitogenesis is cAMP-dependent (42). Because membranes of the nucleus and endoplasmic reticulum (ER) are contiguous (43), perinuclear accumulation of iPLA2β is consistent with its association with a subcellular compartment that is likely to include the ER. Using organelle-specific trackers, association of iPLA2β with β-cell ER and Golgi compartments upon stimulation has been observed (44). The likelihood that iPLA2β might associate with the nucleus is also supported by the presence of a bipartite nuclear localization consensus sequence (511KREFGEHTKMTDVKKPK527) in the deduced amino acid sequence of iPLA2β (45). Taken together, the above findings suggest that iPLA2β has a signaling role in the β-cell, although iPLA2β does not appear to participate in β-cell phospholipid remodeling or in phosphatidylcholine homeostasis.
FIG. 2.
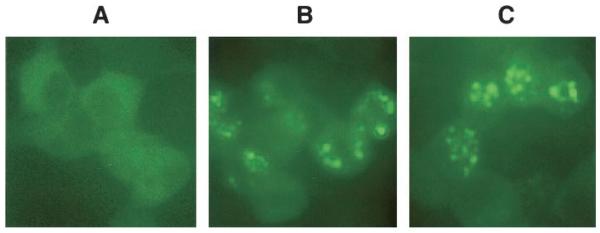
Stimulated translocation of iPLA2β to the perinuclear region of INS-1 cells. INS-1 cells overexpressing iPLA2β as a fusion protein with enhanced green fluorescence protein (EGFP) were stimulated with glucose in the absence and presence of forskolin, as described (41). After a stimulation period of 30 min, the cells were examined by fluorescence microscopy to monitor green fluorescence associated with EGFP. A: Control cells. B: Cells treated with glucose (2 mmol/l) alone. C: Cells treated with both glucose (2 mmol/l) and forskolin (2.5 μmol/l). After stimulation, punctate perinuclear accumulation of fluorescence that reflects the location of iPLA2β-EGFP is evident.
iPLA2β and β-cell proliferation
The iPLA2β-overexpressing INS-1 cell line also proliferates more rapidly than nontransfected parental or empty-vector transfected INS-1 cells. This is reflected by a greater rate of increase in cell number in cultures of iPLA2β-overexpressing INS-1 cells (46). The bases for these phenomena have not been determined, but the presence of a bipartite nuclear localization consensus sequence in iPLA2β raises the possibility that iPLA2β might affect nuclear events involved in cell division (18). Further, iPLA2β activation converts phosphatidic acid to lysophosphatidic acid (LPA) (5), and LPA is a potent mitogen (16,47). Enhanced proliferation might result from a rise in cellular LPA levels that occurs as a consequence of iPLA2β overexpression, and OE cells have been demonstrated to contain higher levels of lysophospholipid species than control cells.
iPLA2β and ER stress–mediated β-cell apoptosis
Apoptosis is involved in β-cell death in type 1 diabetes (48) and might also contribute to β-cell death in type 2 diabetes (49). At present, three apoptotic signaling pathways are recognized (50). These are the extrinsic death receptor pathway involving adaptor molecules, the intrinsic mitochondrial pathway, and the ER stress pathway.
Agents that deplete ER Ca2+ stores, such as thapsigargin (51), induce apoptosis of MIN-6 insulinoma cells by a pathway that does not require increases in [Ca2]i but that does require generation of the arachidonic acid metabolite 12-HETE (52). Thapsigargin has been demonstrated to induce hydrolysis of arachidonic acid from islet membrane phospholipids, and this is suppressed by the iPLA2β inhibitor BEL (53). These findings suggest that ER stress induced by Ca2+ store depletion activates iPLA2β, which then hydrolyzes membrane phospholipids to yield products that promote β-cell death.
In support of this hypothesis are the findings that thapsigargin induces a threefold increase in apoptosis of parental INS-1 cells (control, 4.2 ± 0.2% vs. plus thapsigargin, 12.3 ± 0.7%, P < 0.05), and apoptosis is suppressed by BEL. Whereas the spontaneous incidence of apoptosis in control OE cells (3.7 ± 0.5%) is similar to that in parental cells, the apoptotic effect of thapsigargin is greatly amplified in the OE cells (Fig. 3A), and apoptosis is significantly suppressed by BEL. Thapsigargin also induces higher iPLA2β activity and the generation of a 62-kDa iPLA2β-immunoreactive protein (Fig. 3B). Inhibition of caspase-3 prevents both thapsigargin-induced apoptosis and perinu-clear accumulation of iPLA2β (Fig. 3C). Immunoblotting and immunofluorescence analyses reveal that a 62-kDa iPLA2β-immunoreactive protein accumulates in the perinuclear region of thapsigargin-treated INS-1 cells. These findings suggest that caspase-3 that is activated during apoptosis cleaves the 84-kDa iPLA2β at its consensus sequence site to generate a 62-kDa product in INS-1 cells.
FIG. 3.

ER stress–induced apoptosis of INS-1 cells. A: Suppression of thapsigargin-induced apoptosis of iPLA2β-overexpressing INS-1 cells by BEL. INS-1 cells stably transfected with a vector containing iPLA2β cDNA (OE) construct were treated with thapsigargin (T) (1 μmol/l) for 24 h in the absence or presence of BEL (10 μmol/l). The cells were then harvested for TUNEL (Tdt-mediated dUTP nick end labeling) analyses to assess the magnitude of cell apoptosis (*T + BEL–treated group significantly different from T alone–treated group, P < 0.05). B: Immunoblotting analyses. Aliquots of INS-1 cell protein (50 μg), prepared from OE cells treated with dimethylsulfoxide (lane 1) or thapsigargin (1 μmol/l) (lane 2), were analyzed by SDS-PAGE (7.5%) and transferred onto an Immobolin-P polyvinylidene difluoride membrane. The electroblot was probed with piPLA2β antibodies, and immunoreactive protein bands were visualized by enhanced chemiluminescence, as described (41). C, control. C: Effects of caspase-3 inhibition on iPLA2β subcellular distribution. iPLA2β OE INS-1 cells seeded in glass chambered slides were pretreated with caspase-3 inhibitor (C3-I) (500 nmol/l) or vehicle for 24 h. The cells were then treated with thapsigargin in the absence and presence of C3-I for 24 h and subsequently processed for iPLA2β immunofluorescence analyses by confocal microscopy. Con, control.
Caspase-3–catalyzed cleavage of iPLA2β has been reported to occur in U937 promonocytes induced to undergo apoptosis, and the COOH-terminal product appears to be constitutively activated (23). Overexpression of the full-length iPLA2β or the COOH-terminal caspase-3 proteolysis product of iPLA2β (184aa → COOH-terminal) in human embryonic kidney cells resulted in a higher incidence of apoptotic cell death and greater induction of arachidonic acid release from cells overexpressing the truncated iPLA2β compared with those overexpressing full-length iPLA2β. These findings suggest that the iPLA2β product of a caspase-3–mediated cleavage was more active than the full-length iPLA2β. It was proposed that the more active shorter iPLA2β isoform could stimulate excessive turnover of nuclear membrane phospholipids, disrupt membrane fluidity, and promote apoptotic cell death.
Our findings suggest that induction of ER stress in INS-1 cells by depleting Ca2+ stores with thapsigargin stimulates iPLA2β activity and promotes caspase-3–mediated cleavage of the INS-1 cell iPLA2β. The resultant accumulation of a 62-kDa iPLA2β-immunoreactive product in the perinuclear region of INS-1 cells raises the possibility that this might be one mechanism by which iPLA2β participates in ER stress–induced apoptosis in β-cells.
Our studies also reveal that thapsigargin induces increased generation of ceramides in the OE cells compared with control cells. Ceramides (a family of 2-N-acylsphingosines) are important lipid second messengers that have been implicated as suppressors of cell growth and inducers of apoptosis (54). They can be generated by sphingomyelinase-catalyzed sphingomyelin hydrolysis (55), de novo synthesis (56), or as a consequence of decreased ceramidase activity (57), raising the possibility that any one of these ceramide-generating pathways might also be affected by iPLA2β.
β-Cell iPLA2β is a candidate for posttranslational modification
To examine the possibility that iPLA2β undergoes posttranslational modifications that affect signaling, recombinant 84-kDa iPLA2β was purified from sf9 cells after a sequential anion exchange and ATP affinity and calmodulin affinity chromatography protocol (58). NH2-terminal amino acid sequencing analyses of the purified material yields a sequence that begins with residue 11 of the iPLA2β-deduced amino acid sequence, reflecting loss of the first 10 amino acid residues. Liquid chromatography/electrospray ionization/mass spectrometry (Fig. 4A) analyses of a tryptic digest of the purified iPLA2β reveal a peptide corresponding to residues 12–23 of the deduced amino acid sequence that would not be expected to arise from trypsin digestion of full-length iPLA2β but that would be expected from an iPLA2β variant lacking the first 11 amino acid residues. In the full-length iPLA2β sequence, the first tryptic cleavage site occurs between residues 6 and 7 and the second between residues 23 and 24. Trypsin digestion thus yields peptides 1–6 and 7–23. In an iPLA2β variant that lacks residues 1–11, trypsin digestion yields peptide 12–23. Peptide 12–23 is thus a signature peptide for this NH2-terminally processed iPLA2β variant. The presence of this peptide in the tryptic digest of purified iPLA2β is reflected by the presence of [M + H]+1 and [M + 2H]+2 ions at m/z 1368.6 and 685.3, respectively. In addition, the expected aa7–23 peptide from intact iPLA2β is also present in the tryptic digest (Fig. 4B), as is the aa1–6 peptide modified at the NH2-terminal by acetylation. This NH2-blocked peptide would not yield NH2-terminal sequence upon Edman analyses of the intact iPLA2β protein. Although the mechanisms responsible for generating an iPLA2β protein lacking the first 11 amino acids have not yet been determined, these findings indicate that the β-cell iPLA2β undergoes posttranslational modification by NH2-terminal processing, and this might represent a means to regulate the activity, subcellular location, or protein–protein interactions of iPLA2β.
FIG. 4.

Liquid chromatography/electrospray ionization/mass spectrometry (LC/ESI/MS) of tryptic peptide of recombinant iPLA2β. Recombinant iPLA2β purified by sequential chromatography was digested with trypsin, and an aliquot was analyzed by LC/ESI/MS. A: Evidence for acetylated NH2-terminus and NH2-terminally truncated variant of iPLA2β. B: Evidence for sequence proximal to residue 12.
Multiple isoforms of iPLA2β
The iPLA2β cDNA first cloned from a rat pancreatic library encodes an 84-kDa protein, and human islets and lymphocytes were subsequently shown to express two iPLA2β mRNA species that arise by alternate splicing and encode 84- and 88-kDa proteins (6,59). Recent studies reveal predominant expression of an iPLA2β-immunoreactive protein with an apparent molecular mass of 70 kDa in robustly glucose-responsive 832/13 INS-1 cells (60). These cells continue to express cytosolic iPLA2β activity that is stimulated by ATP and inhibited by BEL. Additionally, inhibition of 832/13 iPLA2β activity with BEL suppresses glucose-stimulated insulin secretion but does not affect arachidonate incorporation into phosphatidylcholine. Therefore, the BEL-sensitive catalytic activity expressed in 832/13 INS-1 cells is most likely attributable to a 70-kDa iPLA2β-immunoreactive protein because it is essentially the only iPLA2β-immunoreactive protein present in the cytosol of these cells.
CONCLUSIONS
A diminished capacity of β-cells to secrete insulin in response to glucose is a prominent characteristic of type 2 diabetes, and this motivates studies to achieve a fuller understanding of glucose-sensing mechanisms within the β-cell. Our findings suggest that iPLA2β participates in β-cell signal transduction. The expression level of iPLA2β appears to affect β-cell proliferation and apoptosis, and the enzyme may thus be an important participant in the life cycle of the β-cell. iPLA2β does not appear to participate in arachidonate acid incorporation into phospholipids or phosphatidylcholine homeostasis in β-cells, although such housekeeping functions of the enzyme have been proposed for other cells. The phospholipase activity of iPLA2β would result in the production of phospholipid-derived mediators including arachidonic acid and arachidonate metabolites, and lysophospholipids and the iPLA2β expression level also affect cellular content of the lipid second messenger ceramide. The finding that multiple iPLA2β-immunoreactive isoforms are expressed in β-cells raises the possibility that different isoforms serve different functions that vary with the stimulation condition, subcellular compartment, phase of cell cycle, levels of interacting proteins, or other factors.
ACKNOWLEDGMENTS
This research was supported by grants from the National Institutes of Health (R37-DK34388, P41-RR00954, P01-HL57278, P60-DK20579, and P30-DK56341) and by an Award (to S.R.) from the American Diabetes Association.
The authors thank Alan Bohrer and Dr. Mary Wohltmann for expert technical assistance.
Glossary
- 12-HETE
12-hydroxy-(5,8,10,14)-eicosatetraenoic acid
- BEL
bromoenol lactone
- CT, CTP
phosphocholine cytidyltransferase
- ER
endoplasmic reticulum
- iNOS
inducible nitric oxide synthase
- iPLA2β
β-isoform of group VIA calcium-independent phospholipase A2
- LPA
lysophosphatidic acid
- OE
over-expressing
- PLA2
phospholipase A2
REFERENCES
- 1.Gijon MA, Leslie CC. Phospholipases A2. Semin Cell Dev Biol. 1977;8:297–303. doi: 10.1006/scdb.1997.0151. [DOI] [PubMed] [Google Scholar]
- 2.Balboa MA, Varela-Nieto I, Lucas KK, Dennis EA. Expression and function of phospholipase A2 in brain. FEBS Lett. 2002;531:12–17. doi: 10.1016/s0014-5793(02)03481-6. [DOI] [PubMed] [Google Scholar]
- 3.Balboa MA, Balsinde J, Jones S, Dennis EA. Identity between the Ca2+-independent phospholipase A2 enzymes from P388D1 macrophages and Chinese hamster ovary cells. J Biol Chem. 1977;27:8576–8590. doi: 10.1074/jbc.272.13.8576. [DOI] [PubMed] [Google Scholar]
- 4.Ma Z, Ramanadham S, Kempe K, Chi XS, Ladenson JL, Turk J. Pancreatic islets express a Ca2+-independent phospholipase A2 enzyme that contains a repeated structural motif homologous to the integral membrane protein binding domain of ankyrin. J Biol Chem. 1997;27:11118–11127. [PubMed] [Google Scholar]
- 5.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra S, Jones S. A novel cytosolic calcium-independent phospholipase A2 contains eight ankyrin motifs. J Biol Chem. 1997;272:8567–8575. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 6.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. Human pancreatic islets express mRNA species encoding two distinct catalytically active isoforms of group VI phospholipase A2 (iPLA2) that arise from an exon-skipping mechanism of alternative splicing of the transcript from the iPLA2 gene on chromosome 22q13.1. J Biol Chem. 1999;27:9607–9616. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mancuso DJ, Jenkins CM, Gross RW. The genomic organization, complete mRNA sequence, cloning, and expression of a novel human intracellular membrane-associated calcium-independent phospholipase A2. J Biol Chem. 2000;275:9937–9945. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka H, Takeya R, Sumimoto H. A novel intracellular membrane-bound calcium-independent phospholipase A2. Biochem Biophys Res Commun. 2000;272:320–326. doi: 10.1006/bbrc.2000.2776. [DOI] [PubMed] [Google Scholar]
- 9.Ma Z, Ramanadham S, Bohrer A, Wohltmann M, Zhang S, Turk J. Studies of insulin secretory responses and of arachidonic acid incorporation into phospholipids of stably transfected insulinoma cells that overexpress group VIA phospholipase A2 (iPLA2β) indicate a signaling rather than a housekeeping role for iPLA2β. J Biol Chem. 2001;276:13198–13208. doi: 10.1074/jbc.M010423200. [DOI] [PubMed] [Google Scholar]
- 10.Balsinde J, Bianco ID, Ackerman EJ, Conde-Friebos K, Dennis EA. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc Natl Acad Sci U S A. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balsinde J, Balboa MA, Dennis EA. Antisense inhibition of group VI Ca2+-independent phospholipase A2 blocks phospholipid fatty acid remodeling in murine P388D1 macrophages. J Biol Chem. 1977;272:29317–29321. doi: 10.1074/jbc.272.46.29317. [DOI] [PubMed] [Google Scholar]
- 12.Chilton FH, Fonteh AN, Suarette ME, Triggiani M, Winkler JD. Control of arachidonate levels within inflammatory cells. Biochim Biophys Acta. 1996;1299:1–15. doi: 10.1016/0005-2760(95)00169-7. [DOI] [PubMed] [Google Scholar]
- 13.Dennis EA. The biosynthesis of phospholipids. Methods Enzymol. 1992;209:1–4. doi: 10.1016/0076-6879(92)09003-l. [DOI] [PubMed] [Google Scholar]
- 14.Barbour SE, Kapur A, Deal CL. Regulation of phosphatidylcholine homeostasis by calcium-independent phospholipase A2. Biochim Biophys Acta. 1999;1439:77–88. doi: 10.1016/s1388-1981(99)00078-5. [DOI] [PubMed] [Google Scholar]
- 15.Anthony ML, Zhao M, Brindle KM. Inhibition of phosphatidylcholine biosynthesis following induction of apoptosis in HL-60 cells. J Biol Chem. 1999;274:19686–19692. doi: 10.1074/jbc.274.28.19686. [DOI] [PubMed] [Google Scholar]
- 16.Roshak AK, Capper EA, Stevenson C, Eichman C, Marshall LA. Human calcium-independent phospholipase A2 mediates lymphocyte proliferation. J Biol Chem. 2000;275:35692–35698. doi: 10.1074/jbc.M002273200. [DOI] [PubMed] [Google Scholar]
- 17.Sanchez T, Moreno JJ. Calcium-independent phospholipase A2 through arachidonic acid mobilization is involved in Caco-2 cell growth. J Cell Physiol. 2002;193:293–298. doi: 10.1002/jcp.10162. [DOI] [PubMed] [Google Scholar]
- 18.Sellmayer A, Danesch U, Weber PC. Effects of different polyunsaturated fatty acids on growth-related early gene expression and cell growth. Lipids. 1996;31:537–540. doi: 10.1007/BF02637048. [DOI] [PubMed] [Google Scholar]
- 19.Atsumi G, Tajima M, Hadano A, Nakatani Y, Murakami M, Kudo I. Fas-induced arachidonic acid release is mediated by Ca2+-independent phospholipase A2 but not cytosolic phospholipase A2, which undergoes proteolytic inactivation. J Biol Chem. 1998;273:13870–13877. doi: 10.1074/jbc.273.22.13870. [DOI] [PubMed] [Google Scholar]
- 20.Bleich D, Chen S, Zipser B, Sun D, Funk C, Nadler J. Resistance to type 1 diabetes induction in 12-lipoxygenase knockout mice. J Clin Invest. 1999;103:1431–1436. doi: 10.1172/JCI5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flodstrom M, Tyrberg B, Eizirik D, Sandler S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to multiple low-dose streptozotocin-induced diabetes. Diabetes. 1999;48:706–713. doi: 10.2337/diabetes.48.4.706. [DOI] [PubMed] [Google Scholar]
- 22.Ma Z, Ramanadham S, Corbett J, Bohrer A, Gross R, McDaniel M, Turk J. Interleukin-1 enhances pancreatic islet arachidonic acid 12-lipoxygenase product generation by increasing substrate availability through a nitric oxide-dependent mechanism. J Biol Chem. 1996;271:1029–1042. doi: 10.1074/jbc.271.2.1029. [DOI] [PubMed] [Google Scholar]
- 23.Atsumi G, Murakami M, Kojima K, Hadano A, Tajima M, Kudo I. Distinct roles of two intracellular phospholipase A2s in fatty acid release in the cell death pathway: proteolytic fragment of type IVA cytosolic phospholipase A2 alpha inhibits stimulus-induced arachidonate release, whereas that of type VI Ca2+-independent phospholipase A2 augments spontaneous fatty acid release. J Biol Chem. 2000;275:18248–18258. doi: 10.1074/jbc.M000271200. [DOI] [PubMed] [Google Scholar]
- 24.Derrickson BH, Mandel LJ. Parathyroid hormone inhibits Na+-K+-ATPase through Gq/G11 and the calcium-independent phospholipase A2. Am J Physiol. 1997;272:F781–F788. doi: 10.1152/ajprenal.1997.272.6.F781. [DOI] [PubMed] [Google Scholar]
- 25.Tithof PK, Peters-Golden M, Ganey PE. Distinct phospholipases A2 regulate the release of arachidonic acid for eicosanoid production and super-oxide anion generation in neutrophils. J Immunol. 1998;160:953–960. [PubMed] [Google Scholar]
- 26.Tithof PK, Olivero J, Ruehle K, Ganey PE. Activation of neutrophil calcium-dependent and -independent phospholipases A2 by organochlorine compounds. Toxicol Sci. 2000;53:40–47. doi: 10.1093/toxsci/53.1.40. [DOI] [PubMed] [Google Scholar]
- 27.Isenovic E, LaPointe MC. Role of Ca2+-independent phospholipase A2 in the regulation of inducible nitric oxide synthase in cardiac myocytes. Hypertension. 2000;35:249–254. doi: 10.1161/01.hyp.35.1.249. [DOI] [PubMed] [Google Scholar]
- 28.Williams SD, Ford DA. Calcium-independent phospholipase A2 mediates CREB phosphorylation and c-fos expression during ischemia. Am J Physiol. 2001;281:H168–H176. doi: 10.1152/ajpheart.2001.281.1.H168. [DOI] [PubMed] [Google Scholar]
- 29.Maggi LB, Jr, Moran JM, Scarim AL, Ford DA, Yoon JW, McHowat J, Buller RM, Corbett JA. Novel role for calcium-independent phospholipase A2 in the macrophage antiviral response of inducible nitric-oxide synthase expression. J Biol Chem. 2002;277:38449–38455. doi: 10.1074/jbc.M206247200. [DOI] [PubMed] [Google Scholar]
- 30.Turk J, Gross R, Ramanadham S. Amplification of insulin secretion by lipid messengers. Diabetes. 1993;42:367–374. doi: 10.2337/diab.42.3.367. [DOI] [PubMed] [Google Scholar]
- 31.Turk J, Mueller M, Bohrer A, Ramanadham S. Arachidonic acid metabolism in isolated pancreatic islets. VI. Carbohydrate insulin secretagogues must be metabolized to induce eicosanoid release. Biochim Biophys Acta. 1992;1125:280–291. doi: 10.1016/0005-2760(92)90057-3. [DOI] [PubMed] [Google Scholar]
- 32.Meglasson MD, Matschinsky F. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2:163–214. doi: 10.1002/dmr.5610020301. [DOI] [PubMed] [Google Scholar]
- 33.Wolf BA, Pasquale SM, Turk J. Free fatty acid accumulation in secretagogue-stimulated pancreatic islets and effects of arachidonate on depolarization-induced insulin secretion. Biochemistry. 1991;30:6372–6379. doi: 10.1021/bi00240a004. [DOI] [PubMed] [Google Scholar]
- 34.Gross RW, Ramanadham S, Kruszka KK, Han X, Turk J. Rat and human pancreatic islet cells contain a calcium ion independent phospholipase A2 activity selective for hydrolysis of arachidonate which is stimulated by adenosine triphosphate and is specifically localized to islet beta-cells. Biochemistry. 1993;32:327–336. doi: 10.1021/bi00052a041. [DOI] [PubMed] [Google Scholar]
- 35.Ramanadham S, Gross RW, Han X, Turk J. Inhibition of arachidonate release by secretagogue-stimulated pancreatic islets suppresses both insulin secretion and the rise in beta-cell cytosolic calcium ion concentration. Biochemistry. 1993;32:337–346. doi: 10.1021/bi00052a042. [DOI] [PubMed] [Google Scholar]
- 36.Ramanadham S, Wolf MJ, Jett PA, Gross RW, Turk J. Characterization of an ATP-stimulatable Ca2+-independent phospholipase A2 from clonal insulin-secreting HIT cells and rat pancreatic islets: a possible molecular component of the beta-cell fuel sensor. Biochemistry. 1994;32:7442–7452. doi: 10.1021/bi00189a052. [DOI] [PubMed] [Google Scholar]
- 37.Ma Z, Ramanadham S, Hu Z, Turk J. Cloning and expression of a group IV cytosolic Ca2+-dependent phospholipase A2 from rat pancreatic islets: comparison of the expressed activity with that of an islet group VI cytosolic Ca2+-independent phospholipase A2. Biochim Biophys Acta. 1998;1391:384–400. doi: 10.1016/s0005-2760(98)00027-7. [DOI] [PubMed] [Google Scholar]
- 38.Ramanadham S, Hsu F-F, Bohrer A, Ma Z, Turk J. Studies of the role of group VI phospholipase A2 in fatty acid incorporation, phospholipid remodeling, lysophosphatidyl-choline generation, and secretagogue-induced arachidonic acid release in pancreatic islets and insulinoma cells. J Biol Chem. 1999;274:13915–13927. doi: 10.1074/jbc.274.20.13915. [DOI] [PubMed] [Google Scholar]
- 39.Sato Y, Henquin J-C. The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: in search of the underlying mechanism. Diabetes. 1998;47:1713–1721. doi: 10.2337/diabetes.47.11.1713. [DOI] [PubMed] [Google Scholar]
- 40.Balsinde J, Dennis EA. Bromoenol lactone inhibits magnesium-dependent phosphatidate phosphohydrolase and blocks triacylglycerol biosynthesis in mouse P388D1 macrophages. J Biol Chem. 1996;271:31937–31941. doi: 10.1074/jbc.271.50.31937. [DOI] [PubMed] [Google Scholar]
- 41.Ma Z, Zhang S, Turk J, Ramanadham S. Stimulation of insulin secretion and associated nuclear accumulation of iPLA2β in INS-1 insulinoma cells. Am J Physiol. 2002;282:E820–E833. doi: 10.1152/ajpendo.00165.2001. [DOI] [PubMed] [Google Scholar]
- 42.Hugl SR, White MF, Rhodes CJ. Insulin-like growth factor I (IGF-I)-stimulated pancreatic beta-cell growth is glucose-dependent: synergistic activation of insulin receptor substrate-mediated signal transduction pathways by glucose and IGF-I in INS-1 cells. J Biol Chem. 1998;273:17771–17779. doi: 10.1074/jbc.273.28.17771. [DOI] [PubMed] [Google Scholar]
- 43.Holz GG, Leech CA, Heller RS, Castonguay M, Habener JF. cAMP-dependent mobilization of intracellular Ca2+-stores by activation of ryanodine receptors in pancreatic beta-cells: a Ca2+ signaling system stimulated by the insulinotropic hormone glucagon-like peptide-1-(7–37) J Biol Chem. 1999;274:14147–14156. doi: 10.1074/jbc.274.20.14147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bao S, Jin C, Zhang S, Turk S, Ma Z, Ramanadham S. The β-cell calcium-independent group VIA phospholipase A2 (iPLA2β): tracking iPLA2β movements in response to stimulation with insulin secretagogues in INS-1 cells. Diabetes. 2004;53(Suppl. 1):xxx–xxx. doi: 10.2337/diabetes.53.2007.s186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma Z, Turk J. The molecular biology of the group VIA Ca2+-independent phospholipase A2. Prog Nucleic Acid Res Mol Biol. 2001;67:1–33. doi: 10.1016/s0079-6603(01)67023-5. [DOI] [PubMed] [Google Scholar]
- 46.Ma Z, Bohrer A, Wohltmann M, Ramanadham S, Hsu F-F, Turk J. Studies of phospholipid metabolism, proliferation, and secretion of stably transfected insulinoma cells that overexpress group VIA phospholipase A2. Lipids. 2001;36:689–700. doi: 10.1007/s11745-001-0774-9. [DOI] [PubMed] [Google Scholar]
- 47.Moolenaar WH. Lysophosphatidic acid, a multifunctional phospholipid messenger. J Biol Chem. 1995;270:12949–12952. doi: 10.1074/jbc.270.22.12949. [DOI] [PubMed] [Google Scholar]
- 48.Mathis D, Vence L, Benoist C. β-Cell death during progression to diabetes. Nature. 2001;414:792–798. doi: 10.1038/414792a. [DOI] [PubMed] [Google Scholar]
- 49.Mandrup-Poulsen T. β-Cell apoptosis: stimuli and signaling. Diabetes. 2001;50(Suppl. 1):S58–S63. doi: 10.2337/diabetes.50.2007.s58. [DOI] [PubMed] [Google Scholar]
- 50.Mehmet H. Caspases find a new place to hide. Nature. 2000;403:29–30. doi: 10.1038/47377. [DOI] [PubMed] [Google Scholar]
- 51.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y-P, Teng D, Dralyuk F, Ostrega D, Roe MW, Philipson L, Polonsky K. Apoptosis in insulin-secreting cells: evidence for the role of intracellular Ca2+ stores and arachidonic acid metabolism. J Clin Invest. 1998;101:1623–1632. doi: 10.1172/JCI1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nowatzke W, Ramanadham S, Ma Z, Hsu F-F, Bohrer A, Turk J. Mass spectrometric evidence that agents that cause loss of Ca2+ from intracellular compartments induce hydrolysis of arachidonic acid from pancreatic islet membrane phospholipids by a mechanism that does not require a rise in cytosolic Ca2+ concentration. Endocrinology. 1998;139:4073–4085. doi: 10.1210/endo.139.10.6225. [DOI] [PubMed] [Google Scholar]
- 54.Obeid LM, Hannun YA. Ceramide: a stress signal and mediator of growth suppression and apoptosis. J Cell Biochem. 1995;58:191–198. doi: 10.1002/jcb.240580208. [DOI] [PubMed] [Google Scholar]
- 55.Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem. 1994;269:3125–3128. [PubMed] [Google Scholar]
- 56.Shimabukuro M, Higa M, Zhou Y-T, Wang M-Y, Newgard CB, Unger RH. Lipoapoptosis in β-cells of obese prediabetic fa/fa rats: role of serine palmitoyl-transferase overexpression. J Biol Chem. 1998;273:32487–32490. doi: 10.1074/jbc.273.49.32487. [DOI] [PubMed] [Google Scholar]
- 57.Franzen R, Fabbro D, Aschrafi A, Pfeilschifter J, Huwiler A. Nitric oxide induces degradation of the neutral ceramidase in rat renal mesangial cells and is counterregulated by protein kinase C. J Biol Chem. 2002;277:46184–46190. doi: 10.1074/jbc.M204034200. [DOI] [PubMed] [Google Scholar]
- 58.Wolf MJ, Gross RW. Expression, purification, and kinetic characterization of a recombinant 80-kDa intracellular calcium-independent phospholipase A2. J Biol Chem. 1996;271:30879–30885. doi: 10.1074/jbc.271.48.30879. [DOI] [PubMed] [Google Scholar]
- 59.Larsson PK, Claesson HE, Kennedy BP. Multiple splice variants of the human calcium-independent phospholipase A2 and their effect on enzyme activity. J Biol Chem. 1998;273:207–214. doi: 10.1074/jbc.273.1.207. [DOI] [PubMed] [Google Scholar]
- 60.Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB. Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. doi: 10.2337/diabetes.49.3.424. [DOI] [PubMed] [Google Scholar]