

Abstract
Pelizaeus-Merzbacher disease (PMD) is a hypomyelinating leukodystrophy caused by mutations of the proteolipid protein 1 gene (PLP1), which is located on the X chromosome and encodes the most abundant protein of myelin in the central nervous sytem. Approximately 60% of PMD cases result from genomic duplications of a region of the X chromosome that includes the entire PLP1 gene. The duplications are typically in a head-to-tail arrangement, and they vary in size and gene content. Although rodent models with extra copies of Plp1 have been developed, none contains an actual genomic rearrangement that resembles those found in PMD patients. We used mutagenic insertion chromosome engineering resources to generate the Plp1dup mouse model by introducing an X chromosome duplication in the mouse genome that contains Plp1 and five neighboring genes that are also commonly duplicated in PMD patients. The Plp1dup mice display progressive gait abnormalities compared with wild-type littermates. The single duplication leads to increased transcript levels of Plp1 and four of the five other duplicated genes over wild-type levels in the brain beginning the second postnatal week. The Plp1dup mice also display altered transcript levels of other important myelin proteins leading to a progressive degeneration of myelin. Our results show that a single duplication of the Plp1 gene leads to a phenotype similar to the pattern seen in human PMD patients with duplications.
Introduction
Pelizaeus-Merzbacher disease (PMD; MIM 312080) is a rare X-linked neurological disorder affecting CNS white matter. It is caused by mutations of the X-linked proteolipid protein 1 gene (PLP1; MIM 300401) that encodes the major protein of myelin, proteolipid protein (PLP). Approximately 60% of cases are caused by a genomic duplication of 56 kb to 11 Mb in size that includes PLP1 and neighboring genes (Inoue et al., 1999; Woodward et al., 2005; Lee et al., 2006; Carvalho et al., 2012). Duplications most commonly occur as direct tandem repeats.
Transgenic rodent models with 2–14 extra copies of Plp1 were made by random integration of the genomic Plp1 sequence from a cosmid clone into the genome (Kagawa et al., 1994; Readhead et al., 1994; Bradl et al., 1999). These models demonstrate that overexpression of Plp1 leads to demyelinating and/or hypomyelinating phenotypes and that Plp1 copy number approximately correlates with phenotypic severity (Anderson et al., 1998, 1999; Karim et al., 2007; Mayer et al., 2011), as in human PMD patients (Wolf et al., 2005). However, the transgenic animals are not ideal models for human PMD caused by duplication. They do not exactly recapitulate the X-linked tandem head-to-tail genomic rearrangement found in PMD patients. In light of a complex relationship between copy number and overexpression of Plp1 in the rodent models (Karim et al., 2007), no clear estimates of the extent of overexpression in human patients can be deduced from the animal model data. Finally, there are differences in the phenotypes between the transgenic models and human PMD. Human duplication patients have a slow progressive demyelinating phenotype, whereas the transgenic rodent models with low Plp1 copy progress more slowly and those with high copy progress more rapidly, dying at a young age. Although the phenotypic difference may simply reflect interspecies difference, it is equally likely to result from the failure to replicate the genomic rearrangement in PMD patients.
We used mutagenic insertion and chromosome engineering resource (MICER) (Liu et al., 2011) reagents to generate a new model of PMD that is a more faithful representation of human PMD duplication than the transgenic models. Our new model has a 260.8 kb genomic duplication at the Plp1 locus, which is highly syntenic with the human PLP1 locus. The duplication is in the size range and the direct repeat arrangement common among PMD patients. Our model is suitable for analysis of the duplication phenotype and the expression of Plp1 and the other genes encoded by the duplication and expression of other genes in the program of myelination. The six other mouse models of genomic duplication diseases that were made using MICER reagents model autosomal rather than X-linked diseases (Walz et al., 2006; Li et al., 2007; Nakatani et al., 2009; Yu et al., 2010; Horev et al., 2011). Our new model will help us understand how an X-linked genomic rearrangement affects phenotype and allow us to pursue therapeutic options for PMD.
Materials and Methods
Generation of the Plp1dup mouse model.
All work was performed in accordance with the U.S. Public Health Service's Policy on Humane Care and Use of Laboratory Animals (Office of Laboratory Animal Welfare, 2013) and the Guide for the Care and Use of Laboratory Animals (Office of Laboratory Animal Welfare, 2013) and with approval and oversight of the Institutional Animal Care and Use Committees of Nemours/Alfred I. duPont Hospital for Children or Thomas Jefferson University as appropriate. The Plp1dup mouse line was created using MICER clones obtained from the Wellcome Trust Sanger Institute (Adams et al., 2004; Liu et al., 2011). Clones were chosen for their insert position relative to the Plp1 gene and the orientation of their loxP site relative to the genomic insert. Because the Plp1 gene is on the X chromosome, only insertion of vector sequences in cis-configuration has to be considered because the constructs were transfected into AB2.2 embryonic stem (ES) cells (a gift from the Wellcome Trust Sanger Institute), which are male cells containing only one X chromosome per cell.
Figure 1 shows steps in the generation of the Plp1dup mouse line. MICER genomic clone MHPN312j24 (NCBIM37, 133123007-133127471) was chosen for insertion proximal of Plp1. DNA prepared from the clone was linearized with NheI enzyme and transferred into ES cells of the Hprt-deficient AB2.2 line by electroporation. Targeted cells were selected with G418 for 10–12 d before colonies were selected for expansion. DNA was isolated from colonies grown in five 96-well plates and digested with XbaI for analysis by Southern blot to determine which samples had the plasmid inserted into the cellular DNA by homologous recombination with the genomic insert of the plasmid rather than by random integration. We obtained 12 positive clones using a 3′ probe. All were confirmed to be positive by long-range PCR amplification across the 5′ integration site. Three clones (4B9, 4F2, and 5H12) were checked by karyotype analysis and subsequently injected into C57BL/6 blastocysts to check for germline transmission. All produced a high percentage of agouti chimeric males when mated with C57BL/6 females, demonstrating germline transmission. Clone 5H12 was chosen for continuation with the second targeting because the line from this clone was the most robust in production of agouti progeny.
Figure 1.

Plp1dup mice were generated by chromosome engineering. A, Strategy for generating Plp1dup mice using two targeting steps and a loxP recombination step. MHPN312j24 and MHPP264b06 are the mouse engineering vectors inserted in the first and second targeting steps at the ends of the region to be duplicated. Boxes with “a–d” represent targeted regions of the genome that were digested in the vectors by NheI and SacI restriction enzymes, respectively. The locations of probes used to check ES cell DNA by Southern blot hybridization after each step are indicated by black squares and restriction fragments analyzed are shown for each step. Representative Southern blot analysis of ES cells after (B) the first engineering step showing one recombinant clone, (C) the second engineering step showing two recombinant clones, and (D) the loxP recombination step showing all recombinant clones. E, Representative interphase nucleus from FISH analysis of blood lymphocytes from a male Plp1dup mouse. The Plp1-containing fosmid probe (red) hybridized to two regions and the control probe that is outside of the duplicated region (green) hybridized to one region. Scores are as follows: Male Plp1dup mouse (n = 100), 24% of nuclei had one red spot, 69% had two, 7% had three or more; normal male mouse (n = 100), 81% of nuclei had one red spot, 15% had two, 4% had 3 or more. Only nuclei with one green spot were scored. F, Diagram of the 260.8 kb duplication in Plp1dup mice showing relative locations and approximate sizes of the genes within the duplicated region in the Plp1dup mouse model. Those genes shown on top are transcribed from centromere to telomere; those on bottom from telomere to centromere. tgt, Targeted genotype.
MICER clone MHPP264b06 (NCBIM37, 133374577-133383807) was chosen for insertion distal of Plp1. DNA prepared from the clone was linearized with SacI enzyme and transferred to clone 5H12 cells by electroporation. Doubly targeted cells were selected using puromycin, and resistant colonies were transferred into 96-well plates. DNA from three 96-well plates was isolated and digested with BsoBI to analyze by Southern blot with a 5′ probe. Five positive clones were obtained; of these, clone 1C7 was selected for the next step.
Doubly targeted clone 1C7 was transfected with the Cre-expressing plasmid pOG231 (O'Gorman et al., 1997) (a gift from Alex Gow, Wayne State University) to catalyze the recombination of the loxP sites within the plasmid sequences incorporated into the genome from the first two targeting events. These loxP sites were located in intron 2 sequence of the 5′ and 3′ parts of HPRT that were inserted with the genomic clones in the first and second targeting steps, respectively. Recombination between the loxP sites generated the genomic duplication that includes Plp1 and the region between the two targeting vectors. It also generated a functional HPRT gene from the 5′ and 3′ parts of HPRT, and selection was performed using HAT medium. HATR colonies were transferred into a 96-well plate for expansion, and DNA was isolated and digested with EcoRI for Southern blot analysis using the rec probe. Most clones showed the expected pattern, but three were chosen for closer analysis: B8, C12, and F11. Clones B8 and F11 were determined to be euploid, but C12 was not. Cells from all three clones were injected into C57BL/6 blastocysts that were then surgically transferred to pseudopregnant female mice. Chimeric progeny were bred to C57BL/6 females to determine germline transmission. All three clones showed germline transmission, but clone B8 had more and the largest litters. Chimeras produced from clone B8 were mated with C57BL/6 mice to produce the F1 generation, which showed germline transmission by agouti coat color. Carrier females were mated with male littermates with black coat color to produce the mixed genetic background F2 generation. Carrier F2 females were mated with mixed background wild-type males, males from either the F1 generation or B6129SF1/J (The Jackson Laboratory), to maintain the Plp1dup line on the mixed background. The mouse colony was maintained in the Life Science Center at the Nemours/Alfred I. duPont Hospital for Children and was provided a diet of water and standard mouse chow ad libitum. Male Plp1dup mice on the mixed background were used in all experiments in this work.
Gait analysis.
Plp1dup mice and their wild-type littermates were analyzed at 2, 4, and 6 months using the CatWalk automated gait analysis system (Noldus Information Technology) according to the manufacturer's instructions. The system includes an elevated glass plate with a fluorescent light source on the side, walls to prevent mice from escaping, and a high-speed camera that connects to a computer and captures video footage. Mice were trained for 2 d to acclimate them to the environment. On the first day, they were allowed to explore the walkway with room lights on, an open lid, and walkway lights off for 10 min. On the second day, the lid was closed, room lights were turned off, and walkway lights were turned on for the same period of time. On the day of trial, three compliant runs were collected with a maximum speed variation of 60% and a maximum time to cross of 8.00 s.
The CatWalk software collects information on a wide variety of gait parameters per run. The software processed the three compliant runs by assigning labels for each paw. We chose to focus on parameters relevant to our mouse model, which are defined in Table 1. Statistical analysis was performed longitudinally using two-way repeated measures ANOVA with GraphPad Prism version 6.0. To obtain cross-sectional analysis at specific time points, individual unpaired t values were performed. A p value <0.05 was considered statistically significant.
Table 1.
CatWalk gait analysis
| Parameter | Paws | Description (units) | Cross-sectional dataa |
6 months | Longitudinal datab |
Interaction | ||
|---|---|---|---|---|---|---|---|---|
| 2 months | 4 months | Group | Time | |||||
| Stand index | Front paws | Speed at which the paw loses contact with the glass plate (arbitrary units) | NS | 4.10* (0.03) | NS | NS | 19.54* (0.0008) | 7.31* (0.01) |
| Hind paws | NS | 2.46* (0.001) | 4.35* (0.002) | 30.75* (0.005) | 31.35* (0.0002) | 25.47* (0.0003) | ||
| Swing | Front paws | Duration of no contact of the paw with the glass plate (seconds) | NS | NS | 0.07* (0.03) | NS | 8.23* (0.01) | NS |
| Hind paws | 0.06* (0.0003) | NS | 0.07* (0.01) | NS | 19.81* (0.0008) | 10.35* (0.006) | ||
| Step cycle | Front paws | Time between two consecutive initial contacts of the same paw (seconds) | NS | NS | 0.14* (0.01) | NS | NS | NS |
| Hind paws | NS | NS | 0.14* (0.004) | NS | NS | NS | ||
| Cadence | Steps per second | NS | NS | 4.30* (0.005) | 25.26* (0.007) | NS | NS | |
| Print area | Front paws | Surface area of the complete paw print (mm2) | NS | NS | NS | NS | NS | NS |
| Hind paws | NS | 3.84* (0.006) | 4.49* (0.01) | NS | 181.00* (<0.0001) | 4.90* (0.04) | ||
| Support single | Percentage of time one paw is in contact with the glass plate | NS | NS | 2.57* (0.02) | NS | NS | NS | |
| Support diagonal | Diagonal paws | Percentage of time diagonal paws are in contact with the glass plate | NS | NS | NS | 11.30* (0.02) | 14.11* (0.002) | NS |
| Support girdle | Front versus hind paws | Percentage of time girdle paws are in contact with the glass plate | NS | NS | NS | NS | NS | NS |
| Support lateral | Left versus right paws | Percentage of time lateral paws are in contact with the glass plate | NS | NS | NS | NS | NS | NS |
| Support three | Percentage of time three paws are in contact with the glass plate | NS | NS | 10.18* (0.003) | 12.41* (0.02) | 20.35* (0.0007) | NS | |
| Support four | Percentage of time all four paws are in contact with the glass plate | NS | NS | NS | NS | NS | NS | |
| Base of support | Front paws | Distance between girdle paws (mm) | NS | NS | NS | NS | 14.54* (0.002) | NS |
| Hind paws | NS | NS | NS | 31.45* (0.005) | 51.36* (<0.0001) | NS | ||
N = 7 for each group (wild-type and Plp1dup) at each time (2, 4, and 6 months). NS, Not significant.
*Significant (p < 0.05).
aCross-sectional data comparing groups at each time point (t test) are presented as difference between means (p value).
bLongitudinal data (two-way repeated measures ANOVA) are presented as F (p value). Interaction is of group and time.
PCR-based genotyping.
DNA for genotyping was isolated from tail snips collected at weaning using the DNeasy Blood and Tissue Kit (QIAGEN) following the manufacturer's protocol for genotyping the mice. DNA was isolated from spleen tissue using the Gentra Puregene Tissue Kit (QIAGEN) for confirmation of the genotype.
Genotyping was performed by semiquantitative multiplex PCR using a Multiplex PCR Kit (QIAGEN) according to the manufacturer's instructions. Primer pairs multiplexed for quantitative analysis were as follows: a region of the Plp1 gene, a region at the proximal end of the duplication, a region of the dystrophin gene (Dmd) that is not within the duplication and served as a single-copy control, a region of the Sry gene on the Y chromosome for gender confirmation, and regions of the bacterial neo and puro genes that were inserted during engineering of the duplication (Table 2). One member of each primer pair was labeled with FAM. PCR products were detected and analyzed by capillary electrophoresis using the 3130 xL Genetic Analyzer (Invitrogen) and Peak Scanner Software (Invitrogen). Copy number was determined using net intensities to distinguish Plp1dup mice from wild-type mice. The ratio of test region to dystrophin of the test animal was normalized by dividing by the average ratio of test region to dystrophin of two wild-type male control mice. If a test region was duplicated, the expected normalized ratio was 2; if it was not duplicated, the expected normalized ratio was 1.
Table 2.
Primer sequences and their uses
| Primer sequence | Use |
|---|---|
| 5′-CCACCTGTTTATTGCTGCGTTTG | Multiplex PCR; detection of Plp1 |
| 5′-GCACACTGCATTGGCACTACATTAAC | |
| 5′-AATGTCTGGGTAGGGTGCTTGTC | Multiplex PCR; detection of proximal region of duplication |
| 5′-AAGAACGGAGATGCTGTTAGATGAG | |
| 5′-GCAGACAGAAAATCCCAAAGAGC | Multiplex PCR; detection of single-copy Dmd |
| 5′-TTGTGCTAACTTCATTCCACCAAG | |
| 5′-GCATTTATGGTGTGGTCCCGTG | Multiplex PCR; detection of Sry |
| 5′-GCTCTACTCCAGTCTTGCCTGTATG | |
| 5′-GATGATCTGGACGAAGAGCATCAG | Multiplex PCR; detection of neo |
| 5′-TAAAGCACGAGGAAGCGGTCAG | |
| 5′-GCCTTCCATCTGTTGCTGCG | Multiplex PCR; detection of puro |
| 5′-GCTGCAAGAACTCTTCCTCACG | |
| 5′-GCTGAGGATTTGGAAAGGGTG | Detection of HPRT |
| 5′-CCTTGAGCACACAGAGGGCTAC | |
| 5′-GTTGTGCTAGATGTCTGGTAGG | RT-PCR; detection of Plp1 and Dm20 isoforms |
| 5′-AGCCATACAACAGTCAGGGCATAG | |
| 5′-GTGACGTTGACATCCGTAAAGACC | RT-PCR; actin control |
| 5′-AAGAAAGGGTGTAAAACGCAGCTC |
For confirmation of the genotype, PCR amplification was performed using the Expand High Fidelity PCR system (Roche) and touchdown PCR (Table 2) (Korbie and Mattick, 2008) on spleen DNA with primers from the HPRT gene, which was produced during generation of the duplication by Cre-mediated recombination. Products were subjected to electrophoresis on 1% agarose gels. Gels were stained with ethidium bromide, and signals were visualized using the Gel Doc XR system (Bio-Rad).
FISH.
Chromosome spreads for interphase FISH were prepared from hemagglutinin- and colcemid-treated lymphocytes obtained from mouse blood by cardiac puncture using heparinized syringes and fixed in methanol:acetic acid (3:1) and stored in fixative at −20°C. Hybridization was performed with Plp1-containing fosmid WI1-1339A16 (BACPAC Resources; NCBI m36 coordinates 133336508-133376654) labeled with biotin-16-dUTP (Roche) and performed with single-copy control probe WI1-403O13 (BACPAC Resources; NCBI m36 coordinates 77435065-77479315) labeled with digoxigenin (Roche) using standard protocols (Pinkel et al., 1988). Cy3-labeled streptavidin (GE Healthcare) was bound to the biotin-labeled probe, amplified with biotinylated antiavidin (Vector Laboratories), and detected with another layer of Cy3-labeled streptavidin. Simultaneously, digoxigenin-labeled probe was coupled with mouse antidigoxigenin (Roche) and detected with rabbit anti-mouse FITC (Jackson ImmunoResearch), further amplified with goat anti-rabbit FITC antibody (Jackson ImmunoResearch). Images were captured using a Leica MTC microscope (Leica Microsystems) and Openlab imaging software (PerkinElmer).
RNA extraction, RT-PCR, semiquantitative RT-PCR, and qRT-PCR.
For semiquantitative RT-PCR and qRT-PCR experiments, total RNA was extracted from half brains cut along the sagittal plane, snap frozen on dry ice with ethanol, and stored at −80°C. RNA was isolated from three wild-type and three Plp1dup half brains at P7, P10, P12, P14, P21, P60, and P120 using QIAzol lysis reagent and the RNeasy Midi Kit (QIAGEN) and treated with DNase using the TURBO DNA-free kit (Invitrogen) following the manufacturers' protocols. Reverse transcription was performed using the Omniscript reverse transcription kit (QIAGEN) following the manufacturer's protocols.
For semiquantitative multiplex RT-PCR of the Plp1 and Dm20 alternative splice variants, PCR amplification was performed with the Multiplex PCR Kit (QIAGEN) according to the manufacturer's protocol. A primer pair was used to amplify Plp1 and Dm20 transcripts as differently sized products, and a β-actin primer pair was used to ensure that equal amounts of RNA were used in the reactions. Primers, one member of each pair labeled with FAM, are listed in Table 2. Products were detected and analyzed by capillary electrophoresis using the 3130 xL Genetic Analyzer (Invitrogen) and Peak Scanner Software (Invitrogen).
Transcript levels of Plp1; the five other genes within the duplication; and the myelin genes, myelin-associated glycoprotein (Mag), myelin basic protein (Mbp), and 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (Cnp), were determined by qRT-PCR in 384-well clear optical reaction plates (Invitrogen) using a QuantiTect SYBR Green PCR kit (QIAGEN) and QuantiTect Primer Assays (QIAGEN) according to the manufacturer's instructions. The following assays were used: Mm_Plp1_1_SG, Mm_Glra4_1_SG, Mm_Tceal1_3_SG, Mm_Tceal3_1_SG, Mm_Morf4l2_1_SG, Mm_BC065397_2_SG, Mm_Mbp_1_SG, Mm_Mag_1_SG, and Mm_Cnp_1_SG. The primer assays on Mus musculus calnexin (Canx) and Mus musculus cytochrome c-1 (Cyc1) were selected as reference genes (PrimerDesign) using geNorm (Vandesompele et al., 2002). Experiments were performed in triplicate reactions on three biological replicates of wild-type and Plp1dup mice at each age by using the ABI 7900HT Fast Real-Time PCR System (Invitrogen) with SDS 2.3 software (Invitrogen). All genes of interest and the endogenous controls were assayed for a single time point on each 384-well plate. Data were normalized by dividing raw Ct values for each sample by the geometric mean of Ct values of the Canx and Cyc1 internal reference genes. Relative quantification (RQ) of RNA transcript levels in the Plp1dup mouse was determined using the average of the three wild-type males as the calibrator and DataAssist Software version 2.0 (Invitrogen) with the 2−ΔΔCt method. The relative amounts of mRNA for each transcript were expressed as a percentage of wild-type at each time point using the average RQ values for Plp1dup mice at each time point. Statistical analyses were performed using a one-way ANOVA. A p value of <0.05 was considered statistically significant.
Protein extraction and Western blot analysis.
Protein experiments by Western blot were performed to detect myelin proteins PLP, myelin basic protein (MBP), 2′,3′-cyclic-nucleotide 3′-phosphodiesterase (CNP), and myelin-associated glycoprotein (MAG). Half-brains cut along the sagittal plane were snap frozen on dry ice with ethanol, stored at −80°C, and thawed on ice. Protein lysates were prepared from three wild-type and three Plp1dup mice at each time point (P7, P10, P12, P14, P21, P60, and P120) by homogenization using a hand-held homogenizer in 1 ml of buffer at 0.1 m NaCl, 0.01 m Tris-Cl, pH 7.6, 0.001 m EDTA, pH 8.0, 10 μl of Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) per milliliter of buffer, and 1 ml of 2× SDS Gel Loading Buffer (100 mm Tris-Cl, pH 6.8, 4% SDS, 20% glycerol, and 200 mm DTT). The homogenate was sonicated with 10 pulses of 1 s each. After centrifugation at 5000 × g for 5 min, the supernatant liquid was removed, stored at −80°C, thawed on ice, and used as the protein lysate. Protein concentration was determined by measuring absorbance at 280 nm using a NanoDrop spectrophotometer (Thermo Fisher Scientific), and 30 μg of protein was separated on 4–20% TGX gel (Bio-Rad) and transferred to a PVDF membrane (Invitrogen). Two gels were used for each experiment to accommodate all of the time points. Membranes were cut appropriately and probed with mouse monoclonal antibodies anti-PLP at a dilution of 1:2000 (AA3 Hybridoma, a gift from Dr. Alexander Gow, Wayne State University), anti-MBP at 1:1000 (Millipore, catalog #09-849), anti-CNP at 1:5000 (Covance, SMI-91R), anti-MAG at 1:1000 (Millipore, catalog #MAB1567), and anti-GAPDH at 1:20000 (Sigma-Aldrich; catalog #G8795). Membranes were then incubated in DyLight-405- (Jackson ImmunoResearch), Cy3-, or Cy5-labeled (GE Healthcare) secondary antibody diluted 1:2500, and labeled protein was detected using the Typhoon Trio imager (GE Healthcare). Bands were quantified using ImageQuant TL version 2003.03 software (GE Healthcare). GAPDH was used as a control for loading accuracy. Relative amounts of protein from Plp1dup mice were expressed as a percentage of wild-type at each time point.
Pathology.
For microscopy of the brain, mice at 1, 3, and 6 months of age were killed by carbon dioxide inhalation followed by cervical dislocation. Brains were removed, fixed in 4% paraformaldehyde in 0.1 m PBS, dissected coronally into 2 mm pieces and submitted to the Histotechnology Core at the Nemours/Alfred I. duPont Hospital for Children for processing and paraffin embedding. Samples were cut in 5 μm sections using a Leica RM2255 microtome (Leica Microsystems). Sections were stained with Luxol Fast Blue, periodic acid-Schiff, and hematoxylin (LFB/PAS) to visualize myelin content. Sections were visualized and imaged using an Olympus BX51 microscope (Olympus) with Image Pro Plus software (Media Cybernetics). Alternatively, immunofluorescent staining was completed using 2% normal goat serum with 0.02% Triton X-100 for blocking and permeabilization, followed by the addition of primary monoclonal antibodies anti-PLP (1:1000, Abcam; AB9311), anti-MBP (1:1000, Covance; SMI-94), and anti-Pan-Axonal neurofilament (1:1000, Covance; SMI-312) and then secondary antibodies goat anti-mouse-IgG2a-Alexa-488, goat anti-mouse-IgG1-Alexa-555, and goat anti-mouse-IgG2b-Alexa-647 (1:1000, Invitrogen). Nuclei were stained with DAPI (Invitrogen). Sections were visualized and imaged using a Zeiss LSM510 microscope (Carl Zeiss) with Zen 2009 software (Carl Zeiss).
For microscopy of the spinal cord, animals were anesthetized and perfusion-fixed transcardially with 4% paraformaldehyde/0.5% glutaraldehyde in 0.1 m PBS. The spinal cords were dissected transversely into 1 mm pieces and processed as follows. Tissues were rinsed in 0.1 m phosphate buffer 3 × 10 min and fixed with 1% osmium tetroxide (Electron Microscopy Sciences) for 2 h. They were rinsed again with 0.1 m phosphate buffer 3 × 10 min and dehydrated with increasing percentages of ethanol for 2 × 5 min up to 100% for 3 × 10 min and then with propylene oxide (Electron Microscopy Sciences) 3 × 10 min. Tissues were then infiltrated overnight with a 1:1 solution of propylene oxide and Araldite 502 (Electron Microscopy Sciences), then embedded the next day with 100% Araldite and cured at 60°C for 48 h. Semithin and ultrathin sections were cut on an Ultracut E microtome (Reichert Technologies) using a glass knife and diamond knife, respectively. The 1 μm semithin sections were stained with toluidine blue, and the ultrathin sections were stained with uranyl acetate (Electron Microscopy Sciences) and lead citrate (Polysciences) and visualized and imaged using a Jeol 1010 electron microscope (Jeol).
Blood–brain barrier integrity.
A 2% solution of Evan's blue dye (Sigma) was prepared in PBS and sterile-filtered. At weaning, male mice received intraperitoneal injections of 4 ml/kg body weight. The dye was allowed to circulate for 4 h. Mice were then killed by carbon dioxide inhalation followed by cervical dislocation, and brains, small and large intestines, liver, and spleen were examined for uptake of dye.
Results
Chromosome engineering of the Plp1dup mouse line
We used MICER reagents to generate a tandem head-to-tail duplication at the Plp1 locus to create the Plp1dup mouse line used in this work (Fig. 1). Figure 1A shows the engineering steps used to generate the duplication. In the two targeting steps, clones selected from MICER libraries were sequentially inserted proximal and then distal to the Plp1 gene in ES cells. These clones contained loxP sites and either the 5′ or the 3′ part of the HPRT gene. In the third step, we expressed Cre in the ES cells for site-specific recombination between the loxP sites, which resulted in direct tandem duplication of the 260.8 kb genomic region between the two sites and a complete HPRT gene at the junction. After each of the three engineering steps, selected clones were analyzed by Southern blot (Fig. 1B–D, examples). After the recombination step, clones were also verified by PCR amplification across the two parts of the HPRT gene (data not shown). The ES cells that were resistant to the three selection reagents used in the engineering steps (G418, puro, and HAT) were used to generate our Plp1dup line of mice as described. Presence of a duplication was confirmed in mice by semiquantitative multiplex PCR (data not shown), Southern blot analysis (data not shown), and FISH (Fig. 1E).
The Plp1dup mouse model is an appropriate model for human duplication disease in several ways. The duplication, diagrammed in Figure 1F, is 260.8 kb in size, which is within the size range of human PMD-causing duplications. It is in a tandem head-to-tail arrangement on the X chromosome, as are most human PMD duplications, and it includes other genes proximal of Plp1. The duplication ends 9.5 kb distal of Plp1. Two PMD patients we have studied, P348 and P116, have duplications that end in the same approximate location distal of PLP1 (Woodward et al., 2005), suggesting that potential regulatory sequences further distal of PLP1 do not need to be included in the duplication to generate the PMD phenotype. The Plp1 gene lies in a region of conserved synteny between mouse and human, and four of the five genes in the duplicated region are present within the region of synteny (Table 3). Further, the Plp1 gene itself is highly conserved from mouse to human. Indeed, the mouse PLP and DM20 proteins are identical in amino acid sequence to their human homologs. In addition, the transcript level of the Plp1 gene in both mice and humans is developmentally regulated with a rapid postnatal increase in transcription and translation as the predominant isoform switches from DM20 to the PLP form.
Table 3.
Locations of genes within the duplication in Plp1dup mouse and their human homologs
| Mouse gene | Locationa | Human homolog | Locationb |
|---|---|---|---|
| Tceal3 | X:133125381-133202917 | TCEAL3 | X:102862379-102884618 |
| TCEAL5 | X:102528619-102531800 | ||
| TCEAL6 | X:101395448-101397942 | ||
| Tceal1 | X:133242521-133246017 | TCEAL1 | X:102883632-102885881 |
| Morf4l2 | X:133267481-133278229 | MORF4L2 | X:102930424-102943086 |
| BC065397 | X:133276397-133277682 | No homologs | |
| Glra4 | X:133292213-133314680 | GLRA4 | X:102962152-102983583 |
| Plp1 | X:133357210-133374272 | PLP1 | X:103028647-103047548 |
aMouse reference sequence, NCBIM37.
bHuman reference sequence, GRCh37.
Observations and behavioral phenotype of the Plp1dup mouse
Plp1dup mice weighed less than wild-type littermates, averaging 35.2 ± 5.5 g compared with wild-type at 41.2 ± 7.7 g (p = 0.001, data not shown) at 6 months. During tail suspension, Plp1dup mice failed to climb or grasp the handler's hands or splay their paws as observed with wild-type mice.
Gait analysis was performed at 2, 4, and 6 months on seven Plp1dup males and seven wild-type littermates using the CatWalk system (Noldus Information Technology). The results for 12 parameters are presented in Table 1 as difference between means (p values) for cross-sectional analyses of individual time points as well as F (p values) for longitudinal studies. Few differences were observed at early time points, but dramatic differences were evident by the 6 month time point. Cross-sectional analyses revealed that Plp1dup mice performed worse on the walkway in terms of the parameters listed. When comparing wild-type and Plp1dup mice longitudinally, Plp1dup mice performed worse in terms of stand index, cadence, diagonal support, three-paw support, and base of support. Parameters significantly influenced by time included stand index, swing, print area, diagonal support, three-paw support, and base of support. Finally, the interaction between group and time was significant in terms of stand index, swing, and print area. The parameters that showed the most pronounced differences were related to speed, fluidity of movement, and support. Cadence, swing, and stand index indicated that Plp1dup mice moved slowly compared with wild-type littermates. An increased print area showed flat-footedness in Plp1dup mice, suggesting weakness. Although it is typical for wild-type mice to alternate diagonal paws, Plp1dup mice tended to rely on three paws at a time for support. Additionally, the hindpaws appeared to be more significantly affected than the front paws, which correlates to PMD patients, whose lower limbs are more severely affected than upper limbs (Golomb et al., 2004).
A chromosomal duplication in the Plp1 locus altered transcript levels of PLP1 mRNA and protein but did not alter the Plp1/Dm20 alternative splice ratio
We performed qRT-PCR on RNA prepared from mouse brains to determine the effect of the chromosomal duplication on the developmental regulation of Plp1 mRNA transcript levels. The time points P7, P10, P12, P14, P21, P60, and P120 were chosen to observe transcript levels throughout the whole myelination program. We first analyzed total Plp1 transcript levels (Plp1 + Dm20) at each time point relative to wild-type at P7, and results are presented in Figure 2A. Our wild-type mice showed the typical pattern of expression that has been reported previously with a peak in transcript levels at approximately the third postnatal week (Wight and Dobretsova, 2004). We also saw another upregulation of transcript levels by P120. Plp1dup mice had a similar pattern of Plp1 transcript levels with a peak at P14. Total Plp1 mRNA levels were not significantly different from wild-type at P7 and P10, but they were elevated to ∼200% of wild-type transcript levels at remaining time points except P21. At P21, there was not a statistically significant difference in Plp1 transcript levels between Plp1dup and wild-type mice, at which point both are decreasing.
Figure 2.
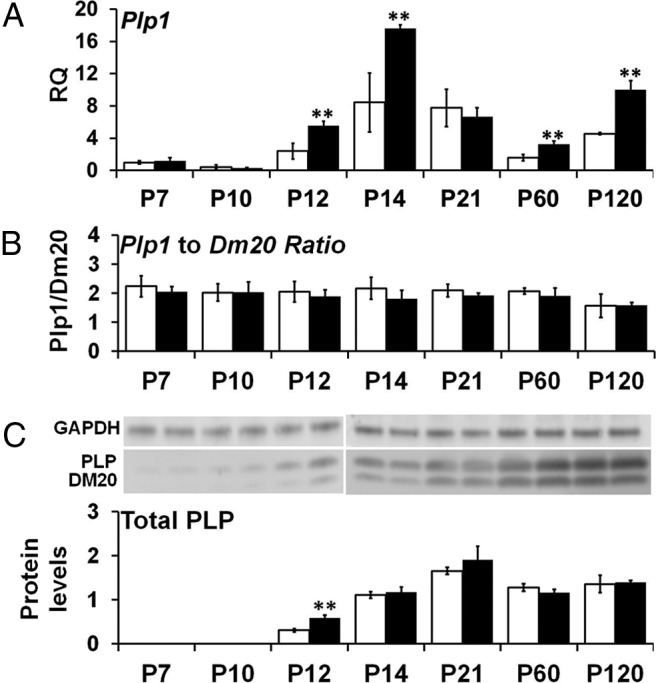
Developmental time course of Plp1 mRNA and PLP protein levels in Plp1dup compared with wild-type mouse brain. A, Levels of Plp1 mRNA measured by qRT-PCR of total RNA prepared from Plp1dup and wild-type brain and expressed as RQ with the average of P7 wild-type used as the calibrator. B, Ratio of alternatively spliced Plp1-specific and Dm20 transcript levels measured by semiquantitative RT-PCR of total RNA prepared from Plp1dup and wild-type brains. C, PLP and DM20 protein levels measured from Western blots, totaled for each time point, and expressed relative to GAPDH. Representative Western blots are shown above the graph in C. Total RNA and protein homogenates were prepared at postnatal days indicated (Plp1dup, n = 3; wild-type, n = 3 at each time point). Open bars represent wild-type; black bars, Plp1dup. Controls are described in Materials and Methods. **p < 0.01.
When we analyzed the isoform ratio of Plp1 to Dm20 at each developmental time point, we found that the ratio for Plp1dup mice were not significantly different from that of wild-type mice at each time point (Fig. 2B).
We also analyzed total PLP protein levels (PLP + DM20) at each developmental time point relative to wild-type at that time point. PLP protein levels were too low to be measured accurately by our methods until P12. Figure 2C shows that, at P12, total PLP was elevated to 191 ± 24% of wild-type levels in the Plp1dup mice but was not elevated at remaining time points. Together, our RNA and protein studies showing overexpression of Plp1 mRNA at data points when the protein is not elevated suggest that Plp1 gene expression is regulated post-transcriptionally.
Duplication at the Plp1 locus altered mRNA transcript levels of four of the five other genes within the duplication
The presence of other genes in addition to PLP1 in the duplications of PMD patients raises the possibility that altered transcript levels of these other genes could contribute to the disease. Thus, we analyzed transcript levels of five other genes within the duplication of Plp1dup mice [transcription elongation factor A (SII-like) 3 (Tceal3), transcription elongation factor A (SII-like) 1 (Tceal1), mortality factor 4-like 2 (Morf4l2), BC065397, and glycine receptor subunit α 4 (Glra4)] at each of the developmental time points relative to wild-type at that time point by qRT-PCR. Figure 3A–D shows that transcript levels of Tceal3, Tceal1, Morf4l2, and BC065397 were elevated to ∼200% in the Plp1dup mice compared with wild-type at all postnatal time points, except for Tceal3 at P10 and Morf4l2 at P7, where some variability among the biological replicates resulted in no statistically significant difference. However, Glra4 mRNA levels were essentially the same in Plp1dup mice compared with wild-type mice at all time points (Fig. 3E). Thus, duplication in the Plp1 locus altered the transcript levels of four of the five genes found within the duplication in addition to Plp1.
Figure 3.
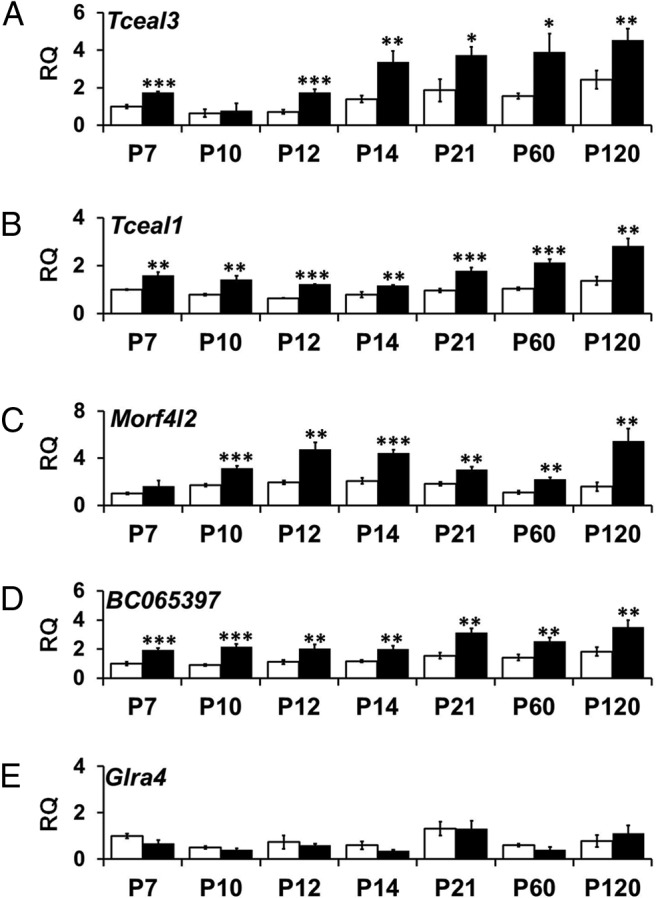
Developmental time course of transcript levels of genes within the duplication in Plp1dup compared with wild-type mouse brain. Levels of mRNA expressed from (A) Tceal3, (B) Tceal1, (C) Morf4l2, (D) BC065397, and (E) Glra4 were measured by qRT-PCR of total RNA prepared from Plp1dup brain and expressed as RQ with the average of P7 wild-type used as the calibrator. Total RNA was prepared at postnatal days indicated (Plp1dup, n = 3; wild-type, n = 3 at each time point). Open bars represent wild-type; black bars, Plp1dup. Controls are described in Materials and Methods. *p < 0.05. **p < 0.01. ***p < 0.001.
Duplication at the Plp1 locus affected transcript and protein levels of other myelin genes
We examined transcripts of oligodendrocyte markers CNP (Fig. 4), MBP (Fig. 5), and MAG (data not shown) at the mRNA and protein levels to determine whether altered expression of Plp1 in the Plp1dup mice altered the myelin program. All three genes were developmentally regulated at the mRNA and protein levels in both Plp1dup and wild-type mice. There was no statistically significant difference in Cnp mRNA levels between Plp1dup mice and wild-type mice, except at P21 when Plp1dup mRNA levels were 72 ± 2.5% of wild-type levels (Fig. 4A). Although Cnp mRNA levels were the same as wild-type at most time points, CNP protein levels were significantly lower at the adult time points, P60 and P120 (Fig. 4B). Mbp mRNA levels were lower in Plp1dup mice than wild-type at P7, P21, and P60 (Fig. 5A). Like CNP, MBP protein levels in Plp1dup mice were significantly lower than in wild-type at the adult time points, P60 and P120, but were only at very low levels at P21, P60, and P120 (Fig. 5B). Mag transcript and MAG protein levels did not show major significant differences from wild-type (data not shown).
Figure 4.
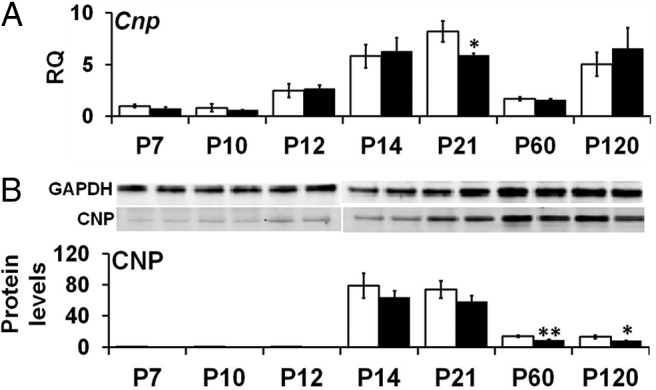
Developmental time course of Cnp mRNA and CNP protein levels in Plp1dup compared with wild-type mouse brain. A, Levels of Cnp mRNA measured by qRT-PCR of total RNA prepared from Plp1dup and wild-type brain and expressed as RQ with the average of P7 wild-type used as the calibrator. B, CNP protein levels measured from Western blots and expressed relative to GAPDH. Representative Western blots are shown above the graph in B. Total RNA and protein homogenates were prepared at postnatal days indicated (Plp1dup, n = 3; wild-type, n = 3 at each time point). Open bars represent wild-type; black bars, Plp1dup. Controls are described in Materials and Methods. *p < 0.05. **< 0.01.
Figure 5.
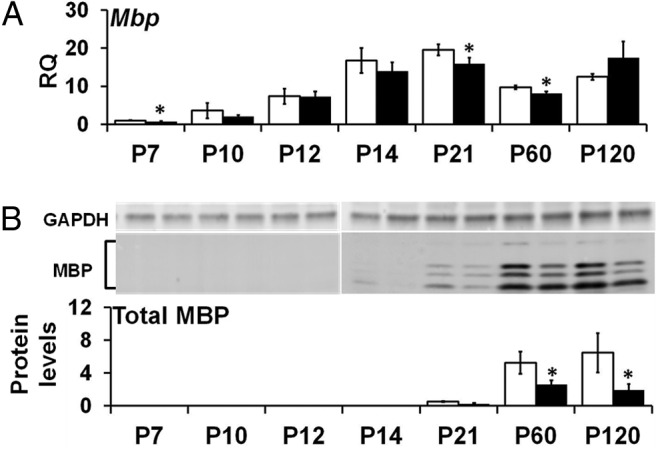
Developmental time course of Mbp mRNA and MBP protein levels in Plp1dup compared with wild-type mouse brain. A, Levels of Mbp mRNA measured by qRT-PCR of total RNA prepared from Plp1dup and wild-type brain and expressed as RQ with the average of P7 wild-type used as the calibrator. B, MBP protein levels measured from Western blots and expressed relative to GAPDH. Representative Western blots are shown above the graph in B. Total RNA and protein homogenates were prepared at postnatal days indicated (Plp1dup, n = 3; wild-type, n = 3 at each time point). Open bars represent wild-type; black bars, Plp1dup. Controls are described in Materials and Methods. *p < 0.05.
Duplication at the Plp1 locus results in degeneration of myelin
To evaluate morphological changes in myelin in Plp1dup mice compared with wild-type, we performed LFB/PAS staining of brain sections at 1, 3, and 6 months (Fig. 6), immunofluorescent studies of brain sections at 6 months (Fig. 7), and toluidine blue staining (Fig. 8) and electron microscopy (Fig. 9) of spinal cord sections at 6 months. At 1 month, the myelin pattern in the corpus callosum of Plp1dup mice was indistinguishable from wild-type mice with LFB/PAS staining (Fig. 6A,B). At 3 months, an uneven and patchy staining pattern of myelin in the corpus callosum was observed in Plp1dup mice; and at 6 months, this pattern became more prominent (Fig. 6C–F). Immunofluorescent staining at 6 months with antibodies against PLP and MBP also showed a reduction of myelin sheaths in the corpus callosum of Plp1dup mice when compared with wild-type (Fig. 7A,B). Whereas PLP and MPB colocalized in the corpus callosum of both wild-type and Plp1dup mice, juxtaposition of PLP with pan-axonal neurofilaments (NF) occurred only in some regions of the corpus callosum (data not shown). In other regions, either PLP was seen with low levels of NF or NF was seen with low levels of PLP (Fig. 7C,D). These observations in the corpus callosum suggest that the prominent process is progressive loss of myelin followed by axonal loss. This conclusion is supported by semithin and ultrathin studies. The ventral funiculus of the wild-type spinal cord (Fig. 8A) showed dense packing of fibers, with larger-diameter fibers having thicker sheaths than smaller-diameter fibers. The ventral funiculi from two different Plp1dup mice (Fig. 8B,C) had large-caliber axons with abnormally thin myelin sheaths. Degenerating fibers were abundant in the Plp1dup mice (Fig. 8B,C) and absent in the wild-type spinal cord. Ventral gray matter of normal mice and Plp1dup mice (Fig. 8D–F) showed numerous dendrites emanating from motor neuron perikarya. Ventral gray matter from Plp1dup mice (Fig. 8E,F) showed numerous thinly myelinated fibers coursing through parenchyma. Large vacuoles, degenerating fibers, and lightly stained degenerating cells were prominent throughout gray matter (Fig. 8F). These vacuoles can also be seen in the brains of 6 month Plp1dup mice (Fig. 6F).
Figure 6.

LFB/PAS-stained coronal sections (5 μm) of the corpus callosum region of Plp1dup compared with wild-type mice. Representative images of brain sections of 1 month (A, B), 3 month (C, D), and 6 month (E, F) wild-type (A, C, E) and Plp1dup (B, D, F) mice. In all pictures, hippocampus is to the right and cerebral cortex to the left. At 3 and 6 months in wild-type mice, LFB staining was homogeneous across the corpus callosum, but in Plp1dup mice at these ages, the staining was uneven and patchy. F, At 6 months, large vacuoles were present in both white and gray matter (*). Plp1dup, n = 3; wild-type, n = 3 at each time point. Scale bar, 50 μm.
Figure 7.

Immunofluorescent staining of coronal sections (5 μm) of the corpus callosum region of Plp1dup compared with wild-type mice at 6 months of age. Representative images of brain sections stained for PLP (green) and MBP (red) (A, B) and PLP (green) and NF (red) (C, D) of wild-type (A, C) and Plp1dup (B, D) mice. In all micrographs, hippocampus is to the upper right and cerebral cortex to the lower left. A and C are the same section; B and D are the same section. At 6 months in wild-type mice, PLP (green) staining was homogeneous across the corpus callosum (A, C) and colocalized with MBP (A, red). Yellow and yellow-orange fluorescence indicated abundant colocalization of PLP and MBP (A). In Plp1dup mice, PLP and MBP staining was patchy with apparently more MBP than PLP staining (B). In some places, PLP was seen with low levels of NF and in others NF was seen with low levels of PLP (D). Plp1dup, n = 3; wild-type, n = 3. Scale bars, 50 μm.
Figure 8.

Toluidine blue-stained plastic sections (1 μm) of spinal cords of Plp1dup compared with wild-type mice at 6 months of age. Representative images of spinal cord sections of the ventral funiculus at ventral fissure (A–C) and the ventral gray matter (D–F) of wild-type (A, D) and Plp1dup (B, C, E, F) mice are shown. B, C, F, Arrows indicate axons of large caliber with abnormally thin myelin sheaths. B, C, E, F, Arrowheads indicate degenerating fibers. E, F, *Large vacuoles that were also seen in the LFB-PAS-stained sections (Fig. 6) and immunofluorescent sections (Fig. 7). Scale bar, 50 μm.
Figure 9.

Electron micrographs of spinal cords from 6 month wild-type and Plp1dup mice. Representative electron micrographs of transition between ventral funiculus and ventral gray matter of wild-type mouse (A) and Plp1dup mouse (B–F) at 6 months of age. Ax, Axon; MG, microglial cell; AG, astrogliosis; DF, large unmyelinated axon (degenerating fiber) packed with neurofilaments; PN/IN with arrow, paranode/internode. B, Arrow points to a degenerating axon being engulfed by a microglial cell. D, Arrows point to a cluster of degenerating fibers. F, The axon (Ax) is myelinated on one side of a node but unmyelinated on the other. Scale bar, 1 μm.
At the electron microscopic level, spinal cords of 6 month Plp1dup mice showed abnormalities in both white and gray matter (Fig. 9). Degenerating fibers were present throughout the spinal cords of Plp1dup mice but were more prominent in gray matter versus white matter (Fig. 9B–E). Myelin sheaths were abnormally thick but often poorly compacted (Fig. 9F). Segments of unmyelinated axons were adjacent to myelinated segments, suggestive of demyelination and/or dysmyelination (Fig. 9E,F). Microglial cells were abundant and contained numerous degenerating fibers within their cytoplasm. Astrogliosis was a prominent feature throughout white and gray matter (Fig. 9B).
The blood–brain barrier of Plp1dup mice remains intact
After intraperitoneal injection of Evan's blue dye into Plp1dup mice, dye was not detected in brain but was detected throughout muscle, intestine, liver, and other tissues (data not shown). This result suggested that the blood–brain barrier of Plp1dup mice is not compromised.
Discussion
Duplication of the Plp1 locus that includes the entire Plp1 gene is the most frequent cause of PMD (Inoue et al., 1999; Woodward et al., 2005; Lee et al., 2006). We used MICER chromosome engineering techniques to generate Plp1dup, a mouse model of the human disease. We demonstrate that the duplication in our Plp1dup mice alters the transcript levels of Plp1 and four of the five other genes within the duplication. It also leads to altered levels of major myelin proteins, abnormal myelin pathology, and gait abnormalities. Thus, our Plp1dup model will be important for insights into how genomic duplication leads to a disease phenotype and for assessing potential treatments.
Several mouse models that harbor extra copies of the Plp1 gene were generated and characterized previously. These transgenic lines were made by random genomic integration of multiple copies of the Plp1-containing segment of a genomic cosmid clone. Within each line, the homozygous animals have very severe hypomyelinating phenotype with seizures and early death, whereas the hemizygous animals have a mild, late-onset demyelinating phenotype, or no abnormal phenotype (Kagawa et al., 1994; Readhead et al., 1994; Inoue et al., 1996; Anderson et al., 1998, 1999; Bradl et al., 1999; Rasband et al., 2003; Karim et al., 2007; Mayer et al., 2011). These results suggest that the copy number of Plp1 correlates with severity of disease, as is the case with human PMD, where three or more copies of Plp1 on the X chromosome in males is more severe than duplication (Wolf et al., 2005). However, it is curious that homozygous mice with six extra copies of Plp1 have a much more severe phenotype than hemizygous mice of another line with seven extra copies, a phenomenon that has not been explained. Given the data from the transgenic animal models, it is also curious that the phenotype of PMD patients is as severe as it is when male PMD patients have only one extra copy of PLP1. In any case, our Plp1dup mouse model displays an intermediate phenotype between the homozygous and hemizygous animals and has both hypomyelination and demyelination. In this respect, our model is more similar than the transgenic models to the human duplication patients who have a combination of hypomyelination and degeneration, seldom develop seizures, and can live into their sixth decade (observation of G.M.H.).
Patients with PMD duplication and the animal models with extra copies of Plp1 have been called “overexpressors” in the literature. This term is misleading because PLP1 transcript levels have not been evaluated in the CNS of PMD duplication patients; and although Plp1 transcript levels have been determined in some of the transgenic animal models, the reported overexpression is modest at best and is clearly not proportionate to the number of extra copies of Plp1 (Kagawa et al., 1994; Readhead et al., 1994; Anderson et al., 1998; Bradl et al., 1999; Karim et al., 2007). In contrast to the transgenic rodents with extra copies of Plp1, our Plp1dup mouse had Plp1 mRNA levels that were approximately double those of wild-type beginning ∼P12 and extending through adulthood. Interestingly, at P21, Plp1 was not elevated in the Plp1dup mouse compared with wild-type at the time when Plp1 levels are rapidly declining from peak levels in both (Fig. 2A). Given that Plp1 expression was studied in the transgenic animals at P19 to P21, our data could explain a lack of overexpression proportionate to copy number in these animals if the extra copies of Plp1 are developmentally regulated.
The PLP protein level was elevated twofold in Plp1dup mouse brains only when first detected at P12, shortly after the onset of developmental increase of Plp1 transcript levels. Our protein assay was not sensitive enough to detect PLP before P12, but elevated PLP was found in spinal cord at P3 in the #66 line homozygotes, with a slight elevation in hemizygotes (Karim et al., 2007). By P20, homozygotes have a small decrease of PLP in spinal cord and hemizygotes have the same level as wild-type, as do our Plp1dup mice. These data suggest that PLP is regulated post-transcriptionally in the brains of our model and in the spinal cords of the transgenic mice. Reduced translation and enhanced degradation of PLP have been demonstrated in cultured oligodendrocytes from the homozygous mice from line #66 (Karim et al., 2010). In rats with multiple copies of Plp1, hemizygotes have elevated levels of PLP at 2 and 3 weeks, whereas the homozygotes have little to no detectable PLP at those time points (Mayer et al., 2011). It is not clear why PLP levels differ between transgenic rats and mice.
Degeneration of myelin resulting from the duplication in the Plp1 locus could account for the decreased transcript and protein levels detected in other myelin proteins, CNP and MBP, in our Plp1dup model compared with wild-type (Figs. 4 and 5). Decreases in other myelin proteins have also been noted in homozygous transgenic overexpressors (Readhead et al., 1994; Karim et al., 2007) and in other Plp1 mutant mice (Delassalle et al., 1981; Al-Saktawi et al., 2003). Structure of myelin in the Plp1dup model resembles the hemizygous animals in the microscopy studies, particularly the 4e line, in which myelin formation proceeded from 10 to 70 d with degeneration beginning at 40 d (Inoue et al., 1996). This formation advanced slowly until 6 months of age, when rapid demyelination occurred. Myelination occurred relatively normally in our Plp1dup mice in the first month, but degeneration began at 3 months and progressed by 6 months. Myelination was much less affected in the hemizygous overexpressors with higher copy numbers than our Plp1dup mice.
Vacuolation seen in Plp1dup mouse brains at 6 months of age (Figs. 6F and 8E,F) was similar to that reported in hemizygous Plp1 overexpressors (Anderson et al., 1998; Bauer et al., 2002) and mice that overexpress CNP (Gravel et al., 1996) or are null for UDP-galactose:ceramide galactosyltransferase (Coetzee et al., 1996). It is unclear what the vacuoles represent, but they seem likely to be processes of degenerating neurons.
The transgenic animal models with multiple extra copies of Plp1 are genetically different from PMD patients in that they have extra copies of Plp1 only, whereas all known human duplications include other genes. Four of the genes duplicated in our model (Tceal1, Tceal3, Morf4l2, and Glra4) are commonly found within the duplicated regions of PMD patients. BC065397 is a hypothetical gene that is not present in the human genome and is thought to be a member of the Ras-like GTPase superfamily. It is unknown to what extent duplication of adjacent genes and regulatory regions contributes to dysregulation of PLP1 expression and pathogenesis of the disease in humans. In that regard, we found that transcript levels of four of the five duplicated genes in our Plp1dup mice were increased approximately twofold (Fig. 3), but we did not detect a phenotype that would be attributed to overexpression of any of these genes. Transcript levels of Glra4 were not shown to be dosage-sensitive (Fig. 3E). However, Glra4 is an expressed pseudogene that is nonfunctional because of a premature stop codon within exon 9 that results in the premature truncation (Muncke et al., 2004).
Although Plp1 mRNA levels were altered in the brain of our Plp1dup mice, we detected no change in the Plp1/Dm20 alternative splicing ratio at any time point (Fig. 2B). This result conflicts with a previous report in which DM20 mRNA is reduced with respect to PLP1 in cultured skin fibroblasts from patients with PMD duplications (Regis et al., 2008). The discrepancy may be the result of either species or tissue difference and requires further study.
Our studies have shown significant differences between Plp1dup and wild-type mice in kinetics, reflected by stand index, swing, step cycle, and cadence, and support, reflected by print area, base of support, and other limb support percentages. Most of the significant parameters are more severe in hind limbs than front limbs, correlating well with human PMD patients who tend to be more severely affected in their lower than upper limbs (Golomb et al., 2004). No studies have been reported regarding the comprehensive gait analysis of Plp1 overexpressor animals.
Our new Plp1dup mouse provides a genomically accurate model for understanding the pathophysiology of PMD resulting from PLP1 duplication. It will also be an important model for preclinical trials of potential treatments for PMD resulting from genomic duplication. Potential treatment strategies include stem-cell therapies and strategies to reduce the amount of PLP, which could be accomplished using RNAi or small molecules identified by high-throughput screening. Notably, knockdown of Plp1 by nucleofection of siRNA into oligodendrocytes cultured from homozygous transgenic animals of an overexpressor line increased formation of myelin and amount of MBP in the membrane (Karim et al., 2010). Recently, a cholesterol-enriched diet was shown to improve the phenotype of overexpressor mice that, unlike Plp1dup, has a compromised blood–brain barrier (Saher et al., 2012).
Footnotes
This work was supported by National Institutes of Health (NIH) Grant P20RR020173 to G.M.H. from the National Center for Research Resources [NCRR; currently NIH Grant P20GM103464 from the National Institute of General Medical Sciences (NIGMS)], NIH Grants R01NS058978 to G.M.H. and R01NS38236 to R.P.S. from the National Institute of Neurological Disorders and Stroke (NINDS), and by a grant from the PMD Foundation to G.M.H. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NCRR, the NIGMS, the NINDS, the NIH, or the PMD Foundation. We thank the Nemours Biomolecular Core, Catherine Kirn-Safran, and Rick Focht of the Transgenic Facility at the University of Delaware for technical assistance with embryonic stem cell manipulations; Jennifer Dunlap, Zuping Qu, and Jan Guz of the Transgenic and Gene Targeting Facility at Thomas Jefferson University for technical assistance with embryonic stem cell manipulations and generation of mutant animals; Dr. Saud A. Sadiq and Dr. Violaine K. Harris of the Tisch MS Center of New York for technical assistance with immunofluorescence studies; and Sherlly Xie of Nemours Biomedical Research for assistance with the statistical analysis.
The authors declare no competing financial interests.
References
- Adams DJ, Biggs PJ, Cox T, Davies R, van der Weyden L, Jonkers J, Smith J, Plumb B, Taylor R, Nishijima I, Yu Y, Rogers J, Bradley A. Mutagenic insertion and chromosome engineering resource (MICER) Nat Genet. 2004;36:867–871. doi: 10.1038/ng1388. [DOI] [PubMed] [Google Scholar]
- Al-Saktawi K, McLaughlin M, Klugmann M, Schneider A, Barrie JA, McCulloch MC, Montague P, Kirkham D, Nave KA, Griffiths IR. Genetic background determines phenotypic severity of the Plp rumpshaker mutation. J Neurosci Res. 2003;72:12–24. doi: 10.1002/jnr.10561. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Schneider A, Barrie JA, Klugmann M, McCulloch MC, Kirkham D, Kyriakides E, Nave KA, Griffiths IR. Late-onset neurodegeneration in mice with increased dosage of the proteolipid protein gene. J Comp Neurol. 1998;394:506–519. doi: 10.1002/(SICI)1096-9861(19980518)394:4<506::AID-CNE8>3.0.CO%3B2-5. [DOI] [PubMed] [Google Scholar]
- Anderson TJ, Klugmann M, Thomson CE, Schneider A, Readhead C, Nave KA, Griffiths IR. Distinct phenotypes associated with increasing dosage of the PLP gene: implications for CMT1A due to PMP22 gene duplication. Ann N Y Acad Sci. 1999;883:234–246. doi: 10.1111/j.1749-6632.1999.tb08585.x. [DOI] [PubMed] [Google Scholar]
- Bauer J, Bradl M, Klein M, Leisser M, Deckwerth TL, Wekerle H, Lassmann H. Endoplasmic reticulum stress in PLP-overexpressing transgenic rats: gray matter oligodendrocytes are more vulnerable than white matter oligodendrocytes. J Neuropathol Exp Neurol. 2002;61:12–22. doi: 10.1093/jnen/61.1.12. [DOI] [PubMed] [Google Scholar]
- Bradl M, Bauer J, Inomata T, Zielasek J, Nave KA, Toyka K, Lassmann H, Wekerle H. Transgenic lewis rats overexpressing the proteolipid protein gene: myelin degeneration and its effect on T cell-mediated experimental autoimmune encephalomyelitis. Acta Neuropathol. 1999;97:595–606. doi: 10.1007/s004010051035. [DOI] [PubMed] [Google Scholar]
- Carvalho CM, Bartnik M, Pehlivan D, Fang P, Shen J, Lupski JR. Evidence for disease penetrance relating to CNV size: Pelizaeus-Merzbacher disease and manifesting carriers with a familial 11 Mb duplication at Xq22. Clin Genet. 2012;81:532–541. doi: 10.1111/j.1399-0004.2011.01716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, Suzuki K, Popko B. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell. 1996;86:209–219. doi: 10.1016/S0092-8674(00)80093-8. [DOI] [PubMed] [Google Scholar]
- Delassalle A, Zalc B, Lachapelle F, Raoul M, Collier P, Jacque C. Regional distribution of myelin basic protein in the central nervous system of quaking, jimpy, and normal mice during development and aging. J Neurosci Res. 1981;6:303–313. doi: 10.1002/jnr.490060305. [DOI] [PubMed] [Google Scholar]
- Golomb MR, Walsh LE, Carvalho KS, Christensen CK, DeMyer WE. Clinical findings in Pelizaeus-Merzbacher disease. J Child Neurol. 2004;19:328–331. doi: 10.1177/088307380401900504. [DOI] [PubMed] [Google Scholar]
- Gravel M, Peterson J, Yong VW, Kottis V, Trapp B, Braun PE. Overexpression of 2′,3′-cyclic nucleotide 3′-phosphodiesterase in transgenic mice alters oligodendrocyte development and produces aberrant myelination. Mol Cell Neurosci. 1996;7:453–466. doi: 10.1006/mcne.1996.0033. [DOI] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M, Mills AA. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A. 2011;108:17076–17081. doi: 10.1073/pnas.1114042108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Imaizumi K, Nezu A, Takanashi J, Arii J, Murayama K, Ono J, Kikawa Y, Mito T, Shaffer LG, Lupski JR. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann Neurol. 1999;45:624–632. doi: 10.1002/1531-8249(199905)45:5<624::AID-ANA11>3.3.CO%3B2-T. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Kagawa T, Matsumura Y, Ikenaka K, Mikoshiba K. Cell death of oligodendrocytes or demyelination induced by overexpression of proteolipid protein depending on expressed gene dosage. Neurosci Res. 1996;25:161–172. doi: 10.1016/0168-0102(96)01039-5. [DOI] [PubMed] [Google Scholar]
- Kagawa T, Ikenaka K, Inoue Y, Kuriyama S, Tsujii T, Nakao J, Nakajima K, Aruga J, Okano H, Mikoshiba K. Glial cell degeneration and hypomyelination caused by overexpression of myelin proteolipid protein gene. Neuron. 1994;13:427–442. doi: 10.1016/0896-6273(94)90358-1. [DOI] [PubMed] [Google Scholar]
- Karim SA, Barrie JA, McCulloch MC, Montague P, Edgar JM, Kirkham D, Anderson TJ, Nave KA, Griffiths IR, McLaughlin M. PLP overexpression perturbs myelin protein composition and myelination in a mouse model of Pelizaeus-Merzbacher disease. Glia. 2007;55:341–351. doi: 10.1002/glia.20465. [DOI] [PubMed] [Google Scholar]
- Karim SA, Barrie JA, McCulloch MC, Montague P, Edgar JM, Iden DL, Anderson TJ, Nave KA, Griffiths IR, McLaughlin M. PLP/DM20 expression and turnover in a transgenic mouse model of Pelizaeus-Merzbacher disease. Glia. 2010;58:1727–1738. doi: 10.1002/glia.21043. [DOI] [PubMed] [Google Scholar]
- Korbie DJ, Mattick JS. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat Protoc. 2008;3:1452–1456. doi: 10.1038/nprot.2008.133. [DOI] [PubMed] [Google Scholar]
- Lee JA, Inoue K, Cheung SW, Shaw CA, Stankiewicz P, Lupski JR. Role of genomic architecture in PLP1 duplication causing Pelizaeus-Merzbacher disease. Hum Mol Genet. 2006;15:2250–2265. doi: 10.1093/hmg/ddl150. [DOI] [PubMed] [Google Scholar]
- Li Z, Yu T, Morishima M, Pao A, LaDuca J, Conroy J, Nowak N, Matsui S, Shiraishi I, Yu YE. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum Mol Genet. 2007;16:1359–1366. doi: 10.1093/hmg/ddm086. [DOI] [PubMed] [Google Scholar]
- Liu C, Szurek PF, Yu YE. MICER targeting vectors for manipulating the mouse genome. Methods Mol Biol. 2011;693:245–256. doi: 10.1007/978-1-60761-974-1_14. [DOI] [PubMed] [Google Scholar]
- Mayer JA, Larsen EC, Kondo Y, Duncan ID. Characterization of a PLP-overexpressing transgenic rat, a model for the connatal form of Pelizaeus-Merzbacher disease. Neurobiol Dis. 2011;44:231–238. doi: 10.1016/j.nbd.2011.07.007. [DOI] [PubMed] [Google Scholar]
- Muncke N, Wogatzky BS, Breuning M, Sistermans EA, Endris V, Ross M, Vetrie D, Catsman-Berrevoets CE, Rappold G. Position effect on PLP1 may cause a subset of Pelizaeus-Merzbacher disease symptoms. J Med Genet. 2004;41:e121. doi: 10.1136/jmg.2004.019141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani J, Tamada K, Hatanaka F, Ise S, Ohta H, Inoue K, Tomonaga S, Watanabe Y, Chung YJ, Banerjee R, Iwamoto K, Kato T, Okazawa M, Yamauchi K, Tanda K, Takao K, Miyakawa T, Bradley A, Takumi T. Abnormal behavior in a chromosome-engineered mouse model for human 15q11–13 duplication seen in autism. Cell. 2009;137:1235–1246. doi: 10.1016/j.cell.2009.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Office of Laboratory Animal Welfare (OLAW) U.S. Department of Health and Human Services. National Institutes of Health. 2013. [Accessed March 21, 2013]. Available at http://grants.nih.gov/grants/olaw/olaw.htm.
- O'Gorman S, Dagenais NA, Qian M, Marchuk Y. Protamine-Cre recombinase transgenes efficiently recombine target sequences in the male germ line of mice, but not in embryonic stem cells. Proc Natl Acad Sci U S A. 1997;94:14602–14607. doi: 10.1073/pnas.94.26.14602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinkel D, Landegent J, Collins C, Fuscoe J, Segraves R, Lucas J, Gray J. Fluorescence in situ hybridization with human chromosome-specific libraries: detection of trisomy 21 and translocations of chromosome 4. Proc Natl Acad Sci U S A. 1988;85:9138–9142. doi: 10.1073/pnas.85.23.9138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Kagawa T, Park EW, Ikenaka K, Trimmer JS. Dysregulation of axonal sodium channel isoforms after adult-onset chronic demyelination. J Neurosci Res. 2003;73:465–470. doi: 10.1002/jnr.10675. [DOI] [PubMed] [Google Scholar]
- Readhead C, Schneider A, Griffiths I, Nave KA. Premature arrest of myelin formation in transgenic mice with increased proteolipid protein gene dosage. Neuron. 1994;12:583–595. doi: 10.1016/0896-6273(94)90214-3. [DOI] [PubMed] [Google Scholar]
- Regis S, Biancheri R, Bertini E, Burlina A, Lualdi S, Bianco MG, Devescovi R, Rossi A, Uziel G, Filocamo M. Genotype-phenotype correlation in five Pelizaeus-Merzbacher disease patients with PLP1 gene duplications. Clin Genet. 2008;73:279–287. doi: 10.1111/j.1399-0004.2007.00961.x. [DOI] [PubMed] [Google Scholar]
- Saher G, Rudolphi F, Corthals K, Ruhwedel T, Schmidt KF, Löwel S, Dibaj P, Barrette B, Möbius W, Nave KA. Therapy of Pelizaeus-Merzbacher disease in mice by feeding a cholesterol-enriched diet. Nat Med. 2012;18:1130–1135. doi: 10.1038/nm.2833. [DOI] [PubMed] [Google Scholar]
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. doi: 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz K, Paylor R, Yan J, Bi W, Lupski JR. Rai1 duplication causes physical and behavioral phenotypes in a mouse model of dup(17) (p11.2p11.2) J Clin Invest. 2006;116:3035–3041. doi: 10.1172/JCI28953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wight PA, Dobretsova A. Where, when and how much: regulation of myelin proteolipid protein gene expression. Cell Mol Life Sci. 2004;61:810–821. doi: 10.1007/s00018-003-3309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf NI, Sistermans EA, Cundall M, Hobson GM, Davis-Williams AP, Palmer R, Stubbs P, Davies S, Endziniene M, Wu Y, Chong WK, Malcolm S, Surtees R, Garbern JY, Woodward KJ. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain. 2005;128:743–751. doi: 10.1093/brain/awh409. [DOI] [PubMed] [Google Scholar]
- Woodward KJ, Cundall M, Sperle K, Sistermans EA, Ross M, Howell G, Gribble SM, Burford DC, Carter NP, Hobson DL, Garbern JY, Kamholz J, Heng H, Hodes ME, Malcolm S, Hobson GM. Heterogeneous duplications in patients with Pelizaeus-Merzbacher disease suggest a mechanism of coupled homologous and nonhomologous recombination. Am J Hum Genet. 2005;77:966–987. doi: 10.1086/498048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T, Liu C, Belichenko P, Clapcote SJ, Li S, Pao A, Kleschevnikov A, Bechard AR, Asrar S, Chen R, Fan N, Zhou Z, Jia Z, Chen C, Roder JC, Liu B, Baldini A, Mobley WC, Yu YE. Effects of individual segmental trisomies of human chromosome 21 syntenic regions on hippocampal long-term potentiation and cognitive behaviors in mice. Brain Res. 2010;1366:162–171. doi: 10.1016/j.brainres.2010.09.107. [DOI] [PMC free article] [PubMed] [Google Scholar]