

Abstract
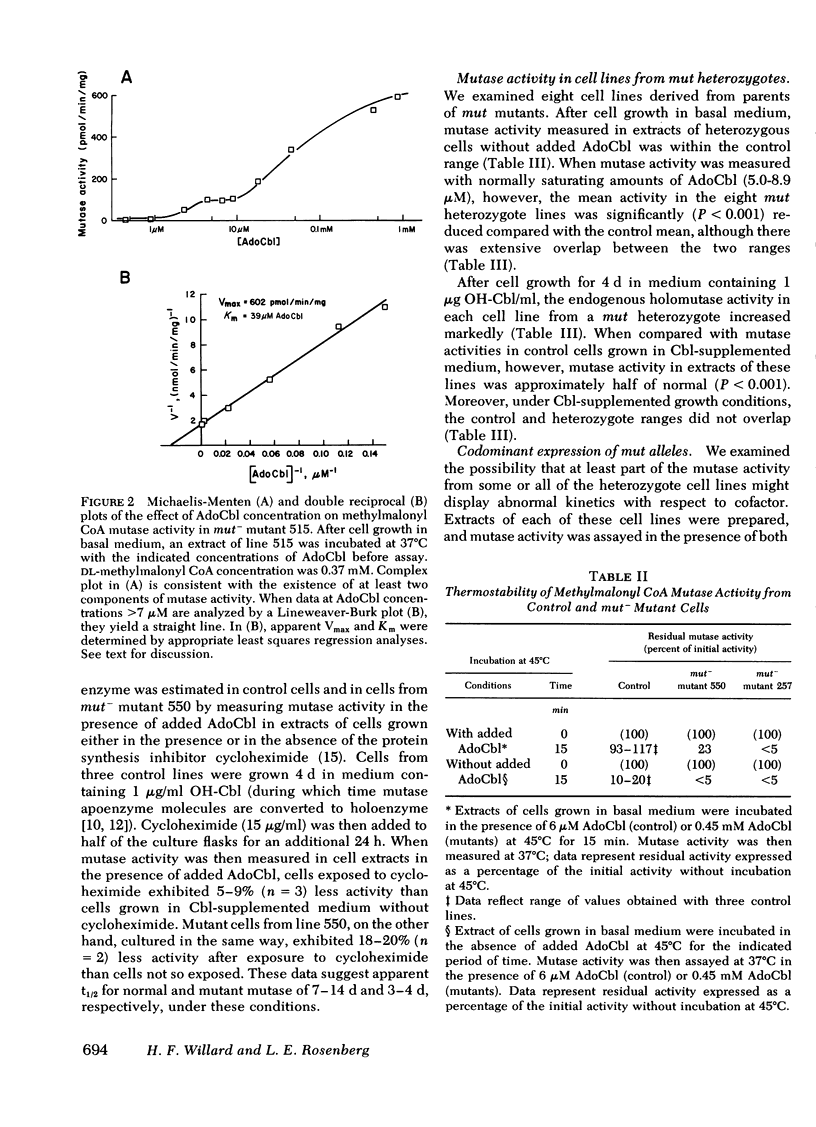
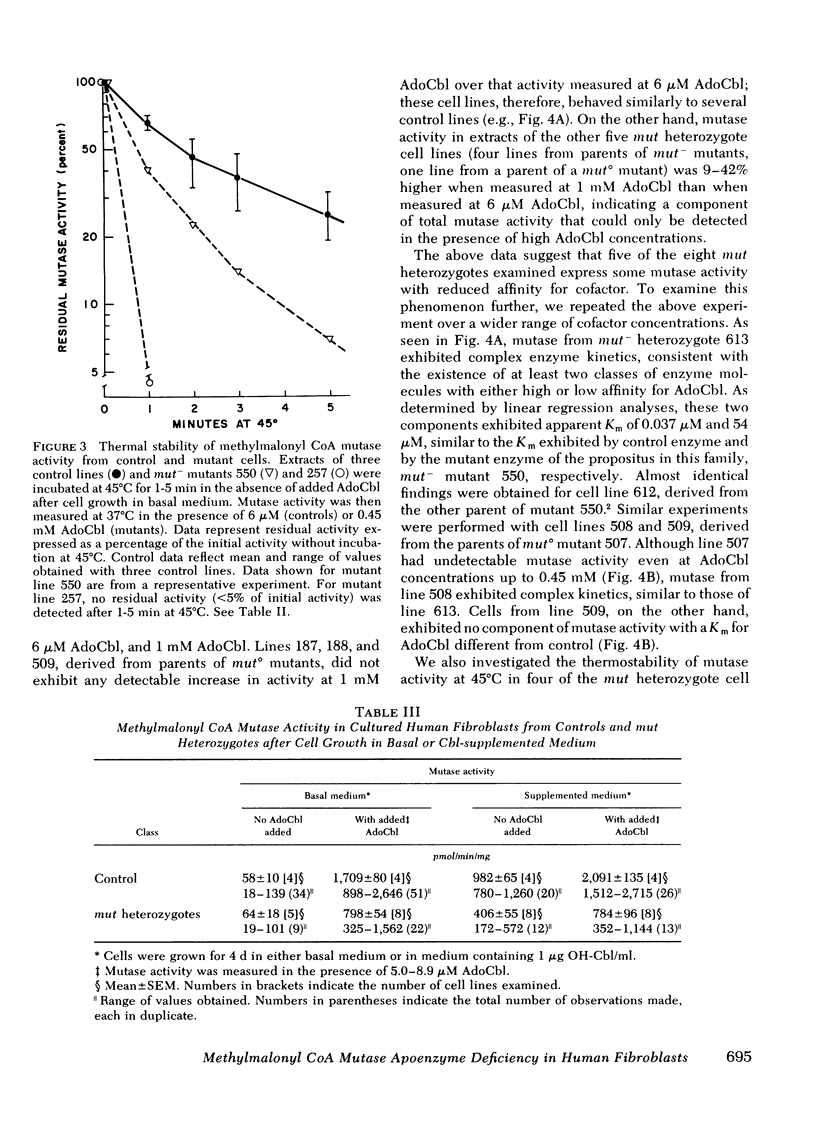
We have measured and characterized methylmalonyl coenzyme A (CoA) mutase activity in extracts of cultured human fibroblasts from 23 patients with inherited deficiency of the mutase apoenzyme and from eight obligate heterozygotes for this defect. The mutant cell lines fall into two categories. Those without detectable residual mutase activity in cell extracts (>0.1% of control), and whose ability to utilize propionate in intact cells is refractory to supplementation of the culture medium with hydroxocobalamin, are designated mut° mutants. Those with detectable residual activity in cell extracts (∼0.5-50% of control), and whose ability to utilize propionate in intact cells in markedly increased by hydroxocobalamin supplementation, are designated mut− mutants. The mutant enzyme in the mut− mutants exhibits a 50- to 5,000-fold elevated Michaelis constant (Km) for adenosylcobalamin in vitro, a normal Km for methylmalonyl CoA, and a strikingly reduced thermal stability at 45°C relative to control. Mutase from one mut− mutant turns over at a rate three to four times that of control enzyme when cells are grown in hydroxocobalamin-supplemented medium.
To detect heterozygous carriers of mutant mut alleles, we compared mutase activity in fibroblast extracts from four controls with that from eight parents of either mut° or mut− mutants. After cell growth in either unsupplemented or hydroxocobalamin-supplemented medium, activity in cell lines from heterozygotes was reduced to 47 or 37% of the mean control activities, respectively. We also examined the effect of adenosylcobalamin concentration on reaction kinetics of mutase from heterozygote cell lines. All four cell lines from parents of mut− mutants exhibited complex enzyme kinetics; ∼80% of mutase activity demonstrated a Km indistinguishable from control, whereas a smaller component of activity exhibited a Km similar to the abnormal Km expressed by the mut− propositus in each family. In two families with a mut° propositus, mutase from three of the four parents exhibited only the normal Km for adenosylcobalamin, whereas mutase from one parent displayed complex kinetics, indicating expression of both a normal allele (mut+) and a mutant allele with an abnormal Km. From these studies, we conclude that mut mutants reflect mutations at the autosomal gene locus for the methylmalonyl CoA mutase apoenzyme; that mut°, mut−, and mut+ alleles at this locus are codominantly expressed; and that some mut mutants may be genetic compounds, inheriting two different mut° or mut− alleles from their parents.
Full text
PDF






Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Böhlen P., Stein S., Dairman W., Udenfriend S. Fluorometric assay of proteins in the nanogram range. Arch Biochem Biophys. 1973 Mar;155(1):213–220. doi: 10.1016/s0003-9861(73)80023-2. [DOI] [PubMed] [Google Scholar]
- Fenton W. A., Rosenberg L. E. Genetic and biochemical analysis of human cobalamin mutants in cell culture. Annu Rev Genet. 1978;12:223–248. doi: 10.1146/annurev.ge.12.120178.001255. [DOI] [PubMed] [Google Scholar]
- Fenton W. A., Rosenberg L. E. Improved techniques for the extraction and chromatography of cobalamins. Anal Biochem. 1978 Oct 1;90(1):119–125. doi: 10.1016/0003-2697(78)90014-3. [DOI] [PubMed] [Google Scholar]
- Gravel R. A., Mahoney M. J., Ruddle F. H., Rosenberg L. E. Genetic complementation in heterokaryons of human fibroblasts defective in cobalamin metabolism. Proc Natl Acad Sci U S A. 1975 Aug;72(8):3181–3185. doi: 10.1073/pnas.72.8.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang E. S., Snodgrass P. J., Gerald P. S. Methylmalonyl coenzyme A racemase defect: another cause of methylmalonic aciduria. Pediatr Res. 1972 Dec;6(12):875–879. doi: 10.1203/00006450-197212000-00004. [DOI] [PubMed] [Google Scholar]
- Linnell J. C., Matthews D. M., Mudd S. H., Uhlendorf B. W., Wise I. J. Cobalamins in fibroblasts cultured from normal control subjects and patients with methylmalonic aciduria. Pediatr Res. 1976 Mar;10(3):179–183. doi: 10.1203/00006450-197603000-00007. [DOI] [PubMed] [Google Scholar]
- Mahoney M. J., Hart A. C., Steen V. D., Rosenberg L. E. Methylmalonicacidemia: biochemical heterogeneity in defects of 5'-deoxyadenosylcobalamin synthesis. Proc Natl Acad Sci U S A. 1975 Jul;72(7):2799–2803. doi: 10.1073/pnas.72.7.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow G., 3rd, Barness L. A., Cardinale G. J., Abeles R. H., Flaks J. G. Congenital methylmalonic acidemia: enzymatic evidence for two forms of the disease. Proc Natl Acad Sci U S A. 1969 May;63(1):191–197. doi: 10.1073/pnas.63.1.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow G., 3rd, Lebowitz J. Studies of methylmalonyl-coenzyme A carbonylmutase activity in methylmalonic acidemia. II. In vitro binding kinetics with adenosylcobalamin. Biochem Med. 1976 Jun;15(3):241–245. doi: 10.1016/0006-2944(76)90054-5. [DOI] [PubMed] [Google Scholar]
- Morrow G., 3rd, Mahoney M. J., Mathews C., Lebowitz J. Studies of methylmalonyl coenzyme A carbonylmutase activity in methylmalonic acidemia. I. Correlation of clinical, hepatic, and fibroblast data. Pediatr Res. 1975 Aug;9(8):641–644. doi: 10.1203/00006450-197508000-00006. [DOI] [PubMed] [Google Scholar]
- Morrow G., 3rd, Revsin B., Clark R., Lebowitz J., Whelan D. T. A new variant of methylmalonic acidemia-defective coenzyme-apoenzyme binding in cultured fibroblasts. Clin Chim Acta. 1978 Apr 3;85(1):67–72. doi: 10.1016/0009-8981(78)90102-x. [DOI] [PubMed] [Google Scholar]
- Narisawa K., Saito T., Hisa S., Suzuki H., Hayasaka K. Methylmalonyl-CoA mutase activity of leukocytes in variants and heterozygotes of methylmalonic acidemia. Tohoku J Exp Med. 1977 Sep;123(1):1–8. doi: 10.1620/tjem.123.1. [DOI] [PubMed] [Google Scholar]
- Willard H. F., Ambani L. M., Hart A. C., Mahoney M. J., Rosenberg L. E. Rapid prenatal and postnatal detection of inborn errors of propionate, methylmalonate, and cobalamin metabolism: a sensitive assay using cultured cells. Hum Genet. 1976 Dec 15;34(3):277–283. doi: 10.1007/BF00295291. [DOI] [PubMed] [Google Scholar]
- Willard H. F., Mellman I. S., Rosenberg L. E. Genetic complementation among inherited deficiencies of methylmalonyl-CoA mutase activity: evidence for a new class of human cobalamin mutant. Am J Hum Genet. 1978 Jan;30(1):1–13. [PMC free article] [PubMed] [Google Scholar]
- Willard H. F., Rosenberg L. E. Inborn errors of cobalamin metabolism: effect of cobalamin supplementation in culture on methylmalonyl CoA mutase activity in normal and mutant human fibroblasts. Biochem Genet. 1979 Feb;17(1-2):57–75. doi: 10.1007/BF00484474. [DOI] [PubMed] [Google Scholar]
- Willard H. F., Rosenberg L. E. Inherited deficiencies of human methylmalonyl CaA mutase activity: reduced affinity of mutant apoenzyme for adenosylcobalamin. Biochem Biophys Res Commun. 1977 Oct 10;78(3):927–934. doi: 10.1016/0006-291x(77)90511-3. [DOI] [PubMed] [Google Scholar]
- Youngdahl-Turner P., Mellman I. S., Allen R. H., Rosenberg L. E. Protein mediated vitamin uptake. Adsorptive endocytosis of the transcobalamin II-cobalamin complex by cultured human fibroblasts. Exp Cell Res. 1979 Jan;118(1):127–134. doi: 10.1016/0014-4827(79)90590-1. [DOI] [PubMed] [Google Scholar]