

Abstract
A series of fused-bicyclic acetals containing a disiloxane ring was investigated to evaluate the source of selectivity in silyl-protected 2-deoxyribose systems. The disiloxane ring unexpectedly enables the diaxial conformer of the cation to be stabilized by an electronegative atom at C-3. This low energy conformer subsequently undergoes stereoelectronically controlled nucleophilic addition to give substituted tetrahydrofurans with high diastereoselectivity.
Introduction
The additions of nucleophiles to five-membered ring oxocarbenium ions are common reactions for the construction of substituted tetrahydrofurans found in furanosides and nucleosides. C-Nucleosides have found use as N-nucleoside mimics, not only to inhibit enzymes,1,2 but also to investigate the factors controlling DNA helix stability.3–5 Furanosides have also become popular synthetic targets as antibacterial6,7 and antiviral agents.8,9 In all cases, the stereochemistry at the anomeric position of the substituted tetrahydrofuran is crucial for the intended activity.10 The broad applicability of substituted tetrahydrofurans as probes in chemistry and biology and as selective inhibitors in medicinal chemistry has highlighted the need for stereoselective methods for the synthesis of such target compounds.
The stereochemical courses of reactions with five-membered ring oxocarbenium ions, particularly in the case of carbon nucleophiles, is governed by stereoelectronic effects.11 Nucleophilic addition to the oxocarbenium ion, which adopts an envelope conformation, may occur from the inside face or from the outside face of the lowest energy envelope conformer (Fig. 1). We proposed that nucleophiles prefer to attack from the inside face of the envelope conformer because unfavorable eclipsing interactions are generated during the transition state associated with outside attack. Evidence for this stereoelectronic preference was provided by experiments involving an oxocarbenium ion fused to an eight-membered ring, which resulted in diastereoselectivity comparable to an unconstrained monocyclic system (eq 1).12 Because the eight-membered ring could not accommodate a diaxial envelope conformer (3-diaxial), the product must have been formed by inside attack on 3-diequatorial (Fig. 1).13
Figure 1.
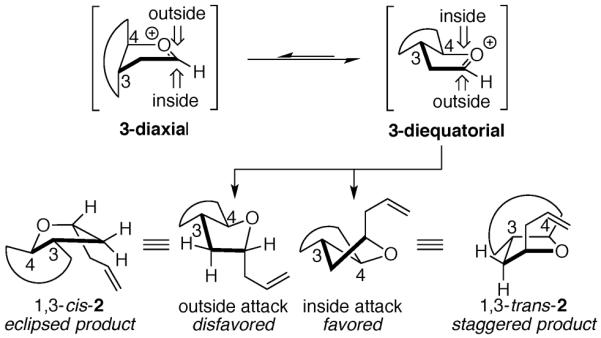
Reactions of fused-ring oxocarbenium ions.
 |
(1) |
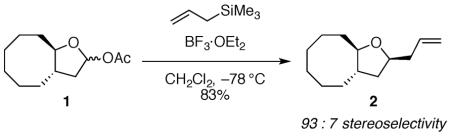
A powerful method for the synthesis of β-C-nucleosides has been developed14 that cannot be readily explained by inside attack on the oxocarbenium ion. The addition of silane to an oxocarbenium ion formed in situ from hemiacetal 414 (eq 2) with a fused eight-membered ring gave the product with the opposite stereochemical sense to the all-carbon analogue shown in eq 1. The difference in this selectivity cannot be attributed to the presence of the substituent at the carbon atom undergoing substitution, because selectivity is independent of substitution at C-1.15 The origin of this high selectivity has not been explained.
 |
(2) |
In this Article, we report reactions of a series of fused-bicyclic acetals containing a disiloxane ring to elucidate the origins of selectivity in silyl-protected 2-deoxyribose systems such as those shown in eq 2. The eight-membered disiloxane ring, although not directly involved in the reaction, exerts powerful influences on the structure and reactivity of the five-membered ring oxocarbenium ion to which it is fused. The three-dimensional structure of this disiloxane ring enables access to a low energy conformer in which the electronegative atom at C-3 is close to the positively charged carbon atom of the oxocarbenium ion.
Background
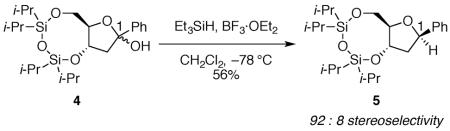
Although the eight-membered ring of disiloxane 4 is necessary to get high yields of product, it is not required to observe 1,3-cis selectivity. Nucleophilic addition to the monocyclic hemiacetal 6 also proceeds with the hydride adding to the same face as the silyloxy group at C-3 (eq 3), but with higher diastereoselectivity than was seen with bicyclic hemiacetal 4.16 The selectivity of nucleophilic addition to hemiacetal 6, unlike disiloxane 4, can be rationalized by the inside attack model.11,12 The substituents at C-3 and C-4 adopt axial orientations to maximize electrostatic stabilization of the oxocarbenium ion (Fig. 2). Inside attack on intermediate 8-diaxial would form the major product.
Figure 2.
The stereoselectivity for 6 can be explained by the inside attack model.
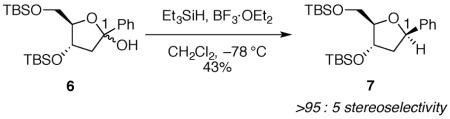 |
(3) |
It is difficult to explain the selectivity observed for disiloxane 4 because different comparisons give different explanations. If an analogy were made to the monocyclic hemiacetal 6, then one would conclude that the fused ring exerts only minor influences on diastereoselectivity of 2-deoxyribose-derived acetals such as 4 (eq 2). Because the eight-membered disiloxane ring has such a different geometry than would be expected if only second-row elements were present in this ring,17 the eight-membered ring disiloxane may be capable of accommodating a diaxial orientation. Diminished selectivity for the fused ring system 4 could result from a diminished preference for the diaxial conformation.
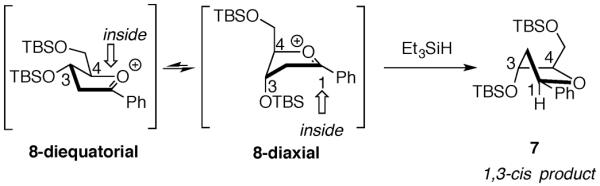
It may not be appropriate to draw an analogy between the unconstrained oxocarbenium ion 8 (Fig. 2) and the oxocarbenium ion derived from disiloxane 4, however. The eight-membered ring disiloxane ring may not be able to accommodate the electrostatically stabilized diaxial conformer 9-axial (Fig. 3). X-ray crystal structures of eight-five fused-bicyclic compounds related to acetate 4 containing a tetraisopropyldisiloxane protecting group exhibit a ring in which the O–Si–O–Si–O linkage places all atoms nearly coplanar with a nearly linear geometry at the central oxygen atom (155°).18 This observation is consistent with the suggestion that the eight-membered disiloxane ring in protected furanoses is constrained in a diequatorial orientation,19 just as observed for the all-carbon fused bicyclic systems 1.13
Figure 3.
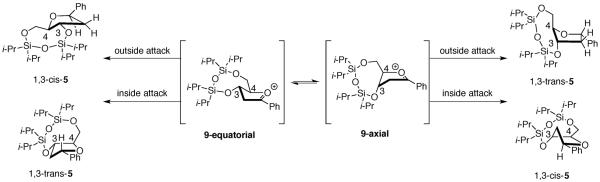
Possible reaction trajectories.
Because the diaxial intermediate 9-axial may not be the favored conformer, the trajectories of attack on the diequatorial conformer 9-equatorial need to be considered more carefully (Fig. 3). The major product 1,3-cis-5 could also be formed by outside attack on diequatorial conformer 9-equatorial. Outside attack, however, is generally disfavored because eclipsing interactions develop between the nucleophile and the substituents at C-2 in the first-formed product.12 Because nucleophilic attack results in significant conformational changes in the five-membered ring, the three-dimensional preference of the flattened disiloxane ring may alter the inherent stereoelectronic selectivity for the inside product.13 The dihedral angle at the ring juncture within the five-membered ring of 1,3-trans-5 is increased compared to the angle in the oxocarbenium ion 9-equatorial (Fig. 3). This increase in dihedral angle could introduce torsional strain into the disiloxane ring and lead to diminished stereoselectivity, as was observed with the system with the cyclohexane fused to the five-membered ring (10, eq 4).13 In contrast, outside attack on 9-equatorial would form the product 1,3-cis-5 with little additional strain because the dihedral angle in the five-membered ring is relatively unchanged (Fig. 3).13
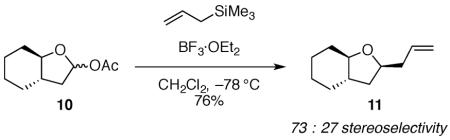 |
(4) |
The explanation that constraints imposed by the eight-membered disiloxane ring reverses the inherent preference for inside attack is also unsatisfying. Selectivity was not reversed in the case of the smaller six-membered ring fused system 10 (eq 4); it was only diminished in that example. Consequently, neither explanation (inside attack on the conformer 9-axial or outside attack on conformer 9-equatorial) is logical given the systems studied.
Experimental Design
To confirm the preference of nucleophilic attack on silyl-protected 2-deoxyribose systems, we envisioned a substrate similar to our previously studied eight-five fused-bicyclic system 1, but replacing the cyclooctane ring with a disiloxane ring (i.e., acetate 12, Fig. 4). We hypothesized that an eight-five fused-bicyclic system containing a disiloxane ring has different conformational preferences than that of an eight-five fused-bicyclic system containing a cyclooctane ring. This hypothesis was derived by examination of X-ray crystal structures of compounds related to bicyclic disiloxane 4 containing the tetraisopropyldisiloxane protecting group.18 In the solid state, this ring system adopted a conformation in which the O–Si–O–Si–O linkage was nearly coplanar. Because the eight-membered disiloxane ring has such a different geometry than would be expected if only second-row elements were present in this ring,17 the eight-membered ring disiloxane may be capable of accommodating a diaxial orientation.
Figure 4.
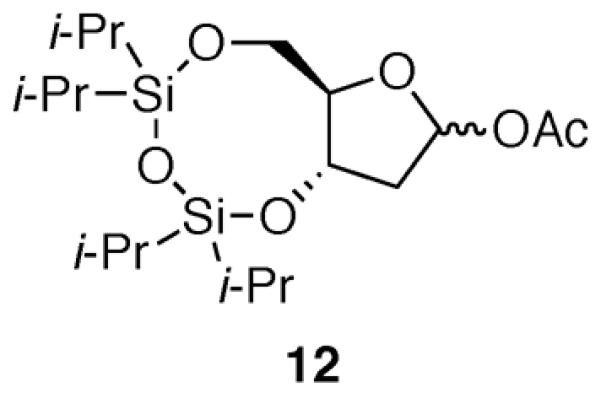
Substrate to test the preferred approach of allyltrimethylsilane.
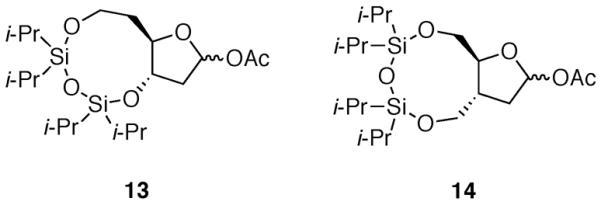
To distinguish between the two possible reaction trajectories for the formation of the 1,3-cis products, substrates with nine-membered rings fused to the five-membered ring were synthesized (Fig. 5). These substrates were designed to give opposite trends in selectivity depending upon which pathway was preferred. If the stereoselectivity observed for acetate 4 were the result of inside attack on a diaxial conformer, then the preference for the 1,3-cis product should improve as the fused ring size was expanded to a nine-membered ring (i.e., acetate 13). The larger size of the ring fusion would presumably allow for easier access to the diaxial conformer in which the electronegative oxygen atom at C-3 can form a favorable electrostatic interaction with the oxocarbenium ion. Conversely, if constraints imposed by a diequatorially fused disiloxane ring disfavored the inside attack pathway, then nucleophilic substitution of acetate 13 with the larger ring should result in lower selectivity for the 1,3-cis product.
Figure 5.
Substrates to test the two possible reaction trajectories.
A related substrate was designed to probe the importance of electrostatic effects in disiloxane-fused oxocarbenium ions (Fig. 5). Acetate 14 possesses a nine-membered ring just as acetate 13 does, so the nine-membered disiloxane ring should impose similar conformational constraints on the five-membered ring to which it is fused. On the other hand, the diequatorial form should be favored because no oxygen atom is present at C-3 that might shift the equilibrium to the diaxial conformer. The outcome of nucleophilic substitution of acetate 14 will reflect the inherent ability of the disiloxane to accommodate the changing conformation of the five-membered ring. If selectivity were high for the 1,3-trans product, then it would indicate that the nine-membered disiloxane ring could allow for inside attack when that ring was fused equatorially, just as observed for the cyclooctane ring of acetal 1 (eq 1).
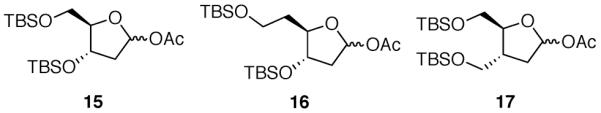
A series of monocyclic acetates 15–17 would serve as direct comparisons to the bicyclic acetates 12–14 (Fig. 6). Without a ring fusion to constrain the conformations of the five-membered ring, acetates 15–17 are expected to undergo nucleophilic substitution in accordance with the inside attack model. Allylation of acetates 15 and 16 should proceed with high stereoselectivity for the 1,3-cis product because the oxocarbenium ion can be stabilized by the silyloxy group in a pseudoaxial position.11,12 On the other hand, the cation derived from acetate 17 is expected to favor the conformation in which the C3 substituent resides in the pseudoequatorial position. Inside attack on this conformer should generate 1,3-trans products with high selectivity.11,12 The substitution products of acetates 12–14 and acetates 15–17 can be correlated directly by standard deprotection and protection reactions.
Figure 6.
Monocyclic substrates to serve as direct comparisons to bicyclic substrates.
A number of experimental constraints were applied to provide results that could be compared easily. All substitution reactions were conducted under similar conditions to ensure that any differences in selectivity could be correlated to structure. Allyltrimethylsilane was used as the nucleophile in these substitution reactions because attack of carbon π-nucleophiles is irreversible.20 In addition, this small nucleophile does not experience strongly destabilizing steric interactions in the transition state for nucleophilic attack, and its reactivity is representative of many nucleophiles.21 The Lewis acid BF3·OEt2 was used to facilitate dissociation of the acetate leaving group because these conditions involve reactions that appear to proceed through oxocarbenium ions.22,23
Substrate Synthesis
The deoxy-ribose acetates 12 and 15 were prepared in a few steps from D-deoxyribose 18 (Scheme 1).14,24 Acetates 13 and 16 were synthesized from 1-methoxycyclohexa-1,4-diene (22) in several steps (Schemes 2 and 3).25 Acetates 14 and 17 were produced following five linear steps each from cis-2-butene-1,4-diol (30) (Scheme 4).
Scheme 1a.
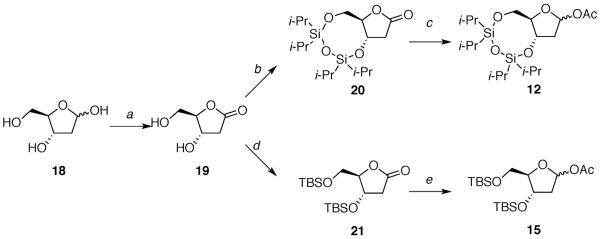
aReagents: (a) Br2, H2O, 68%; (b) (Cli-Pr2Si)2O, 2,6-lutidine, 28%; (c) i-Bu2AlH, pyr, DMAP, Ac2O, 76%; (d) t-BuMe2SiCl, imidazole, 78%; (e) i-Bu2AlH, pyr, DMAP, Ac2O, 92%.
Scheme 2a.
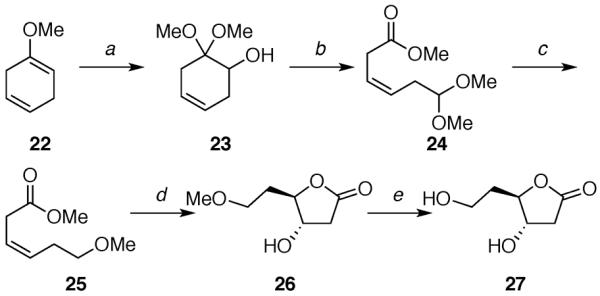
aReagents: (a) m-CPBA, MeOH, 91%; (b) H5IO6, HC(OMe)3, MeOH, 74%; (c) Et3SiH, BF3·OEt2, 90%; (d) OsO4, NMO, 83%; (e) BBr3, 70%.
Scheme 3a.
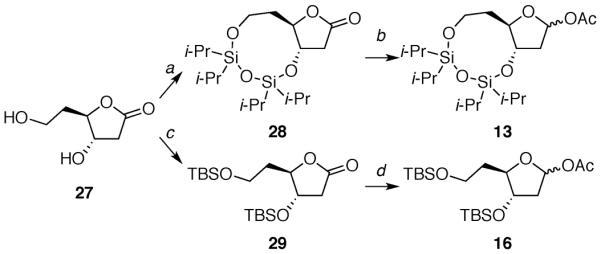
aReagents: (a) (Cli-Pr2Si)2O, 2,6-lutidine, 28%; (b) i-Bu2AlH, pyr, DMAP, Ac2O, 88%; (c) t-BuMe2SiCl, imidazole, 75%; (d) i-Bu2AlH, pyr, DMAP, Ac2O, 93%.
Scheme 4a.
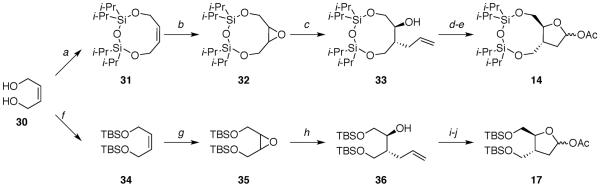
aReagents: (a) (Cli-Pr2Si)2O, 2,6-lutidine, 28%; (b) m-CPBA, 98%; (c) allylMgBr, 92%; (d) OsO4, NMO, NaIO4, 92%; (e) Ac2O, Et3N, 99%; (f) t-BuMe2SiCl, imidazole, 75%; (g) m-CPBA, 97%; (h) allylMgBr, 97%; (i) OsO4, NMO, NaIO4, 87%; (j) Ac2O, Et3N, 94%.
Results
Initial experiments verified that the use of allyltrimethylsilane as a nucleophile gave similar results to those observed for hydride attack (eq 2). The sense of selectivity exhibited by the hydride addition is shared by addition of allyltrimethylsilane: nucleophilic substitution of the bicyclic acetate 12 proceeded with 1,3-cis selectivity (eq 5). This observation provides a direct contrast with observations of the corresponding acetal 1 with the cyclooctane ring fused to the five-membered ring, which gave the 1,3-trans product 2 with high diastereoselectivity (eq 1).13 The diastereoselectivity of neither reaction (eq 1 or eq 5) depended upon the starting ratio of anomeric acetates,13 suggesting that oxocarbenium ion intermediates were involved in both cases. The use of Me3SiOTf as a Lewis acid also did not change the diastereoselectivity, indicating that ion-pairing effects or direct displacement reactions are not responsible for selectivity.21,22
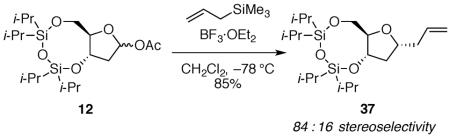 |
(5) |
The nucleophilic substitution reactions of bicyclic acetates 13 and 14 with fused nine-membered disiloxane rings proceeded with high diastereoselectivity (eqs 6 and 7), therefore clarifying the origin of diastereoselectivity in these systems. The acetate 13 possessing a nine-membered ring exhibited even higher selectivity for the 1,3-cis product than observed for acetate 12 with an eight-membered disiloxane ring. This increased selectivity suggests that the corresponding oxocarbenium ion had a greater ability to adopt a diaxial conformation due to its larger ring size. Had the selectivity decreased, then it would have been likely that the product resulted from a diminished preference for outside attack on the diequatorial conformer. An important observation was that the nucleophilic substitution of acetate 13 and acetate 14 formed products with opposite diastereoselectivity, despite having the same size disiloxane ring. While the substitution of both acetate 13 and acetate 14 proceeded through the preferred inside attack pathway, the nucleophile must have encountered different oxocarbenium ion envelope conformers to form the products of opposite diastereoselectivity. This result is consistent with observations of unconstrained systems (Fig. 2) that require the two different oxocarbenium ions to adopt different low energy conformations (diaxial for the cation derived from acetate 13, and diequatorial for the cation derived from acetate 14).12
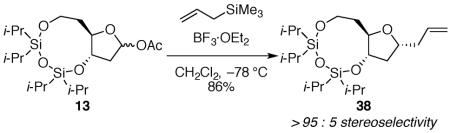 |
(6) |
 |
(7) |
The nucleophilic substitution reactions of the monocyclic acetals 15–17 analogous to acetates 12–14 also resulted in similar diastereoselectivity, respectively (eqs 8–10). The high diastereoselectivity obtained in all of these substitution reactions is consistent with inside attack on the cations. It is important to note that the level of stereoselectivity of addition to bicyclic acetate 13 is similar to the unconstrained acetate 16. The diminished 1,3-cis stereoselectivity observed for acetate 12 (eq 5) suggests that the eight-membered ring disiloxane ring had a decreased preference for the diaxial conformation.
 |
(8) |
 |
(9) |
 |
(10) |
X-ray Crystallographic Data
Crystallographic data was necessary to confirm the relative configurations of the substitution products 37–42 because the Nuclear Overhauser Effect for five-membered rings is too small to be conclusive. Each substitution product was derivatized to provide a solid that could be recrystallized for X-ray diffraction studies (Schemes 5–7). In addition, the bicyclic substitution products 37–39 underwent additional transformations for chemical correlation to the corresponding monocyclic substitution products 40–42.
Scheme 5a.
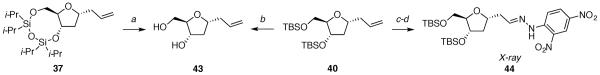
aReagents: (a) n-Bu4NF, 86%; (b) n-Bu4NF, 99%; (c) OsO4, NMO, 92%; (d) NaIO4, 88%; (e) C6H4(NO2)2NHNH2, 83%.
Scheme 7.
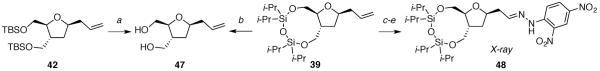
aReagents: (a) n-Bu4NF, 99%; (b) n-Bu4NF, 99%; (c) O3, PPh3, 93%; (d) C6H4(NO2)2NHNH2, 92%.
Discussion
The results presented here indicate that there are significant differences in the conformational preferences between the two eight-five fused bicyclic substrates 1 and 12. In the all-carbon substrate 1, the cyclooctane ring can adopt several low-energy conformations, but all of these possibilities orient the fused ring equatorially.26 On the other hand, the disiloxane rings of acetate 12 and the nine-five fused system 13, which possess an electronegative oxygen atom at C-3 that can engage in stabilizing electrostatic interactions with the oxocarbenium ion intermediate,12 enable the cation to accommodate the diaxial conformer.
Preliminary computational analysis of the low energy conformers of the oxocarbenium ions derived from acetates 12 and 13 indicate that there are low-energy conformers that resemble the diaxial conformer, although the five-membered ring is somewhat flattened as compared to unconstrained substrates (Fig. 7). Eligible low-energy conformers were found by a systematic search of the conformational space using semi-empirical methods (PM3) in Spartan10,27 and energies were minimized with low-level ab initio methods (HF/3-21G). Conformations within 10 kcal/mol of the lowest energy conformer were selected to undergo further optimization with ab initio calculations, first using a larger basis set (HF/6-31G*) and later using density functional methods (B3LYP/6-31G*). The relative energy difference between the lowest energy diaxial conformer 49-axial and the lowest energy diequatorial conformer 49-equatorial for the oxocarbenium ion derived from acetate 12 was too small (0.3 kcal/mol) to draw any significant conclusion. Instead, the presence of multiple low-energy conformers within 1 kcal/mol implies a low preference for any particular conformation. Because nucleophiles can react with any of these low-energy conformers, the major product could be the product of a conformation that is not the lowest energy oxocarbenium ion.28 By expanding the eight-membered disiloxane ring to the nine-membered one, however, the lowest energy diaxial conformer 50-axial was favored over the lowest energy diequatorial conformer 50-equatorial by 5.2 kcal/mol, suggesting a potential for high selectivity. Although preliminary, these calculations support the stereoselectivity seen in our experimental results.
Figure 7.
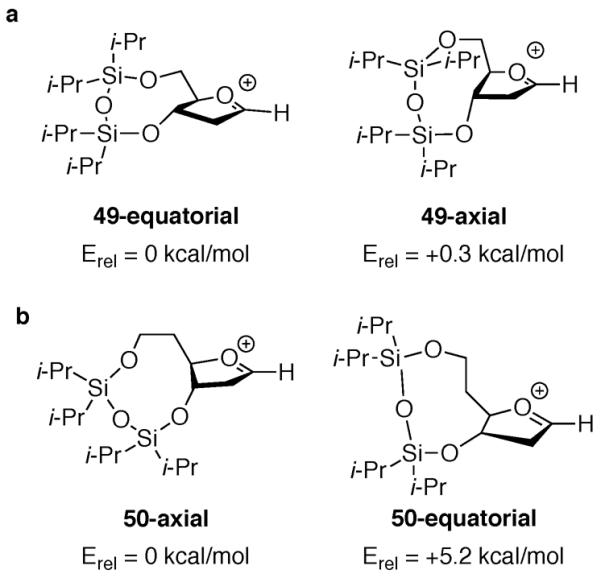
Calculated (B3LYP/6–31G*) enthalpies of fused-bicyclic oxocarbenium ions.
Conclusion
Experiments with closely related substrates indicate that the disiloxane ring permits greater flexibility to a ring to which it is fused than its all-carbon analogue does. The shape of the tetraisopropyldisiloxane protecting group, likely the longer bond lengths as compared to an all-carbon system in combination with more shallow vibrational and torsional minima,17 enables the cation to adopt a low-energy envelope conformation in which the electronegative atom at C-3 provides a stabilizing interaction. Expansion of the eight-membered disiloxane ring to a nine- membered ring increases the preference for the diaxial conformer, leading to an increase in diastereoselectivity. Without the electronegative oxygen atom at C-3, however, the nine-membered ring disiloxane biases the five-membered ring oxocarbenium ion to the diequatorial conformer.
Experimental Section
(4S,5R)-4-hydroxy-5-(hydroxymethyl)dihydrofuran-2(3H)-one 19
To a solution of 2-deoxy-d-ribose (1.0 g, 7.5 mmol) in distilled water (6 mL) was added Br2 (2.0 mL, 39 mmol) by syringe. The reaction was allowed to stir at room temperature for 5 days. Excess bromine was extracted from the reaction mixture with Et2O until the yellow color faded. The pH of the aqueous layer was adjusted to pH 7 with the addition of Ag2CO3. The aqueous layer was then filtered and concentrated in vacuo to afford a brown oil.24 The product was purified by flash column chromatography on silica gel (1:1 acetone:hexanes) to give 19 as a light brown oil (0.68 g, 68%): [α]24D +4.11 (c 1.61, MeOH); 1H NMR (400 MHz, CD3OD) δ 4.43 (dt, J = 6.7, 2.3, 1H), 4.37 (dd, J = 5.5, 3.3, 1H), 3.76 (dd, J = 12.4, 3.3, 1H), 3.69 (dd, J = 12.4, 3.6, 1H), 2.92 (dd, J = 18.0, 6.8, 1H), 2.37 (dd, J = 18.0, 2.5, 1H); 13C NMR (125 MHz, CD3OD) δ 178.9, 90.2, 69.8, 62.5, 39.1; IR (thin film) 3383, 2941, 1767 cm−1; HRMS (TOF MS ES+) m / z calcd for C5H8NaO4 (M + Na)+ 155.0320, found 155.0315.
(6aR,9aS)-2,2,4,4-tetraisopropyltetrahydro-8H-furo[3,2-f][1,3,5,2,4]trioxadisilocin-8-one 20
To a solution of 2-Deoxy-d-ribonolactone 19 (0.062 g, 0.47 mmol) and imidazole (0.080 g, 1.2 mmol) in DMF (1.5 mL) was added 1,3-dichlorotetraisopropyldisiloxane (0.23 mL, 0.71 mmol) dropwise. The reaction mixture was stirred overnight at room temperature. The reaction mixture was then extracted with Et2O (2 × 10 mL) and washed with distilled water (10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a colorless oil.14 The product was purified by flash column chromatography on silica gel (15:85 EtOAc:hexanes) to afford 20 as a colorless oil (0.049 g, 28%). The spectral data obtained matched the reported values for this compound:14 [α]24D +16.7 (c 1.87, MeOH); 1H NMR (500 MHz, CDCl3) δ 4.64 (app q, J = 8.0, 1H), 4.22 (td, J = 6.7, 3.6, 1H), 4.14 (dd, J = 12.3, 3.6, 1H), 3.93 (dd, J = 12.3, 6.6, 1H), 2.86 (dd, J = 17.3, 8.0, 1H), 2.72 (dd, J = 17.3, 9.2, 1H), 1.11–0.93 (m, 28H); 13C NMR (125 MHz, CDCl3) δ 173.2, 85.0, 69.8, 62.5, 38.0, 17.6, 17.51, 17.48, 17.4, 17.2, 17.1, 17.0, 13.5, 13.3, 13.0, 12.7; IR (thin film) 2943, 2870, 1795, 1034 cm−1; HRMS (TOF MS ES+) m / z calcd for C17H34NaO5Si2 (M + Na)+ 397.1843, found 397.1849.
(6aR,9aS)-2,2,4,4-tetraisopropyltetrahydro-6H-furo[3,2-f][1,3,5,2,4]trioxadisilocin-8-yl acetate 12
A solution of protected ribonolactone 20 (0.51 g, 1.4 mmol) in CH2Cl2 (8 mL) was cooled to −78 °C. Diisobutylaluminum hydride (1.5 M in toluene, 1.8 mL, 2.7 mmol) was added dropwise by syringe. After the reaction mixture was stirred for 6 h at −78 °C, a solution of dimethylaminopyridine (0.33 g, 2.7 mmol) in dry CH2Cl2 (3 mL), pyridine (0.33 mL, 4.1 mmol), and acetic anhydride (0.77 mL, 8.1 mmol) were added sequentially by syringe. The reaction mixture was left to stir overnight and warm to room temperature. After the reaction mixture was cooled to 0 °C, saturated aqueous NH4Cl (8 mL) and saturated aqueous sodium potassium tartrate (8 mL) were added. The mixture was allowed to warm to room temperature and was stirred until the layers were completely separated. The reaction mixture was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo to give a dark yellow oil.29 The crude mixture was purified by flash column chromatography on silica gel (1:7:92 Et3N:EtOAc:hexanes) to afford 12 as a colorless oil (0.43 g, 76% yield). 1H and 13C NMR of a 30:70 (1,3-cis-12:1,3-trans-12) mixture of diastereomers is given. IR, HRMS, and elemental analysis of a 58:42 (1,3-cis-12:1,3-trans-12) mixture of diastereomers is provided: 1H NMR (500 MHz, C6D6) δ 6.33 (dd, J = 5.6, 2.6, 1H), 4.74 (dt, J = 8.9, 6.8, 0.3H), 4.29 (dt, J = 8.6, 5.6, 0.7H), 4.20–4.16 (m, 0.7H), 4.12 (dd, J = 11.3, 3.8, 0.3H), 4.07 (dd, J = 11.7, 3.6, 0.7H), 4.01–3.97 (m, 0.3H), 3.88 (dd, J = 11.2, 8.8, 0.3H), 3.70 (dd, J = 11.7, 7.9, 0.7H), 2.20–2.13 (m, 1H), 2.06–2.01 (m, 1H), 1.65 (s, 2.1H), 1.58 (s, 0.9H), 1.14–0.90 (m, 28H); 13C NMR (125 MHz, C6D6) δ 169.8, 169.2, 97.6, 97.4, 85.9, 85.3, 73.9, 73.8, 66.2, 64.2, 41.7, 41.3, 21.18, 21.15, 18.13, 18.10, 18.05, 18.02, 18.00, 17.96, 17.9, 17.71, 17.68, 17.60, 17.57, 14.1, 14.0, 13.9, 13.7, 13.6, 13.4, 13.3; IR (thin film) 2943, 1738, 1035 cm−1; HRMS (TOF MS ES+) m / z calcd for C19H38NaO6Si2 (M + Na)+ 441.2105, found 441.2107. Anal. Calcd for C19H38O6Si2: C, 54.51; H, 9.15. Found: C, 54.67; H, 9.16. A 44:56 (1,3-cis-12:1,3-trans-12) mixture of diastereomers gave [α]24D −11.3 (c 1.74, MeOH). A 36:64 (1,3-cis-12:1,3-trans-12) mixture of diastereomers gave [α]24D −0.485 (c 2.99, MeOH). The optical rotation of the pure diastereomer 1,3-cis-12 was calculated to be −86.8, and the optical rotation of the pure diastereomer 1,3-trans-12 was calculated to be +48.1.
(4S,5R)-4-((tert-butyldimethylsilyl)oxy)-5-(((tertbutyldimethylsilyl)oxy)methyl)dihydrofuran-2(3H)-one 21
To a solution of 2-Deoxy-l-ribonolactone 19 (0.112 g, 0.848 mmol) in DMF (4 mL) at 0 °C was added imidazole (0.289 g, 4.24 mmol) and tert-butyldimethylsilyl chloride (0.268 g, 1.78 mmol). The reaction mixture was stirred overnight at room temperature. The reaction mixture was then extracted with EtOAc (2 × 20 mL) and washed with distilled water (10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give a colorless oil. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to afford 21 as a white solid (0.238 g, 78%). The spectral data obtained matched the reported values for this compound:30 1H NMR (500 MHz, CDCl3) δ 4.49 (dt, J = 6.5, 2.2, 1H), 4.32 (dt, J = 2.5, 2.3, 1H), 3.80 (dd, J = 11.6, 3.3, 1H), 3.75 (dd, J = 11.5, 2.5, 1H), 2.81 (dd, J = 17.7, 6.7, 1H), 2.37 (dd, J = 17.7, 2.5, 1H), 0.87 (s, 18H), 0.08 (s, 6H), 0.06 (s, 3H), 0.05 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 176.0, 88.3, 69.8, 62.7, 39.2, 26.0, 25.8, 18.4, 18.1, −4.56, −4.64, −5.3, −5.5.
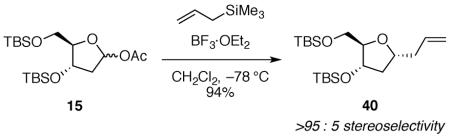
(4S,5R)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl acetate 15
A solution of bis-TBS protected ribonolactone 21 (0.377 g, 1.04 mmol) in CH2Cl2 (10 mL) was cooled to −78 °C. Diisobutylaluminum hydride (1.5 M in toluene, 2.1 mL, 3.1 mmol) was added dropwise by syringe. After the reaction mixture was stirred for 1 h at −78 °C, a solution of dimethylaminopyridine (0.255 g, 2.09 mmol) in dry CH2Cl2 (3 mL), pyridine (0.253 mL, 3.13 mmol), and acetic anhydride (0.592 mL, 6.26 mmol) were added sequentially by syringe. The reaction mixture was left to stir overnight and warm to room temperature. After the reaction mixture was cooled to 0 °C, saturated aqueous NH4Cl (5 mL) and saturated aqueous sodium potassium tartrate (5 mL) were added. The mixture was allowed to warm to room temperature and was stirred until the layers were completely separated. The reaction mixture was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (10 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. The crude mixture was purified by flash column chromatography on silica gel (1:9:90 Et3N:EtOAc:hexanes) to afford 15 as a colorless oil (0.39 g, 92% yield). Characterization was performed on a 28:72 (1,3-cis-15:1,3-trans-15) mixture of diastereomers. The spectral data obtained matched the reported values for this compound:31 [α]24D 0.165 (c 4.45, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 6.28 (dd, J = 5.2, 2.8, 0.72H), 6.24 (dd, J = 5.6, 1.0, 0.28H), 4.45 (td, J = 6.1, 4.6, 0.72H), 4.33 (dt, J = 6.7, 2.7, 0.28H), 4.10 (dt, J = 4.2, 3.4, 0.28H), 3.87 (q, J = 4.7, 0.72H), 3.67–3.56 (m, 2H), 2.30 (ddd, J = 14.0, 6.7, 5.8, 0.28H), 2.20–2.11 (m, 1.44H), 2.02 (s, 0.84H), 2.00 (s, 2.16H), 1.95 (ddd, J = 14.0, 2.2, 1.1, 0.28H), 0.88–0.86 (m, 18H), 0.05–0.02 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 170.7, 170.3, 99.3, 98.4, 88.7, 87.9, 71.6, 71.2, 62.9, 62.8, 41.5, 41.2, 26.00, 25.97, 25.81, 25.75, 21.45, 18.5, 18.4, 18.03, 17.95, −4.6, −4.67, −4.71, −4.8, −5.25, −5.31, −5.33, −5.4; IR (thin film) 2943, 1741, 1240 cm−1; HRMS (TOF MS ES+) m / z calcd for C19H40NaO5Si2 (M + Na)+ 427.2312, found 427.2307. Anal. Calcd for C19H40O5Si2: C, 56.39; H, 9.96. Found: C, 56.88; H, 10.01. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
6,6-dimethoxycyclohex-3-en-1-ol 23
To a solution of 1-methoxycyclohexa-1,4-diene (1.00 g, 9.11 mmol) in MeOH (30 mL) at −10 °C was added dropwise a solution of m-chloroperoxybenzoic acid (2.1 g, 75%, 9.1 mmol) in MeOH (20 mL). The mixture was stirred at −10 °C for 1 h, then at room temperature for 4 h. The reaction mixture was washed with saturated aqueous NaHCO3 (80 mL) and extracted with CH2Cl2 (4 × 80 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo to give an amber oil. The product was purified by flash column chromatography on silica gel (40:60 ether:hexanes) to give 23 as a colorless oil (1.3 g, 91%). The spectral data obtained matched the reported values for this compound:25 1H NMR (400 MHz, CDCl3) δ 5.63–5.54 (m, 2H), 4.02 (s, 1H), 3.30 (s, 3H), 3.25 (s, 3H), 2.47–2.23 (m, 5H); 13C NMR (100 MHz, CDCl3) δ 123.7, 122.8, 100.4, 66.8, 48.6, 48.2, 31.2, 29.8; IR (thin film) 3454, 3030, 2944, 1658, 1422 cm−1; HRMS (TOF MS ES+) m / z calcd for C8H14NaO3 (M + Na)+ 181.0841, found 181.0839. Anal. Calcd for C8H14O3: C, 60.74; H, 8.92. Found: C, 60.82; H, 9.06.
Methyl (Z)-6,6-dimethoxyhex-3-enoate 24
To a solution of 23 (0.41 g, 2.6 mmol) in MeOH (9 mL) at −10 °C was added dropwise a solution of periodic acid (0.70 g, 3.1 mmol) in MeOH (4 mL). The reaction mixture was stirred at −10 °C for 1 h, then at room temperature for 5 h. Trimethyl orthoformate (0.84 mL, 7.7 mmol) was added all at once, and the reaction mixture was left to stir overnight. The reaction mixture was washed with brine (50 mL) and extracted with EtOAc (2 × 40 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a yellow oil. The product was purified by flash column chromatography on silica gel (25:75 EtOAc:hexanes) to afford 24 as a yellow oil (0.36 g, 74%). The spectral data obtained matched the reported values for this compound:25 1H NMR (600 MHz, CDCl3) δ 5.70 (dtt, J = 10.6, 7.2, 1.6, 1H), 5.60 (dtt, J = 10.5, 7.2, 1.6, 1H), 4.39 (t, J = 5.7, 1H), 3.69 (s, 3H), 3.34 (s, 3H and s, 3H), 3.12 (dt, J = 7.2, 0.7, 2H), 2.38 (tt, J = 6.4, 0.8, 2H); 13C NMR (150 MHz, CDCl3) δ 172.2, 127.4, 123.6, 104.0, 53.3, 52.0, 33.1, 31.4; IR (thin film) 3032, 2953, 1741, 1661, 1437 cm−1; HRMS (TOF MS ES+) m / z calcd for C9H16NaO4 (M + Na)+ 211.0947, found 211.0945. Anal. Calcd for C9H16O4: C, 57.43; H, 8.57. Found: C, 57.14; H, 8.42.
Methyl (Z)-6-methoxyhex-3-enoate 25
To a solution of 24 (0.788 g, 4.19 mmol) in CH2Cl2 (42 mL) at −78 °C was added triethylsilane (2.68 mL, 16.8 mmol) and boron trifluoride diethyl etherate (0.620 mL, 5.02 mmol). After 6 h, saturated aqueous sodium bicarbonate (100 mL) was added. The reaction mixture was extracted with CH2Cl2 (2 × 100 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo to give a colorless oil. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to afford 25 as a colorless oil (0.598 g, 90%): 1H NMR (600 MHz, CDCl3) δ 5.67 (dtt, J = 10.7, 7.1, 1.5, 1H), 5.61 (dtt, J = 10.4, 7.1, 1.5, 1H), 3.69 (s, 3H), 3.41 (t, J = 6.8, 2H), 3.34 (s, 3H), 3.12 (dd, J = 7.1, 1.3, 2H), 2.34 (qd, J = 6.8, 1.3, 2H); 13C NMR (150 MHz, CDCl3) δ 172.4, 129.6, 123.1, 72.1, 58.9, 52.1, 33.1, 28.2; IR (ATR) 3028, 2927, 1737, 1436 cm−1; HRMS (TOF MS ES+) m / z calcd for C8H14NaO3 (M + Na)+ 181.0841, found 181.0840. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
(4R*,5S*)-4-hydroxy-5-(2-methoxyethyl)dihydrofuran-2(3H)-one 26
To a solution of 25 (0.341 g, 2.15 mmol) in acetone and water (1:1, 22 mL) was added 4-methylmorpholine N-oxide (0.757 g, 6.46 mmol). The reaction mixture was cooled to 0 °C, and osmium tetroxide (4.0% in H2O, 0.68 mL, 0.11 mmol) was added by syringe. The reaction mixture was allowed to warm to room temperature overnight, then was extracted with EtOAc (6 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a grey oil. The product was purified by flash column chromatography on silica gel (EtOAc) to afford 26 as a colorless oil (0.32 g, 83%): 1H NMR (500 MHz, CDCl3) δ 4.28–4.23 (m, 2H), 3.59 (dd, J = 3.8, 0.6, 1H), 3.58 (d, J = 3.9, 1H), 3.56 (br s, 1H), 3.40 (s, 3H), 2.84 (dd, J = 17.6, 7.0, 1H), 2.60 (dd, J = 17.5, 7.8, 1H), 2.08–1.95 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 174.3, 85.8, 72.7, 69.2, 59.2, 37.3, 33.5; IR (thin film) 3419, 2930, 1778 cm−1; HRMS (TOF MS ES+) m / z calcd for C7H12NaO4 (M + Na)+ 183.0633, found 183.0638. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
(4R*,5S*)-4-hydroxy-5-(2-hydroxyethyl)dihydrofuran-2(3H)-one 27
To a solution of 26 (0.26 g, 1.5 mmol) in CH2Cl2 (15 mL) at −78 °C was added boron tribromide (0.41 mL, 4.4 mmol). The reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was cooled to −78 °C before adding MeOH (1 mL) and concentrating in vacuo to give a brown oil. The product was purified by flash column chromatography on silica gel (2.5:97.5 MeOH:EtOAc) to give 27 as a colorless oil (0.15 g, 70%): 1H NMR (400 MHz, CD3OD) δ 4.48 (ddd, J = 8.8, 4.8, 2.8, 1H), 4.27 (ddd, J = 6.4, 3.4, 2.9, 1H), 3.74–3.65 (m, 2H), 2.90 (dd, J = 17.9, 6.5, 1H), 2.40 (dd, J = 17.9, 3.5, 1H), 1.90 (dddd, J = 14.3, 7.9, 6.5, 4.9, 1H), 1.77 (ddt, J = 14.3, 9.0, 5.3, 1H); 13C NMR (100 MHz, CD3OD) δ 178.1, 87.0, 72.5, 59.0, 38.1, 36.9; IR (ATR) 3360, 2925, 1748 cm−1; HRMS (TOF MS ES+) m / z calcd for C6H10NaO4 (M + Na)+ 169.0477, found 169.0477. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
(7aR*,10aS*)-2,2,4,4-tetraisopropyltetrahydrofuro[3,2-f][1,3,5,2,4]trioxadisilonin-9(6H)-one 28
To a solution of 27 (0.186 g, 1.27 mmol) in dichloroethane (13 mL) at 0 °C was added 2,6-lutidine (0.592 mL, 5.09 mmol) and 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (0.813 mL, 2.54 mmol). The reaction mixture was stirred at room temperature overnight, then concentrated in vacuo. The product was purified by flash column chromatography on silica gel (10:90 EtOAc/hexanes) to give 28 as a colorless oil (0.137 g, 28%): 1H NMR (600 MHz, CDCl3) δ 4.67 (dt, J = 7.4, 3.3, 1H), 4.57 (ddd, J = 11.8, 3.5, 2.9, 1H), 4.02–3.96 (m, 2H), 2.87 (dd, J = 18.3, 7.7, 1H), 2.59 (dd, J = 18.3, 3.5, 1H), 2.23–2.21 (m, 1H), 1.63–1.57 (m, 1H), 1.17–0.91 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 175.3, 87.7, 70.3, 58.6, 37.8, 35.2, 17.6, 17.53, 17.46, 17.4, 17.28, 17.26, 17.24, 17.19, 13.6, 13.5, 13.3, 13.0; IR (ATR) 2945, 1782, 1464, 1081 cm−1; HRMS (TOF MS ES+) m / z calcd for C18H37O5Si2 (M + H)+ 389.2179, found 389.2174. Anal. Calcd for C18H36O5Si2: C, 55.63; H, 9.34. Found: C, 55.45; H, 9.51.
(7aR*,10aS*)-2,2,4,4-tetraisopropylhexahydrofuro[3,2-f][1,3,5,2,4]trioxadisilonin-9-yl acetate 13
To a solution of 28 (0.137 g, 0.352 mmol) in CH2Cl2 at −78 °C was added diisobutylaluminum hydride (1.0 M in toluene, 0.70 mL, 0.70 mmol). After 2 h at −78 °C, a solution of dimethylaminopyridine (0.0860 g, 0.704 mmol) in CH2Cl2 (0.5 mL), pyridine (0.0854 mL, 1.06 mmol), and acetic anhydride (0.200 mL, 2.11 mmol) were added. After 2 h at −78 °C, the reaction mixture was warmed to 0 °C and saturated aqueous sodium potassium tartrate (1 mL) and saturated aqueous ammonium chloride (1 mL) were added. The mixture was allowed to warm to room temperature and was stirred until the layers were completely separated. The reaction mixture was extracted with CH2Cl2 (2 × 15 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo to give a colorless oil. The product was purified by flash column chromatography on silica gel (1:5:94 Et3N:EtOAc:hexanes) to give 13 as a colorless oil (0.14 g, 88%). 1H and 13C NMR of the separated diastereomers is given. IR, HRMS, and elemental analysis of a mixture of diastereomers is given. High Rf diastereomer: 1H NMR (600 MHz, CDCl3) δ 6.31 (dd, J = 5.2, 1.2, 1H), 4.71 (td, J = 6.9, 4.0, 1H), 4.19 (dt, J = 11.5, 4.0, 1H), 3.99–3.91 (m, 2H), 2.38 (ddd, J = 13.7, 7.5, 1.2, 1H), 2.25 (ddd, J = 13.7, 6.4, 5.3, 1H), 2.19 (ddt, J = 13.7, 9.5, 3.9, 1H), 2.03 (s, 3H), 1.63 (dddd, J = 13.9, 11.5, 5.3, 2.3, 1H), 1.10–0.90 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 170.4, 98.8, 86.7, 73.6, 59.2, 42.5, 37.6, 21.6, 17.7, 17.6, 17.51, 17.46, 17.40, 17.38, 17.37, 13.6, 13.4, 13.1, 13.0. Low Rf diastereomer: 1H NMR (600 MHz, CDCl3) δ 6.25 (d, J = 5.0, 1H), 4.49 (ddd, J = 7.5, 3.0, 1.6, 1H), 4.34 (dt, J = 11.5, 3.4, 1H), 3.96–3.94 (m, 2H), 2.33 (ddd, J = 14.3, 7.6, 5.2, 1H), 2.17–2.09 (m, 2H), 2.05 (s, 3H), 1.49–1.43 (m, 1H), 1.18–0.90 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 171.0, 98.6, 87.4, 73.3, 59.5, 41.7, 35.8, 21.5, 17.7, 17.6, 17.5, 17.43, 17.38, 17.3, 13.61, 13.56, 13.10, 13.06; IR (ATR) 2944, 1746, 1464, 1086 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H40NaO6Si2 (M + Na)+ 455.2261, found 455.2264. Anal. Calcd for C20H40O6Si2: C, 55.52; H, 9.32. Found: C, 55.79; H, 9.45.
(4R*,5S*)-4-((tert-butyldimethylsilyl)oxy)-5-(2-((tertbutyldimethylsilyl)oxy)ethyl)dihydrofuran-2(3H)-one 29
To a solution of 27 (0.046 g, 0.20 mmol) in DMF (1 mL) at 0 °C were added imidazole (0.067 g, 0.99 mmol) and tert-butyldimethylsilyl chloride (0.075 g, 0.49 mmol). The reaction mixture was allowed to warm to room temperature overnight before concentrating in vacuo to give a yellow oil. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to give 29 as a colorless oil (0.056 g, 75%): 1H NMR (600 MHz, CDCl3) δ 4.46 (dt, J = 8.8, 3.9, 1H), 4.28 (ddd, J = 6.5, 4.0, 3.6, 1H), 3.78–3.66 (m, 2H), 2.76 (dd, J = 17.6, 6.6, 1H), 2.44 (dd, J = 17.6, 4.1, 1H), 1.87 (dddd, J = 14.1, 8.0, 6.1, 4.1, 1H), 1.70 (ddt, J = 14.1, 9.0, 4.9, 1H), 0.89 (s, 9H), 0.88 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H), 0.06 (s, 3H); 13C NMR (150 MHz, CDCl3) δ 175.3, 85.2, 72.6, 58.9, 38.3, 36.3, 26.1, 25.8, 18.4, 18.1, −4.5, −4.6, −5.21, −5.22; IR (ATR) 2929, 1784, 1078 cm−1; HRMS (MS TOF ES+) m / z calcd for C18H39O4Si2 (M + H)+ 375.2386, found 375.2387. Anal. Calcd for C18H38O4Si2: C, 57.70; H, 10.22. Found: C, 58.00; H, 10.22.
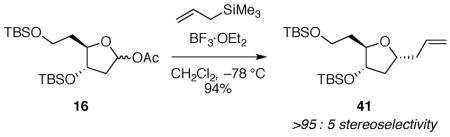
(4R*,5S*)-4-((tert-butyldimethylsilyl)oxy)-5-(2-((tert-butyldimethylsilyl)oxy)ethyl)tetrahydrofuran-2-yl acetate 16
To a solution of 29 (0.151 g, 0.403 mmol) in CH2Cl2 (4 mL) at −78 °C was added diisobutylaluminum hydride (0.81 mL, 1.0 M in toluene, 0.81 mmol). After 1 h at −78 °C, a solution of dimethylaminopyridine (0.098 g, 0.81 mmol) in CH2Cl2 (1 mL), pyridine (0.098 mL, 1.2 mmol), and acetic anhydride (0.228 mL, 2.42 mmol) were added sequentially, and the reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was cooled to 0 °C before adding saturated aqueous sodium potassium tartrate (2 mL) and saturated aqueous ammonium chloride (2 mL). The reaction mixture was allowed to warm to room temperature and was stirred until the layers were completely separated. The reaction mixture was extracted with CH2Cl2 (3 × 20 mL), and the combined organic layers were filtered through a cotton plug and concentrated in vacuo to give a colorless oil. The product was purified by flash column chromatography on silica gel (1:5:94 Et3N:EtOAc:hexanes) to give 16 as a colorless oil (0.15 g, 93%). Characterization was performed on a 44:56 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 6.29 (dd, J = 5.3, 2.0, 0.56H), 6.22 (dd, J = 5.8, 2.0, 0.44H), 4.23 (td, J = 6.4, 4.7, 0.56H), 4.09 (dt, J = 8.5, 4.5, 0.44H), 4.03 (ddd, J = 7.5, 5.0, 4.4, 0.44H), 3.96 (dt, J = 9.1, 4.4, 0.56H), 3.76–3.68 (m, 2H), 2.45 (ddd, J = 13.9, 7.4, 5.8, 0.44H), 2.21 (ddd, J = 13.5, 6.4, 2.0, 0.56H), 2.15 (ddd, J = 13.5, 6.3, 5.5, 0.56H), 2.05 (s, 1.32H), 2.03 (s, 1.68H), 1.94 (ddd, J = 14.1, 4.2, 2.0, 0.44H), 1.87–1.80 (m, 1H), 1.70 (dddd, J = 13.9, 9.1, 6.5, 5.0, 0.56H), 1.63 (ddt, J = 13.9, 8.5, 5.7, 0.44H), 0.89–0.88 (m, 18H), 0.08–0.05 (m, 12H); 13C NMR (150 MHz, CDCl3) δ 170.9, 170.4, 98.8, 98.3, 84.5, 83.4, 75.4, 75.2, 60.2, 60.0, 41.7, 41.4, 38.0, 36.4, 26.1, 25.91, 25.88, 21.62, 21.56, 18.5, 18.14, 18.09, −4.41, −4.42, −4.6, −5.11, −5.13, −5.14, −5.15; IR (thin film) 2954, 1750, 1251, 1089 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H42NaO5Si2 (M + Na)+ 441.2467, found 441.2462. Anal. Calcd for C20H42O5Si2: C, 57.37; H, 10.11. Found: C, 57.62; H, 10.18.
(Z)-2,2,4,4-tetraisopropyl-6,9-dihydro-1,3,5,2,4-trioxadisilonine 31
To a solution of cis-2-butene-1,4-diol (0.214 g, 2.43 mmol) in CH2Cl2 (48 mL) at 0 °C was added 2,6-lutidine (0.850 mL, 7.30 mmol) and 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (0.778 mL, 2.43 mmol). The reaction mixture was stirred overnight at room temperature, then was washed with saturated aqueous sodium bicarbonate (100 mL). The aqueous layer was extracted with CH2Cl2 (2 × 50 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (5:95 EtOAc:hexanes) to give 31 as a colorless oil (0.484 g, 60%): 1H NMR (600 MHz, CDCl3) δ 5.82 (ddd, J = 6.7, 4.8, 1.9, 2H), 4.39 (d, J = 6.7, 2H and dd, J = 4.8, 1.9, 2H), 1.06–1.04 (m, 24H), 1.00–0.94 (m, 4H); 13C NMR (150 MHz, CDCl3) δ 130.8, 57.9, 17.6, 17.5, 13.7; IR (ATR) 3023, 2943, 1464, 1082 cm−1; HRMS (TOF MS ES+) m / z calcd for C16H35O3Si2 (M + H)+ 331.2124, found 331.2125. Anal. Calcd for C16H34O3Si2: C, 58.13; H, 10.37. Found: C, 58.38; H, 10.31.
4,4,6,6-tetraisopropyl-3,5,7,10-tetraoxa-4,6-disilabicyclo[7.1.0]decane 32
To solution of 31 (0.484 g, 1.47 mmol) in CH2Cl2 (15 mL) at 0 °C was added m-chloroperoxybenzoic acid (0.66 g, 77%, 2.9 mmol). The reaction mixture was stirred room temperature overnight before washing sequentially with saturated sodium bisulfite (50 mL) and saturated aqueous sodium bicarbonate (100 mL) and extracting with CH2Cl2(4 × 50 mL). The combined organic layers were filtered through a cotton plug concentrated in vacuo. The product was purified by flash column chromatography silica gel to give 32 as a colorless oil (0.50 g, 98%): 1H NMR (600 MHz, CDCl3) δ 4.17–4.15 (m, 2H), 3.65–3.61 (m, 2H), 3.22–3.19 (m, 2H), 1.08–0.91 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 58.8, 55.8, 17.6, 17.38, 17.37, 17.3, 13.5, 13.3; IR (ATR) 2944, 1083 cm−1; HRMS (TOF MS ES+) m / z calcd for C16H35O4Si2 (M + H)+ 347.2073, found 347.2075. Two attempts to obtain satisfactory combustion analysis data unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
(7R*,8S*)-8-allyl-2,2,4,4-tetraisopropyl-1,3,5,2,4-trioxadisilonan-7-ol 33
To a solution of 32 (0.495 g, 1.43 mmol) in Et2O (15 mL) at 0 °C was added allylmagnesium bromide (4.3 mL, 1.0 M in Et2O, 0.43 mmol). After 2 h at room temperature, saturated aqueous ammonium chloride (50 mL) was added and the layers were separated. The aqueous layer was extracted with EtOAc (2 × 40 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo to give a light yellow oil. The product was purified by flash column chromatography (1:5:94 Et3N:EtOAc:hexanes) to give 33 as a light yellow oil (0.51 g, 92%): 1H NMR (600 MHz, CDCl3) δ 5.81 (dddd, J = 17.0, 10.1, 7.5, 6.7, 1H), 5.08 (ddt, J = 17.0, 2.0, 1.5, 1H), 5.03 (ddt, J = 10.1, 2.0, 1.0, 1H), 4.27 (dd, J = 10.6, 1.1, 1H), 4.01 (dd, J = 11.4, 2.5, 1H), 3.93 (dd, J = 10.7, 4.1, 1H), 3.69 (dd, J = 11.5, 8.4, 1H), 3.45 (dddd, J = 9.1, 7.3, 4.1, 1.1, 1H), 2.85 (d, J = 9.2, 1H), 2.33 (dddt, J = 14.0, 6.1, 4.6, 1.4, 1H), 2.03 (dddt, J = 14.0, 8.7, 7.6, 1.1, 1H), 1.95 (tddd, J = 8.6, 7.2, 4.5, 2.5, 1H), 1.09–0.92 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 136.8, 116.8, 73.9, 64.3, 64.2, 44.1, 33.5, 17.70, 17.67, 17.59, 17.56, 17.53, 17.50, 13.8, 13.4, 13.2, 12.8; IR (ATR) 3452, 3077, 2944, 1640, 1464, 1025 cm−1; HRMS (TOF MS ES+) m / z calcd for C19H40NaO4Si2 (M + Na)+ 411.2362, found 411.2359. Anal. Calcd for C19H40O4Si2: C, 58.71; H, 10.37. Found: C, 58.56; H, 10.33.
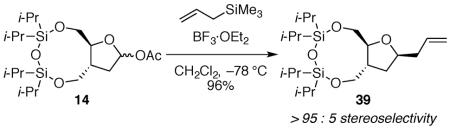
(3aR*,10aS*)-6,6,8,8-tetraisopropylhexahydrofuro[2,3-g][1,3,5,2,4]trioxadisilonin-2-yl acetate 14
To a solution of 33 (0.511 g, 1.31 mmol) in acetone and water (10:1, 13.2 mL) at 0 °C was added 4-methylmorpholine N-oxide (0.500 g, 4.27 mmol) and osmium tetroxide (0.42 mL, 4.0% in water, 0.066 mmol). The reaction mixture was stirred overnight at room temperature before adding sodium periodate (0.562 g, 2.63 mmol). The reaction mixture was stirred overnight before washing with saturated aqueous sodium bicarbonate (60 mL). The aqueous layer was extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo to give a grey oil. The product was purified by flash column chromatography on silica gel (20:80 EtOAc:hexanes) to give a lactol as a colorless oil (0.47 g, 92%). Characterization was performed on a 56:44 mixture of diastereomers: 1H NMR (600 MHz, CDCl3) δ 5.56 (dt, J = 5.5, 3.5, 0.44H), 5.40 (t, J = 4.9, 0.56H), 4.18 (dd, J = 11.3, 3.7, 0.56H), 4.13–4.09 (m, 1.32H), 4.06 (dd, J = 11.8, 2.3, 0.56H), 4.00 (dd, J = 11.8, 3.2, 0.56H), 3.98–3.94 (m, 1H), 3.75 (dd, J = 11.3, 9.9, 0.44H), 3.66 (dd, J = 11.3, 9.8, 0.56H), 3.00 (d, J = 3.4, 0.44H), 2.88 (d, J = 5.0, 0.56H), 2.87–2.82 (m, 0.56H), 2.58 (ttd, J = 9.5, 9.2, 3.8, 0.44H), 2.28 (ddd, J = 13.4, 9.2, 5.5, 0.44H), 1.92 (dd, J = 12.2, 6.5, 0.56H), 1.68 (td, J = 12.3, 4.6, 0.56H), 1.52 (ddd, J = 13.2, 9.5, 3.7, 0.44H), 1.12–0.92 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 97.9, 97.5, 85.3, 82.7, 65.43, 65.39, 64.1, 63.5, 41.6, 38.8, 37.8, 37.1, 17.67, 17.66, 17.62, 17.61, 17.59, 17.55, 17.51, 17.48, 17.47, 17.44, 17.42, 17.40, 13.74, 13.69, 13.29, 13.27, 13.01, 12.96, 12.9; IR (ATR) 3419, 2944, 1464, 1040 cm−1; HRMS (TOF MS ES+) m / z calcd for C18H42NO5Si2(M + NH4)+ 408.2601, found 408.2605. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
To a solution of the lactol (0.134 g, 0.344 mmol) in CH2Cl2 (1.4 mL) at 0 °C was added triethylamine (0.288 mL, 2.06 mmol) and acetic anhydride (0.0651 mL, 0.689 mmol). The reaction mixture was stirred at room temperature overnight before washing with saturated aqueous sodium bicarbonate (20 mL). The layers were separated and the aqueous layer was extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (1:10:89 Et3N:EtOAc:hexanes) to give 14 as a colorless oil (0.148 g, 99%). 1H and 13C NMR of the separated diastereomers is given. IR, HRMS, and elemental analysis of a mixture of diastereomers is given. High Rf diastereomer: 1H NMR (500 MHz, CDCl3) δ 6.26 (dd, J = 5.9, 2.9, 1H), 4.16 (ddd, J = 8.3, 5.3, 2.9, 1H), 4.14–4.11 (m, 2H), 3.91 (dd, J = 11.6, 5.3, 1H), 3.78 (dd, J = 11.4, 8.6, 1H), 2.55 (quintd, J = 8.8, 3.6, 1H), 2.42 (ddd, J = 13.7, 9.7, 5.9, 1H), 2.04 (s, 3H), 1.75 (ddd, J = 13.7, 8.6, 3.0, 1H), 1.10–0.92 (m, 28H); 13C NMR (100 MHz, CDCl3) δ 170.6, 98.2, 82.9, 64.3, 63.8, 42.2, 35.5, 21.5, 17.62, 17.58, 17.54, 17.51, 17.46, 17.4, 13.5, 13.21, 13.16, 13.0. Low Rf diastereomer: 1H NMR (600 MHz, CDCl3) δ 6.22 (d, J = 4.9, 1H), 4.18 (dd, J = 11.4, 3.8, 1H), 4.09 (dd, J = 11.9, 2.5, 1H), 4.03 (dd, J = 11.9, 3.8, 1H), 3.98 (ddd, J = 9.3, 3.5, 2.8, 1H), 3.66 (dd, J = 11.4, 9.2, 1H), 2.81 (dtdd, J = 12.5, 9.4, 6.4, 3.7, 1H), 2.03 (s, 3H), 2.01 (dd, J = 13.1, 6.4, 1H), 1.87 (td, J = 12.8, 4.9, 1H), 1.13–0.93 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 170.7, 97.8, 86.1, 64.7, 63.8, 39.3, 36.2, 21.7, 17.63, 17.61, 17.56, 17.5, 17.43, 17.38, 13.6, 13.3, 13.0, 12.9; IR (ATR) 2944, 1748, 1464, 1040 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H40NaO6Si2(M + Na)+ 455.2261, found 455.2259. Anal. Calcd for C20H40O6Si2: C, 55.52; H, 9.32. Found: C, 55.64; H, 9.24.
(Z)-2,2,3,3,10,10,11,11-octamethyl-4,9-dioxa-3,10-disiladodec-6-ene 34
To a solution of cis-2-butene-1,4-diol (0.429 g, 4.87 mmol) and imidazole (1.66 g, 24.3 mmol) in DMF (10 mL) at 0 °C was added tert-butyldimethylchlorosilane (1.83 g, 12.2 mmol). The reaction mixture was stirred at room temperature overnight before washing with saturated aqueous sodium bicarbonate (40 mL) and extracting with EtOAc (2 × 40 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (1:99 EtOAc:hexanes) to give 34 as a colorless oil (1.48 g, 96%). The spectral data obtained matched the reported values for this compound:32 1H NMR (600 MHz, CDCl3) δ 5.55 (t, J = 4.2, 2H), 4.23 (d, J = 4.2, 4H), 0.90 (s, 18H), 0.07 (s, 12H); 13C NMR (150 MHz, CDCl3) δ 130.4, 59.9, 26.2, 18.6, −5.0; HRMS (TOF MS ES+) m / z calcd for C16H37O2Si2 (M + H)+ 317.2332, found 317.2327.
2,3-bis(((tert-butyldimethylsilyl)oxy)methyl)oxirane 35
To a solution of 34 (1.47 g, 4.66 mmol) in CH2Cl2 (47 mL) at 0 °C was added m-chloroperoxybenzoic acid (2.1 g, 77%, 9.2 mmol). The reaction mixture was stirred at room temperature overnight before washing sequentially with saturated aqueous sodium bisulfite (150 mL) and saturated aqueous sodium bicarbonate (200 mL) and extracting with CH2Cl2 (3 × 50 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (1:5:94 Et3N:EtOAc:hexanes) to give 35 as a colorless oil (1.5 g, 97%). The spectral data obtained matched the reported values for this compound:32 1H NMR (600 MHz, CDCl3) δ 3.80 (dd, J = 11.8, 4.2, 2H), 3.72 (dd, J = 11.8, 6.1, 2H), 3.15–3.12 (m, 2H), 0.91 (s, 18H), 0.09 (s, 6H), 0.08 (s, 6H); 13C NMR (150 MHz, CDCl3) δ 62.0, 57.1, 26.1, 18.5, −5.0, −5.2; HRMS (TOF MS ES+) m / z calcd for C16H37O3Si2 (M + H)+ 333.2281, found 333.2284.
(6R*,7S*)-7-allyl-2,2,3,3,10,10,11,11-octamethyl-4,9-dioxa-3,10-disiladodecan-6-ol 36
To a solution of 35 (1.48 g, 4.44 mmol) in Et2O (44 mL) at 0 °C was added allylmagnesium bromide (6.7 mL, 1.0 M in Et2O, 6.7 mmol). After 3.5 h at room temperature, saturated aqueous ammonium chloride (100 mL) was added and the layers were separated. The aqueous layer was extracted with EtOAc (2 × 100 mL). The combined organic layers were dried over anhydrous sodium sulfate, filtered, and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (5:95 EtOAc:hexanes) to give 36 as a colorless oil (1.6 g, 97%):1H NMR (500 MHz, CDCl3) δ 5.80 (dddd, J = 16.9, 10.1, 8.0, 6.3, 1H), 5.08–5.00 (m, 2H), 3.82 (dtd, J = 7.4, 4.7, 2.9, 1H), 3.68–3.67 (m, 2H), 3.67 (dd, J = 10.0, 5.1, 1H), 3.60 (dd, J = 10.1, 7.3, 1H), 2.95 (d, J = 2.9, 1H), 2.31–2.12 (m, 2H), 1.72 (tt, J = 9.0, 4.5, 1H), 0.90 (s, 9H), 0.89 (s, 9H), 0.07 (s, 6H), 0.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 137.6, 116.3, 73.9, 65.2, 63.6, 42.3, 30.5, 26.12, 26.06, 18.5, 18.3, −5.1, −5.4; IR (ATR) 3511, 3077, 2928, 1640, 1089 cm−1; HRMS (TOF MS ES+) m / z calcd for C19H42NaO3Si2 (M + Na)+ 397.2570, found 397.2565. Anal. Calcd for C19H42O3Si2: C, 60.90; H, 11.30. Found: C, 61.13; H, 11.16.
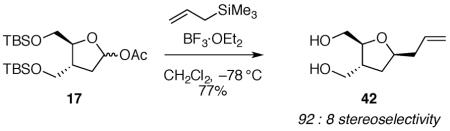
(4R*,5S*)-4,5-bis(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl acetate 17
To a solution of 36 (1.61 g, 4.30 mmol) in acetone and water (10:1, 44 mL) at 0 °C was added 4-methylmorpholine-N-oxide (1.64 g, 14.0 mmol) and osmium tetroxide (1.4 mL, 4.0% in water). The reaction mixture was stirred overnight at room temperature before adding sodium periodate (1.84 g, 8.59 mmol). After 8 h at room temperature, the reaction mixture was washed with saturated aqueous sodium bicarbonate (100 mL) and extracted with CH2Cl2 (2 × 125 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo to give a grey oil. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to give a lactol as a colorless oil (1.4 g, 87%). Characterization was performed on a 50:50 mixture of diastereomers: 1H NMR (500 MHz, CDCl3) δ 5.37 (t, J = 4.6, 0.5H), 5.35 (dd, J = 4.6, 2.4, 0.5H), 4.81 (d, J = 10.4, 0.5H), 4.08–4.00 (m, 1.5H), 3.84 (dd, J = 10.5, 2.5, 0.5H), 3.74–3.68 (m, 2H), 3.62 (dd, J = 10.5, 2.0, 0.5H), 3.59 (dd, J = 10.4, 5.7, 0.5H), 3.51 (dd, J = 9.4, 8.4, 0.5H), 2.61–2.54 (m, 0.5H), 2.39–2.34 (m, 0.5H), 2.25 (td, J = 11.4, 5.1, 0.5H), 1.96 (dd, J = 12.7, 7.8, 0.5H), 1.75–1.68 (m, 1H), 0.93 (s, 4.5H and s, 4.5H), 0.89 (s, 4.5H), 0.88 (s, 4.5H), 0.12 (s, 6H), 0.06 (s, 1.5H), 0.05 (s, 1.5H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 98.8, 98.6, 83.8, 80.2, 65.50, 65.46, 65.3, 41.3, 39.3, 38.6, 37.7, 26.13, 26.12, 26.09, 26.06, 18.7, 18.6, 18.5, 18.4, −5.1, −5.18, −5.23, −5.28, −5.30, −5.37, −5.39; IR (ATR) 3422, 2953, 1253, 1085 cm−1; HRMS (TOF MS ES+) m / z calcd for C18H40NaO4Si2 (M + Na)+ 399.2363, found 399.2360. Anal. Calcd for C18H40O4Si2: C, 57.40; H, 10.70. Found: C, 57.94; H, 10.77. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
To a solution of the lactol (0.200 g, 0.531 mmol) in CH2Cl2 (5 mL) at 0 °C was added triethylamine (0.89 mL, 6.4 mmol) and acetic anhydride (0.20 mL, 2.1 mmol). The reaction mixture was stirred at room temperature overnight before washing with saturated aqueous sodium bicarbonate (20 mL) and extracting with CH2Cl2 (2 × 20 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (1:5:94 Et3N:EtOAc:hexanes) to give 17 as a colorless oil (0.21 g, 94%). The spectral data obtained matched the reported values for this compound.33 Characterization was performed on a 50:50 mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 6.29 (dd, J = 5.4, 1.0, 0.5H), 6.24 (d, J = 4.7, 0.5H), 4.07 (app q, J = 4.2, 0.5H), 3.93 (dt, J = 7.7, 5.5, 0.5H), 3.77–3.60 (m, 4H), 2.46–2.36 (m, 1H), 2.29 (ddd, J = 13.7, 10.0, 5.4, 0.5H), 2.11–1.96 (m, 1H), 2.03 (s, 1.5H), 2.02 (s, 1.5H), 1.85 (ddd, J = 13.7, 3.4, 1.0, 0.5H), 0.90 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H and s, 3H), 0.05 (s, 3H), 0.04 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 170.6, 99.8, 99.2, 83.9, 83.8, 66.4, 65.1, 65.0, 63.9, 41.8, 41.7, 36.2, 35.2, 26.14, 26.10, 21.7, 21.6, 18.6, 18.5, −5.10, −5.13, −5.17, −5.19, −5.22, −5.24; IR (ATR) 2929, 1750, 1092 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H42NaO5Si2 (M + Na)+ 441.2469, found 441.2467. Anal. Calcd for C20H42O5Si2: C, 57.37; H, 10.11. Found: C, 57.38; H, 9.90.
General procedure for Allyltrimethylsilane Addition to Bicyclic Acetates
A solution of acetate (0.10 M) in dry CH2Cl2 was cooled to −78 °C. Allyltrimethylsilane (4 equiv) was added to the reaction mixture, followed by dropwise addition of boron trifluoride etherate (1.6 equiv). The reaction mixture was allowed to come slowly to room temperature overnight. A solution of 1:1:1 dry CH2Cl2:MeOH:Et3N was added to the reaction mixture at −78 °C. The reaction mixture was extracted with CH2Cl2. The combined organic layers were washed with saturated aqueous NaHCO3, dried over anhydrous Na2SO4, and concentrated in vacuo.
(6aR,8R,9aS)-8-allyl-2,2,4,4-tetraisopropyltetrahydro-6H-furo[3,2-f][1,3,5,2,4]trioxadisilocine 37
The general allyltrimethylsilane addition procedure was followed with acetate 12 (0.038 g, 0.090 mmol). Purification by flash column chromatography on silica gel (5:95 EtOAc:hexanes) afforded 37 as a light yellow oil (0.031 g, 85%). Characterization was performed on a 84:16 (1,3-cis-37:1,3-trans-37) mixture of diastereomers: [α]24D −10.1 (c 1.18, MeOH); 1H NMR (500 MHz, C6D6) δ 5.82–5.74 (m, 1H), 5.06–5.00 (m, 2H), 4.42 (dd, J = 13.9, 7.6, 1H), 4.19 (dd, J = 11.3, 3.6, 0.16H), 4.11 (dd, J = 11.5, 3.6, 0.84H), 4.08–4.03 (m, 0.16H), 3.95 (ddd, J = 7.7, 6.4, 3.6, 0.84H), 3.92–3.85 (m, 1H), 3.82–3.77 (m, 1H), 2.39 (dt, J = 13.9, 6.6, 0.84H), 2.27 (dt, J = 13.5, 6.8, 0.16H), 2.22–2.13 (m, 1H), 2.07 (ddd, J = 12.1, 7.2, 6.1, 0.84H), 1.94 (ddd, J = 12.8, 6.4, 3.8, 0.16H), 1.78 (dt, J = 12.1, 8.6, 0.84H), 1.67 (dt, J = 12.8, 8.2, 0.16H), 1.18–0.93 (m, 28H); 13C NMR (125 MHz, C6D6) δ 135.3, 135.2, 117.5, 117.4, 87.1, 84.1, 77.6, 77.1, 75.5, 75.3, 65.3, 65.0, 41.1, 41.00, 40.97, 40.4, 18.2, 18.09, 18.05, 18.02, 17.97, 17.82, 17.79, 17.72, 17.69, 14.3, 14.2, 14.1, 14.0, 13.8, 13.7, 13.4; IR (thin film) 3078, 2945, 1643, 1036 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H40NaO4Si2 (M + Na)+ 423.2363, found 423.2361. Anal. Calcd for C20H40O4Si2: C, 59.95; H, 10.06. Found: C, 60.21; H, 10.15.
(((2R,3S,5R)-5-allyl-2-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane 40
The general allyltrimethylsilane addition procedure was followed with acetate 15 (0.148 g, 0.366 mmol). Purification by flash column chromatography on silica gel (5:95 EtOAc:hexanes) afforded 40 as a colorless oil (0.129 g, 91%): [α]24D 34.9 (c 1.45, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 5.80 (ddt, J = 17.2, 10.2, 7.0, 1H), 5.10–5.03 (m, 2H), 4.35 (ddd, J = 6.4, 5.0, 4.1, 1H), 4.09 (quint, J = 6.6, 1H), 3.84 (app q, J = 4.1, 1H), 3.61 (dd, J = 10.9, 3.9, 1H), 3.56 (dd, J = 10.9, 5.0, 1H), 2.47 (dt, J = 14.0, 6.6, 1H), 2.32 (dt, J = 14.0, 7.0, 1H), 2.17 (dt, J = 12.6, 6.7, 1H), 1.66 (ddd, J = 12.7, 6.2, 5.2, 1H), 0.89 (s, 9H), 0.88 (s, 9H), 0.06 (s, 6H), 0.05 (s, 3H and s, 3H); 13C NMR (125 MHz, CDCl3) δ 135.4, 116.9, 86.5, 78.5, 73.5, 63.7, 41.1, 40.2, 26.2, 26.0, 18.6, 18.2, -4.5, −4.6, −5.1, −5.2; IR (thin film) 3078, 2954, 1643, 1471, 1255, 1111 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H42NaO3Si2 (M + Na)+ 409.2570, found 409.2562. Anal. Calcd for C20H42O3Si2: C, 62.12; H, 10.95. Found: C, 62.42; H, 11.07.
(7aR*,9R*,10aS*)-9-allyl-2,2,4,4-tetraisopropylhexahydrofuro[3,2-f][1,3,5,2,4]trioxadisilonine 38
The general allyltrimethylsilane addition procedure was followed with acetate 13 (0.056 g, 0.13 mmol). Purification by flash column chromatography on silica gel (5:95 EtOAc:hexanes) afforded 38 as a colorless oil (0.458 g, 86%): 1H NMR (600 MHz, CDCl3) δ 5.81 (ddt, J = 17.2, 10.2, 7.0, 1H), 5.09 (ddt, J = 17.2, 2.0, 1.5, 1H), 5.06 (ddt, J = 10.2, 2.1, 1.0, 1H), 4.48 (dt, J = 7.1, 3.9, 1H), 4.09 (dt, J = 11.3, 3.7, 1H), 4.05 (dt, J = 12.6, 6.7, 1H), 3.98–3.92 (m, 2H), 2.50 (dtt, J = 13.9, 6.8, 1.3, 1H), 2.35 (dtt, J = 13.9, 7.1, 1.2, 1H), 2.22 (dt, J = 12.6, 7.0, 1H), 2.09–2.03 (m, 1H), 1.77 (ddd, J = 12.8, 5.8, 4.1, 1H), 1.53–1.47 (m, 1H), 1.19–0.87 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 135.4, 117.1, 84.6, 77.6, 75.6, 59.5, 40.9, 39.9, 36.2, 17.8, 17.7, 17.59, 17.56, 17.47, 17.46, 17.45, 13.60, 13.57, 13.11, 13.10; IR (ATR) 2943, 2867, 1642, 1464 cm−1; HRMS (TOF MS APCI+) m / z calcd for C21H43O4Si2 (M + H)+ 415.2692, found 415.2693. Anal. Calcd for C21H42O4Si2: C, 60.82; H, 10.21. Found: C, 60.70; H, 9.99.
(((2R*,3S*,5R*)-5-allyl-2-(2-((tert-butyldimethylsilyl)oxy)ethyl)tetrahydrofuran-3-yl)oxy)(tert-butyl)dimethylsilane 41
The general allyltrimethylsilane addition procedure was followed with acetate 16 (0.080 g, 0.19 mmol). The products were purified by flash column chromatography on silica gel (5:95 – 25:75 EtOAc:hexanes) to give 41 (0.031 g, 40%) and a mono-TBS protected allyl product (0.030 g, 54%) as colorless oils: 1H NMR (600 MHz, C6D6) δ 5.85 (dddd, J = 17.2, 10.2, 7.3, 6.7, 1H), 5.09–5.02 (m, 2H), 4.07 (ddd, J = 8.9, 5.1, 3.5, 1H), 3.97 (tt, J = 6.8, 6.7, 1H), 3.91 (td, J = 6.3, 5.3, 1H), 3.84 (dd, J = 7.6, 5.4, 2H), 2.49 (dtd, J = 13.4, 6.5, 1.4, 1H), 2.29 (dtd, J = 14.0, 7.1, 1.0, 1H), 1.96 (dt, J = 12.4, 6.8, 1H), 1.89 (dtd, J = 13.5, 7.5, 3.5, 1H), 1.67–1.59 (m, 2H), 0.99 (s, 9H), 0.95 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H), 0.04 (s, 3H), 0.02 (s, 3H); 13C NMR (150 MHz, C6D6) δ 135.6, 116.8, 81.5, 77.8, 77.0, 60.6, 41.3, 40.6, 37.4, 26.2, 26.0, 18.5, 18.2, −4.4, −4.6, −5.12, −5.14; IR (ATR) 3078, 2929, 1642, 1472, 1251, 1085 cm−1; HRMS (MS TOF ES+) m / z calcd for C21H44NaO3Si2 (M + Na)+ 423.2726, found 423.2728. Anal. Calcd for C21H44O3Si2: C, 62.94; H, 11.07. Found: C, 63.15; H, 11.01.
2-((2R*,3S*,5R*)-(5-allyl-3-((tert-butyldimethylsilyl)oxy)tetrahydrofuran-2-yl)ethan-1-ol
1H NMR (600 MHz, C6D6) δ 5.74 (ddt, J = 17.2, 10.2, 7.0, 1H), 5.05–4.99 (m, 2H), 3.92 (ddd, J = 9.2, 5.6, 3.6, 1H), 3.87 (tt, J = 7.0, 6.6, 1H), 3.80 (td, J = 6.7, 5.8, 1H), 3.73 (m, 2H), 2.47 (br s, 1H), 2.34 (dtt, J = 13.7, 6.6, 1.3, 1H), 2.17 (dtt, J = 13.9, 7.0, 1.0, 1H), 1.88 (dt, J = 12.4, 6.8, 1H), 1.69 (dtd, J = 14.0, 5.1, 3.6, 1H), 1.61–1.55 (m, 1H), 1.50 (ddd, J = 12.4, 7.3, 6.9, 1H), 0.92 (s, 9H), −0.01 (s, 3H), −0.02 (s, 3H); 13C NMR (150 MHz, C6D6) δ 135.1, 117.1, 84.2, 77.4, 77.1, 61.3, 41.0, 40.2, 35.8, 26.0, 18.1, −4.5, −4.7; IR (ATR) 3421, 3077, 2929, 1642 cm−1; HRMS (MS TOF ES+) m / z calcd for C15H31O3Si (M + H)+ 287.2042, found 287.2044. Anal. Calcd for C15H30O3Si: C, 62.89; H, 10.55. Found: C, 62.67; H, 10.35.
(2R*,3aS*,10aR*)-2-allyl-6,6,8,8-tetraisopropylhexahydrofuro[2,3-g][1,3,5,2,4]trioxadisilonine 39
The general allyltrimethylsilane addition procedure was followed with acetate 14 (0.115 g, 0.266 mmol). Purification by flash column chromatography on silica gel (5:95 EtOAc:hexanes) afforded 39 as a colorless oil (0.106 g, 96%): 1H NMR (500 MHz, CDCl3) δ 5.82 (ddt, J = 17.2, 10.2, 7.0, 1H), 5.09–5.03 (m, 2H), 4.12–4.07 (m, 2H), 4.02 (tt, J = 7.2, 5.8, 1H), 3.95 (dd, J = 11.7, 4.2, 1H), 3.77 (tt, J = 3.6, 3.1, 1H), 3.64 (dd, J = 11.2, 9.8, 1H), 2.52 (quintd, J = 9.1, 3.6, 1H), 2.37 (dt, J = 13.2, 6.6, 1H), 2.23 (dt, J = 13.9, 7.0, 1H), 1.75–1.65 (m, 2H), 1.11–0.88 (m, 28H); 13C NMR (125 MHz, CDCl3) δ 135.0, 117.2, 84.1, 77.1, 65.3, 64.1, 41.4, 41.0, 33.5, 17.69, 17.67, 17.64, 17.59, 17.50, 17.47, 13.7, 13.3, 13.1; IR (thin film) 3076, 2945, 1642, 1465, 1045 cm−1; HRMS (TOF MS ES+) m / z calcd for C21H46NO4Si2 (M + NH4)+ 432.2965, found 432.2962. Anal. Calcd for C21H42O4Si2: C, 60.82; H, 10.21. Found: C, 60.85; H, 10.07.
((((2R*,3S*,5R*)-5-allyltetrahydrofuran-2,3-diyl)bis(methylene))bis(oxy))bis(tert-butyldimethylsilane) 42
The general allyltrimethylsilane addition procedure was followed with acetate 17 (0.103 g, 0.246 mmol). Purification by flash column chromatography on silica gel (5:95 EtOAc:hexanes) afforded 42 as a colorless oil (0.076 g, 77%). Characterization was performed on a 8:92 (1,3-cis-42:1,3-trans-42) mixture of diastereomers: 1H NMR (400 MHz, CDCl3) δ 5.81 (ddt, J = 17.2, 10.2, 7.0, 1H), 5.10–5.01 (m, 2H), 4.02 (ddd, J = 11.8, 9.7, 6.2, 0.08H), 3.95 (dt, J = 14.4, 6.3, 0.92H), 3.80 (dt, J = 6.8, 4.6, 0.08H), 3.72–3.54 (m, 4.92H), 2.39–2.18 (m, 3H), 2.07 (ddd, J = 12.5, 7.7, 5.4, 0.08H), 1.81 (ddd, J = 12.4, 6.3, 4.6, 0.92H), 1.68–1.61 (m, 0.92H), 1.35 (dt, J = 12.1, 9.7, 0.08H), 0.90 (s, 9H), 0.89 (s, 9H), 0.06 (s, 6H), 0.04 (s, 6H); 13C NMR (100 MHz,) δ 135.4, 116.8, 82.4, 78.4, 65.9, 65.0, 43.6, 40.6, 34.4, 26.2, 18.6, 18.5, −5.0, −5.1, −5.2; IR (ATR) 3078, 2928, 1643, 1092 cm−1; HRMS (TOF MS ES+) m / z calcd for C21H44NaO3Si2 (M + Na)+ 423.2726, found 423.2726. Anal. Calcd for C21H44O3Si2: C, 62.94; H, 11.07. Found: C, 63.14; H, 11.28.
Stereochemical Proofs of Nucleophilic Substitution Reactions
In all cases, the diastereoselectivities were determined by GC analysis of the unpurified reaction mixture and confirmed by 1H NMR spectroscopy. The relative stereochemistry of the nucleophilic substitution products was proven by X-ray structure determination of crystalline derivatives and subsequent chemical correlation.
(E)-1-(2-((2R,4S,5R)-4-((tert-butyldimethylsilyl)oxy)-5-(((tert-butyldimethylsilyl)oxy)methyl)tetrahydrofuran-2-yl)ethylidene)-2-(2,4-dinitrophenyl)hydrazine 44
To a solution of 40 (0.102 g, 0.263 mmol) in acetone and water (2.2 mL, 10:1) at 0 °C was added 4-methylmorpholine N-oxide (0.100 g, 0.854 mmol) and osmium tetroxide (0.17 mL, 2.5% in H2O, 0.013 mmol). After stirring overnight at room temperature, the reaction mixture was washed with saturated aqueous sodium thiosulfate (4 mL) and extracted with Et2O (2 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (Et2O) to give a diol as a colorless oil (0.102 g, 92%). Characterization was performed on a 50:50 mixture of diastereomers: [α]24D 34.9 (c 1.35, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 4.38–4.29 (m, 2H), 3.98–3.90 (m, 1H), 3.88–3.83 (m, 1.5H), 3.65–3.48 (m, 4H), 3.02 (d, J = 3.8, 0.5H), 2.39–2.36 (m, 0.5H), 2.31–2.23 (m, 1.5H), 1.91–1.64 (m, 3H), 0.89 (s, 9H), 0.088 (s, 9H), 0.06 (s, 6H), 0.05 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 86.5, 86.4, 78.6, 76.4, 73.1, 72.9, 71.7, 70.0, 67.0, 66.7, 63.4, 63.3, 41.5, 40.8, 39.2, 38.5, 26.0, 25.9, 18.4, 18.1, −4.6, −4.7, −5.2, −5.3; IR (thin film) 3417, 2929, 1647, 1464 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H44NaO5Si2 (M + Na)+ 443.2625, found 443.2622. Anal. Calcd for C20H44O5Si2: C, 57.09; H, 10.54. Found: C, 56.89; H, 10.60.
To a solution of the diol (0.079 g, 0.19 mmol) in acetone and water (2 mL, 1:1) was added sodium periodate (0.048 g, 0.23 mmol). After stirring overnight at room temperature, the reaction mixture was washed with saturated aqueous NaHCO3 and extracted with CH2Cl2 (3 × 15 mL). The combined organic layers were filtered through a cotton plug and concentrated in vacuo. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to give an aldehyde as a colorless oil (0.065 g, 88%): [α]24D 38.8 (c 0.98, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 9.81 (t, J = 1.9, 1H), 4.56 (tt, J = 7.3, 5.4, 1H), 4.38 (tt, J = 3.4, 3.0, 1H), 3.90 (dt, J = 5.4, 3.7, 1H), 3.61 (dd, J = 10.8, 3.9, 1H), 3.51 (dd, J = 10.8, 5.4, 1H), 2.90 (ddd, J = 16.7, 7.3, 2.2, 1H), 2.66 (ddd, J = 16.7, 5.5, 1.6, 1H), 2.32 (tt, J = 7.2, 6.2, 1H), 1.67 (ddd, J = 12.7, 4.8, 3.9, 1H), 0.89 (s, 9H), 0.088 (s, 9H), 0.07 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H and s, 3H); 13C NMR (125 MHz, CDCl3) δ 201.9, 87.1, 74.1, 73.7, 63.7, 50.6, 40.6, 26.1, 25.9, 18.5, 18.1, −4.65, −4.68, −5.2, −5.3; IR (thin film) 2954, 2719, 1728, 1471 cm−1; HRMS (TOF MS ES+) m / z calcd for C19H40NaO4Si2 (M + Na)+ 411.2363, found 411.2359. Anal. Calcd for C19H40O4Si2: C, 58.71; H, 10.37. Found: C, 58.80; H, 10.46.
To a solution of aldehyde (0.041 g, 0.11 mmol) in EtOH (1 mL) was added 2,4-dinitrophenylhydrazine (0.020 g 0.10 mmol). The reaction mixture was stirred at 80 °C overnight, then concentrated in vacuo to give an orange oil. The product was purified by flash column chromatography on silica gel (10:90 EtOAc/hexanes) to give hydrazone 44 as an orange oil (0.050 g, 83%): [α]24D +24 (c 0.55, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 11.45 (s, 0.27H), 11.05 (s, 0.73H), 9.12 (d, J = 2.6, 0.27H), 9.11 (d, J = 2.6, 0.73H), 8.31 (d, J = 2.5, 0.27H), 8.28 (dd, J = 9.8, 2.5, 0.73H), 7.95 (d, J = 9.7, 0.27H), 7.92 (d, J = 9.6, 0.73H), 7.60 (t, J = 5.6, 0.73H), 7.15 (t, J = 5.8, 0.27H), 4.46–4.36 (m, 2H), 4.05 (dt, J = 4.6, 3.5, 0.27H), 3.94 (dt, J = 4.5, 3.7, 0.73H), 3.63 (dd, J = 10.9, 3.9, 1H), 3.57–3.53 (m, 1H), 2.89–2.80 (m, 1H), 2.68 (dt, J = 15.0, 5.4, 0.73H), 2.56 (ddd, J = 15.6, 5.4, 3.5, 0.27H), 2.40–2.30 (m, 1H), 1.80–1.72 (m, 1H), 0.90 (s, 6.57H), 0.89 (s, 6.57H), 0.88 (s, 2.43H), 0.86 (s, 2.43H), 0.08 (s, 3H and s, 3H), 0.06 (s, 2.19H), 0.05 (s, 2.19H), 0.04 (s, 0.81H), 0.03 (s, 0.81H); 13C NMR (125 MHz, CDCl3) δ 150.6, 149.0, 145.8, 145.3, 138.3, 138.0, 130.1, 129.9, 129.0, 124.7, 123.7, 123.6, 116.9, 116.7, 87.4, 87.3, 77.4, 76.5, 73.9, 73.4, 63.8, 63.5, 40.9, 40.5, 39.6, 35.3, 26.12, 26.08, 25.98, 25.95, 18.5, 18.2, −4.55, −4.58, −4.61, −5.1, −5.19, −5.22, −5.3; IR (thin film) 3302, 3109, 2954, 1620 cm−1; HRMS (TOF MS ES+) m / z calcd for C25H44N4NaO7Si2 (M + Na)+ 591.2646, found 591.2643. Anal. Calcd for C25H44N4O7Si2: C, 52.79; H, 7.80. Found: C, 53.08; H, 7.89.
(E)-1-(2-((2R*,4S*,5R*)-4-((tert-butyldimethylsilyl)oxy)-5-(2-((tert-butyldimethylsilyl)oxy)ethyl)tetrahydrofuran-2-yl)ethylidene)-2-(2,4-dinitrophenyl)hydrazine 46
To a solution of 41 (0.051 g, 0.13 mmol) in CH2Cl2 (2 mL) at −78 °C was added a stream of ozone until a blue color persisted. The reaction mixture was purged with oxygen for 30 min and triphenylphosphine (0.066 g, 0.25 mmol) was added. The reaction mixture was stirred at room temperature overnight before concentrating in vacuo. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to give an aldehyde as a colorless oil (0.043 g, 83%): 1H NMR (600 MHz, CDCl3) δ 9.81 (t, J = 2.0, 1H), 4.52 (tt, J = 7.4, 5.6, 1H), 4.09 (dt, J = 6.2, 4.1, 1H), 3.92 (d, J = 8.8, 4.0, 1H), 3.72 (ddd, J = 10.2, 7.0, 5.2, 1H), 3.67 (ddd, J = 10.2, 7.7, 6.1, 1H), 2.88 (ddd, J = 16.7, 7.3, 2.2, 1H), 2.65 (ddd, J = 16.7, 5.7, 1.8, 1H), 2.34 (ddd, J = 12.9, 7.4, 6.3, 1H), 1.71 (dtd, J = 13.7, 7.3, 4.3, 1H), 1.63 (ddd, J = 13.0, 5.6, 4.4, 1H), 1.57 (dddd, J = 13.9, 8.8, 6.1, 5.2, 1H), 0.89 (s, 9H), 0.88 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H and s, 3H and s, 3H); 13C NMR (150 MHz, CDCl3) δ 201.9, 82.8, 77.1, 72.8, 60.2, 50.6, 40.7, 36.9, 26.1, 26.0, 18.5, 18.1, −4.5, −4.6, −5.1; IR (ATR) 2929, 2722, 1726, 1084 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H43O4Si2 (M + H)+ 403.2699, found 403.2699. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
To a solution of the aldehyde (0.035 g, 0.088 mmol) in EtOH (1 mL) was added 2,4-dinitrophenylhydrazine (0.017 g, 0.083 mmol). The reaction mixture was heated at 80 °C overnight, then concentrated in vacuo. The product was purified by flash column chromatography (5:95 EtOAc:hexanes) to give 46 as a yellow solid (0.049 g, 96%): mp 116–128 °C; 1H NMR (600 MHz, CDCl3) δ 11.4 (s, 0.2H), 11.1 (s, 0.8H), 9.13 (d, J = 2.6, 0.2H), 9.12 (d, J = 2.6, 0.8H), 8.31 (ddd, J = 9.6, 2.6, 0.6, 0.2H), 8.29 (ddd, J = 9.6, 2.6, 0.8, 0.8H), 7.96 (d, J = 9.5, 0.2H), 7.92 (d, J = 9.5, 0.8H), 7.60 (t, J = 5.5, 0.8H), 7.16 (t, J = 5.7, 0.2H), 4.38 (tdd, J = 7.7, 6.0, 3.8, 0.2H), 4.34 (tt, J = 7.4, 5.2, 0.8H), 4.15 (ddd, J = 6.1, 4.4, 3.8, 0.2H), 4.12 (dt, J = 6.3, 3.9, 0.8H), 4.05 (dt, J = 8.5, 4.1, 0.2H), 3.97 (dt, J = 8.6, 4.0, 0.8H), 3.73 (ddd, J = 10.2, 6.8, 5.3, 1H), 3.69 (ddd, J = 10.2, 7.7, 6.1, 0.8H), 3.65 (ddd, J = 10.3, 7.4, 6.5, 0.2H), 2.83 (ddd, J = 14.9, 7.5, 5.3, 0.8H), 2.79 (ddd, J = 15.6, 8.0, 5.9, 0.2H), 2.68 (dt, J = 14.9, 5.5, 0.8H), 2.59 (ddd, J = 15.7, 5.5, 3.9, 0.2H), 2.36 (ddd, J = 13.1, 7.3, 6.2, 0.2H), 2.33 (ddd, J = 13.1, 7.4, 6.4, 0.8H), 1.76–1.69 (m, 2H), 1.61–1.57 (m, 1H), 0.90 (s, 7.2H), 0.89 (s, 7.2H), 0.88 (s, 1.8H), 0.86 (s, 1.8H), 0.08 (s, 2.4H and s, 1.2H), 0.07 (s, 2.4H), 0.06 (s, 2.4H), 0.05 (s, 2.4H), 0.02 (s, 0.6H), 0.01 (s, 0.6H); 13C NMR (150 MHz, CDCl3) δ 150.6, 145.3, 138.1, 130.1, 123.7, 116.7, 83.1, 77.1, 75.5, 60.2, 40.4, 39.4, 36.8, 35.0, 26.2, 26.1, 26.0, 25.9, 18.5, 18.2, −4.45, −4.51, −5.1; 3282, 3062, 2951, 1615, 1095 cm−1; HRMS (TOF MS ES+) m / z calcd for C26H47N4O7Si2 (M + H)+ 583.2983, found 583.2986. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
(E)-1-(2,4-dinitrophenyl)-2-(2-((2R*,3aR*,10aS*)-6,6,8,8-tetraisopropylhexahydrofuro[2,3-g][1,3,5,2,4]trioxadisilonin-2-yl)ethylidene)hydrazine 48
To a solution of 39 (0.162 g, 0.390 mmol) in CH2Cl2 (4 mL) at −78 °C was added a stream of ozone until a blue color persisted. The reaction mixture was purged with oxygen for 30 min and triphenylphosphine (0.205 g, 0.780 mmol) was added. The reaction mixture was stirred at room temperature overnight before concentrating in vacuo. The product was purified by flash column chromatography on silica gel (10:90 EtOAc:hexanes) to give an aldehyde as a colorless oil (0.151 g, 93%): 1H NMR (600 MHz, CDCl3) δ 9.81 (t, J = 1.9, 1H), 4.42 (dtd, J = 8.2, 6.3, 4.9, 1H), 4.13 (dd, J = 11.4, 3.8, 1H), 4.08 (dd, J = 11.8, 2.6, 1H), 3.97 (dd, J = 11.8, 3.9, 1H), 3.79 (ddd, J = 8.8, 3.7, 2.7, 1H), 3.66 (dd, J = 11.4, 9.5, 1H), 2.77 (ddd, J = 16.6, 6.7, 1.9, 1H), 2.60–2.53 (m, 2H), 1.87 (ddd, J = 12.7, 9.5, 8.3, 1H), 1.71 (ddd, J = 12.6, 8.9, 4.8, 1H), 1.11–0.92 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 201.4, 84.6, 72.7, 65.2, 63.8, 50.5, 41.1, 34.5, 17.67, 17.66, 17.64, 17.62, 17.58, 17.56, 17.52, 17.45, 13.7, 13.3, 13.07, 13.05; IR (ATR) 2955, 2725, 1726, 1464, 1040 cm−1; HRMS (TOF MS ES+) m / z calcd for C20H40O5Si2 (M + H)+ 417.2493, found 417.2489. Anal. Calcd for C20H40O5Si2: C, 57.65; H, 9.68. Found: C, 57.38; H, 9.71.
To a solution of the aldehyde (0.0909 g, 0.218 mmol) in EtOH (2 mL) was added 2,4-dinitrophenylhydrazine (0.0411 g, 0.207 mmol). The reaction mixture was heated at 80 °C overnight and cooled to room temperature before concentrating in vacuo to give an orange solid. The product was purified by flash column chromatography on silica gel (15:85 EtOAc:hexanes) to give 48 as an orange solid (0.114 g, 92%): mp 116–120 °C; 1H NMR (600 MHz, CDCl3) δ 11.05 (s, 1H), 9.12 (d, J = 2.5, 1H), 8.30 (ddd, J = 9.5, 2.6, 0.6, 1H), 7.92 (d, J = 9.6, 1H), 7.60 (t, J = 5.5, 1H), 4.26 (tt, J = 7.6, 4.9, 1H), 4.15 (dd, J = 11.4, 3.7, 1H), 4.11 (dd, J = 11.9, 2.5, 1H), 3.99 (dd, J = 11.8, 3.7, 1H), 3.81 (ddd, J = 8.9, 3.5, 2.7, 1H), 3.67 (dd, J = 11.3, 9.5, 1H), 2.70–2.57 (m, 3H), 1.85 (ddd, J = 12.6, 9.7, 8.2, 1H), 1.80 (ddd, J = 12.5, 8.9, 4.7, 1H), 1.11–0.91 (m, 28H); 13C NMR (150 MHz, CDCl3) δ 150.0, 145.3, 138.1, 130.1, 129.1, 123.7, 116.7, 84.8, 75.4, 65.2, 63.7, 41.1, 39.5, 34.2, 17.67, 17.65, 17.62, 17.55, 17.4, 13.7, 13.3, 13.1, 13.0, 12.8; IR (ATR) 3303, 2941, 1611, 1320, 1037 cm−1; HRMS (TOF MS ES+) m / z calcd for C26H45N4O8Si2 (M + H)+ 597.2776, found 597.2776. Anal. Calcd for C26H44N4O8Si2: C, 52.32; H, 7.43. Found: C, 52.50; H, 7.58.
General procedure for Deprotection
A solution of tetraisopropyldisiloxane protected diol was dissolved in tetrahydrofuran (0.1M). Tetrabutylammonium fluoride (3 equiv) was added dropwise at room temperature and the reaction mixture was allowed to stir overnight. Aqueous NH4HCO3 (5%, 3 mL) was added to the reaction mixture, which was then extracted with EtOAc (2 × 10 mL). The combined organics were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo.
(2R,3S,5R)-5-allyl-2-(hydroxymethyl)tetrahydrofuran-3-ol 43
The general deprotection procedure was followed with compound 37 (0.15 g, 0.38 mmol). The resulting oil was purified by flash column chromatography on silica gel (4:1 EtOAc:hexanes) to afford diol 43 as a light yellow oil (0.053 g, 86%). Characterization was performed on a 86:14 (1,3-cis-43:1,3-trans-43) mixture of diastereomers: [α]24D +34 (c 0.89, MeOH); 1H NMR (500 MHz, CD3OD) δ 5.89–5.78 (m, 1H), 5.09 (dd, J = 7.2, 1.6, 1H), 5.03 (dt, J = 10.3, 1.0, 1H), 4.22–4.10 (m, 1.16H), 4.09 (tt, J = 7.0, 6.5, 0.84H), 3.79–3.75 (m, 1H), 3.61 (dd, J = 11.7, 3.8, 0.84H), 3.53 (dd, J = 11.5, 5.3, 1.16H), 2.46–2.26 (m, 2.86H), 1.88 (dd, J = 13.2, 5.4, 0.14H), 1.74–1.63 (m, 1H); 13C NMR (125 MHz, CD3OD) δ 136.2, 136.1, 117.54, 117.49, 88.8, 86.9, 79.4, 79.2, 74.2, 73.5, 64.2, 63.5, 41.8, 41.4, 41.04, 40.95; IR (thin film) 3375, 3078, 2931, 1643 cm1; HRMS (TOF MS ES+) m / z calcd for C8H14NaO3 (M + Na)+ 181.0841, found 181.0837.
(2R*,3S*,5R*)-5-allyl-2-(2-hydroxyethyl)tetrahydrofuran-3-ol 45
The general deprotection procedure was followed with compound 38 (0.046 g, 0.11 mmol). The resulting oil was purified by flash column chromatography on silica gel (EtOAc) to afford diol 45 as a colorless oil (0.018 g, 97%): 1H NMR (600 MHz, CD3OD) δ 5.82 (ddt, J = 17.2, 10.3, 7.0, 1H), 5.11–5.03 (m, 2H), 4.05 (tt, J = 7.7, 6.5, 1H), 4.01 (td, J = 6.7, 5.3, 1H), 3.80 (dt, J = 8.4, 5.0, 1H), 3.69 (ddd, J = 10.8, 6.9, 5.4, 1H), 3.64 (ddd, J = 10.8, 7.6, 6.3, 1H), 2.41 (dtt, J = 13.5, 7.0, 1.3, 1H), 2.33–2.27 (m, 2H), 1.78 (dddd, J = 13.9, 7.6, 7.0, 4.8, 1H), 1.67 (dddd, J = 14.0, 8.3, 6.3, 5.4, 1H), 1.62 (ddd, J = 12.7, 7.7, 6.6, 1H); 13C NMR (150 MHz, CD3OD) δ 136.0, 117.4, 83.2, 78.0, 77.2, 60.3, 41.7, 40.5, 37.1; IR (thin film) 3351, 3077, 2923, 1642, 1050 cm−1; HRMS (TOF MS ES+) m / z calcd for C9H16NaO3 (M + Na)+ 195.0998, found 195.1001. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
((2R*,3S*,5R*)-(5-allyltetrahydrofuran-2,3-diyl)dimethanol 47
The general deprotection procedure was followed with compound 42 (0.071 g, 0.18 mmol). The resulting oil was purified by flash column chromatography on silica gel (EtOAc) to afford diol 47 as a colorless oil (0.030 g, 99%). Characterization was performed on a 8:92 (1,3-cis-47:1,3-trans-47) mixture of diastereomers: 1H NMR (600 MHz, MeOD) δ 5.87–5.80 (m, 1H), 5.10–5.02 (m, 2H), 4.05 (dq, J = 9.7, 6.0, 0.1H), 3.97 (dq, J = 7.7, 6.4, 0.9H), 3.79 (ddd, J = 7.3, 5.6, 4.3, 0.1H), 3.68 (td, J = 6.0, 4.4, 0.9H), 3.62 (dd, J = 11.3, 4.4, 0.9H), 3.59–3.54 (m, 2.2H), 3.52 (dd, J = 10.8, 7.0, 0.9H), 2.38–2.33 (m, 1H), 2.26–2.21 (m, 1.1H), 2.20–2.16 (m, 0.9H), 2.13 (ddd, J = 12.8, 7.6, 5.5, 0.1H), 1.82 (ddd, J = 12.5, 6.5, 5.2, 0.9H), 1.72 (ddd, J = 12.6, 9.2, 7.9, 0.9H), 1.37 (dt, J = 12.2, 9.8, 0.1H); 13C NMR (150 MHz, MeOD) δ 136.3, 117.4, 84.5, 79.7, 65.7, 64.8, 44.8, 41.5, 35.3; IR (ATR) 3335, 3076, 2929, 1641 cm−1; HRMS (TOF MS ES+) m / z calcd for C9H16NaO3 (M + Na)+ 195.0997, found 195.0995. Two attempts to obtain satisfactory combustion analysis data were unsuccessful, likely because minor impurities were present that could be detected by 1H NMR spectroscopy.
Supplementary Material
Scheme 6.
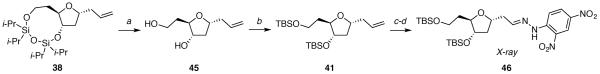
aReagents: (a) n-Bu4NF, 97%; (b) t-BuMe2SiCl, imidazole, 56%; (c) O3, PPh3, 83%; (d) C6H4(NO2)2NHNH2, 96%.
Acknowledgement
This research was supported by the National Institutes of Health, National Institute of General Medical Science (GM-61066). We thank Dr. Phil Dennison (University of California, Irvine) and Dr. Chin Lin (NYU) for assistance with NMR spectroscopy, Dr. John Greaves (UCI) and Ms. Shirin Sorooshian (UCI) for mass spectrometric data, and Dr. Joe Ziller (UCI) and Dr. Chunhua Hu (NYU) for the X-ray crystallographic analysis. We also thank the Molecular Design Institute of NYU for purchasing a single-crystal diffractometer.
Footnotes
Supporting Information Available: Complete experimental procedures, product characterization, stereochemical proofs, X-ray crystallographic data (CIF) and spectral data for all compounds are available in the online version of the paper. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Jarchow-Choy SK, Sjuvarsson E, Sintim HO, Eriksson S, Kool ET. J. Am. Chem. Soc. 2009;131:5488–5494. doi: 10.1021/ja808244t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Huang H, Stivers JT, Greenberg MM. J. Am. Chem. Soc. 2009;131:1344–1345. doi: 10.1021/ja807705z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Matsuda S, Henry AA, Schultz PG, Romesberg FE. J. Am. Chem. Soc. 2003;125:6134–6139. doi: 10.1021/ja034099w. [DOI] [PubMed] [Google Scholar]
- (4).Lai JS, Qu J, Kool ET. Angew. Chem. Int. Ed. 2003;42:5973–5977. doi: 10.1002/anie.200352531. [DOI] [PubMed] [Google Scholar]
- (5).Haraguchi K, Delaney MO, Wiederholt CJ, Sambandam A, Hantosi Z, Greenberg MM. J. Am. Chem. Soc. 2002;124:3263–3269. doi: 10.1021/ja012135q. [DOI] [PubMed] [Google Scholar]
- (6).D'Souza FW, Ayers JD, McCarren PR, Lowary TL. J. Am. Chem. Soc. 2000;122:1251–1260. [Google Scholar]
- (7).Ishiwata A, Ito Y. J. Am. Chem. Soc. 2011;133:2275–2291. doi: 10.1021/ja109932t. [DOI] [PubMed] [Google Scholar]
- (8).Migawa MT, Drach JC, Townsend LB. J. Med. Chem. 2005;48:3840–3851. doi: 10.1021/jm0402014. [DOI] [PubMed] [Google Scholar]
- (9).Li P, Xu Z, Liu H, Wennefors CK, Dobrikov MI, Ludwig J, Shaw BR. J. Am. Chem. Soc. 2005;127:16782–16783. doi: 10.1021/ja055179y. [DOI] [PubMed] [Google Scholar]
- (10).Hocek M. Chem. Rev. 2009;109:6729–6764. doi: 10.1021/cr9002165. [DOI] [PubMed] [Google Scholar]
- (11).Larsen CH, Ridgway BH, Shaw JT, Smith DM, Woerpel KA. J. Am. Chem. Soc. 2005;127:10879–10884. doi: 10.1021/ja0524043. [DOI] [PubMed] [Google Scholar]
- (12).Larsen CH, Ridgway BH, Shaw JT, Woerpel KA. J. Am. Chem. Soc. 1999;121:12208–12209. [Google Scholar]
- (13).Smith DM, Tran MB, Woerpel KA. J. Am. Chem. Soc. 2003;125:14149–14152. doi: 10.1021/ja0375176. [DOI] [PubMed] [Google Scholar]
- (14).Wichai U, Woski SA. Org. Lett. 1999;1:1173–1175. doi: 10.1021/ol990813w. [DOI] [PubMed] [Google Scholar]
- (15).Smith DM, Woerpel KA. Org. Lett. 2004;6:2063–2066. doi: 10.1021/ol0492647. [DOI] [PubMed] [Google Scholar]
- (16).Wichai U, Woski SA. Bioorg. Med. Chem. Lett. 1998;8:3465–3468. doi: 10.1016/s0960-894x(98)00632-5. [DOI] [PubMed] [Google Scholar]
- (17).Weinhold F, West R. Organometallics. 2011;30:5815–5824. [Google Scholar]
- (18).Verdegaal CHM, Dekok AJ, Westerink HP, Van Boom JH, Romers C. Acta Crystallogr., Sect. B: Struct. Sci. 1981;37:1924–1926. [Google Scholar]
- (19).Ishiwata A, Akao H, Ito Y. Org. Lett. 2006;8:5525–5528. doi: 10.1021/ol062198j. [DOI] [PubMed] [Google Scholar]
- (20).Shimomura N, Saitoh M, Mukaiyama T. Chem. Lett. 1994:433–436. [Google Scholar]
- (21).Krumper JR, Salamant WA, Woerpel KA. J. Org. Chem. 2009;74:8039–8050. doi: 10.1021/jo901639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Krumper JR, Salamant WA, Woerpel KA. Org. Lett. 2008;10:4907–4910. doi: 10.1021/ol8019956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Friestad GK, Lee HJ. Org. Lett. 2009;11:3958–3961. doi: 10.1021/ol901613k. [DOI] [PubMed] [Google Scholar]
- (24).Deriaz RE, Overend WG, Stacey M, Teece EG, Wiggins LF. J. Chem. Soc. 1949:1879–1883. [Google Scholar]
- (25).Sandri J, Viala J. Synthesis. 1995:271–275. [Google Scholar]
- (26).Hendrickson JB. J. Am. Chem. Soc. 1967;89:7036–7043. [Google Scholar]
- (27).Shao Y, Molnar LF, Jung Y, Kussmann J, Ochsenfeld C, Brown ST, Gilbert ATB, Slipchenko LV, Levchenko SV, O'Neill DP, DiStasio RA, Jr, Lochan RC, Wang T, Beran GJO, Besley NA, Herbert JM, Yeh Lin C, Van Voorhis T, Hung Chien S, Sodt A, Steele RP, Rassolov VA, Maslen PE, Korambath PP, Adamson RD, Austin B, Baker J, Byrd EFC, Dachsel H, Doerksen RJ, Dreuw A, Dunietz BD, Dutoi AD, Furlani TR, Gwaltney SR, Heyden A, Hirata S, Hsu C-P, Kedziora G, Khalliulin RZ, Klunzinger P, Lee AM, Lee MS, Liang W, Lotan I, Nair N, Peters B, Proynov EI, Pieniazek PA, Min Rhee Y, Ritchie J, Rosta E, Sherrill CD, Simmonett AC, Subotnik JE, Woodcock HL, III, Zhang W, Bell AT, Chakraborty AK, Chipman DM, Keil FJ, Warshel A, Hehre WJ, Schaefer HF, III, Kong J, Krylov AI, Gill PMW, Head-Gordon M. Phys. Chem. Chem. Phys. 2006;8:3172–3191. doi: 10.1039/b517914a. [DOI] [PubMed] [Google Scholar]
- (28).Rhoad JS, Cagg BA, Carver PW. J. Chem. Phys. A. 2010;114:5180–5186. doi: 10.1021/jp9100448. [DOI] [PubMed] [Google Scholar]
- (29).Kopecky D, Rychnovsky S. J. Org. Chem. 2000;65:191–198. doi: 10.1021/jo9914521. [DOI] [PubMed] [Google Scholar]
- (30).Urban M, Pohl R, Klepetářová B, Hocek M. J. Org. Chem. 2006;71:7322–7328. doi: 10.1021/jo061080d. [DOI] [PubMed] [Google Scholar]
- (31).Rothman JH. J. Org. Chem. 2007;72:3945–3948. doi: 10.1021/jo062206+. [DOI] [PubMed] [Google Scholar]
- (32).Liang Y, Hnatiuk N, Rowley JM, Whiting BT, Coates GW, Rablen PR, Morton M, Howell AR. J. Org. Chem. 2011;76:9962–9974. doi: 10.1021/jo201565h. [DOI] [PubMed] [Google Scholar]
- (33).Gould JHM, Mann J. Nucleosides Nucleotides. 1997;16:193–213. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.