

Abstract
Transcription-coupled nucleotide excision repair (TC-NER) specifically removes transcription-blocking lesions from our genome. Defects in this pathway are associated with two human disorders: Cockayne syndrome (CS) and UV-sensitive syndrome (UVSS). Despite a similar cellular defect in the UV DNA damage response, patients with these syndromes exhibit strikingly distinct symptoms; CS patients display severe developmental, neurological, and premature aging features, whereas the phenotype of UVSS patients is mostly restricted to UV hypersensitivity. The exact molecular mechanism behind these clinical differences is still unknown; however, they might be explained by additional functions of CS proteins beyond TC-NER. A short overview of the current hypotheses addressing possible molecular mechanisms and the proteins involved are presented in this review. In addition, we will focus on two new players involved in TC-NER which were recently identified: UV-stimulated scaffold protein A (UVSSA) and ubiquitin-specific protease 7 (USP7). UVSSA has been found to be the causative gene for UVSS and, together with USP7, is implicated in regulating TC-NER activity. We will discuss the function of UVSSA and USP7 and how the discovery of these proteins contributes to a better understanding of the molecular mechanisms underlying the clinical differences between UVSS and the more severe CS.
Introduction
Alterations in the DNA structure that hinder the progression of RNA polymerases during transcription are highly toxic for the cell and, if not properly resolved, can lead to cellular apoptosis or senescence (Ljungman and Zhang 1996; Ljungman and Lane 2004). Helix-distorting lesions located in the transcribed strand of active genes initiate the transcription-coupled nucleotide excision repair (TC-NER) pathway to resolve the transcription-blocking DNA damage (Hanawalt and Spivak 2008). Lesion stalled elongating RNA polymerase II (RNA Pol II) triggers the recruitment of several TC-NER-specific factors to form a functional TC-NER complex, including the Cockayne syndrome A and B (CSA, CSB) proteins (Fousteri et al. 2006) (Fig. 1a).
Fig. 1.
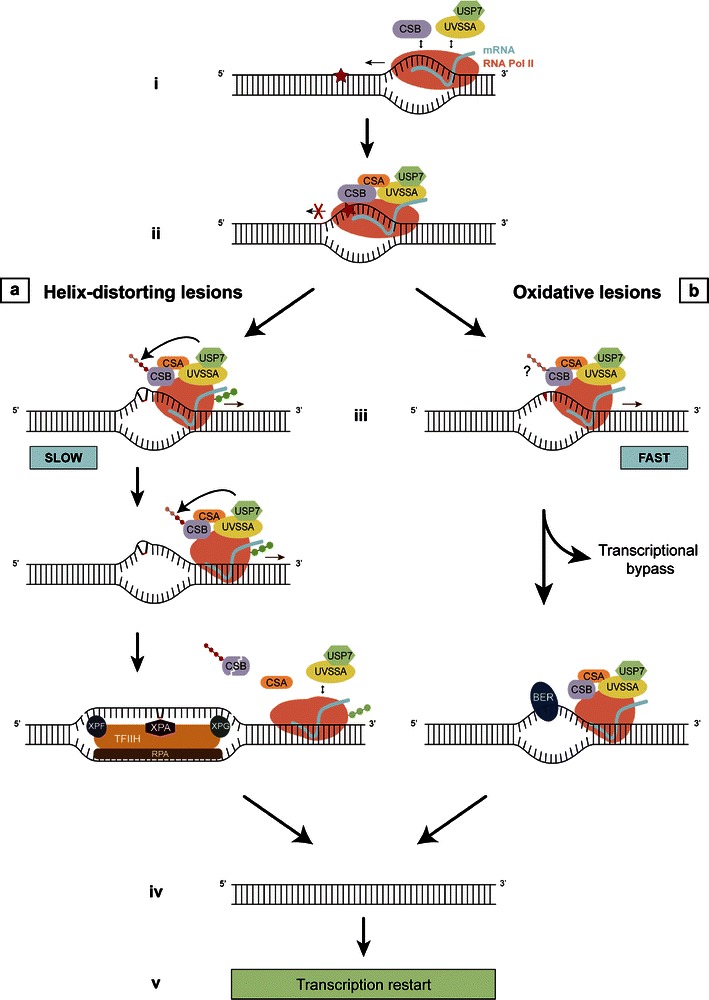
Model for resolving transcription-blocking lesions. i During transcription, UVSSA, USP7, and CSB interact transiently with elongating RNA Pol II. ii Upon encountering a lesion (indicated by star), stalled RNA Pol II stabilizes its binding with these proteins and triggers the recruitment of several other DNA repair factors, including CSA. Since RNA Pol II shields the DNA lesion in its active pocket, the stalled transcription complex must be remodeled to enable access of repair proteins to the lesion. In the case of helix-distorting lesions (iiia), TC-NER is initiated for which extensive modulation of stalled RNA Pol II is needed, causing a prolonged transcriptional arrest. During this slow process, CSB assembles a functional TC-NER complex and assists in the remodeling of RNA Pol II. In response to DNA damage, CSB is ubiquitinated and eventually degraded. In order to allow sufficient time for CSB to perform its function in TC-NER, its presence is protected by the concerted action of UVSSA and USP7 by counteracting the ubiquitination-dependent degradation. When the required remodeling of stalled RNA Pol II is completed, the TC-NER complex is destabilized and CSB is degraded by the proteasome. XPA, RPA, and the TFIIH complex are subsequently recruited. Following further helix unwinding and lesion verification, XPG and XPF are required for the double incision of the damaged DNA strand. The repair reaction is completed with DNA polymerase gap filling of the repair patch and sealing of the nicks. Oxidative lesions (iiib) have a minor effect on the DNA helix structure, which therefore requires relatively little remodeling of RNA Pol II for efficient repair. Consequently, RNA Pol II is only slowed down or transiently arrested in response to these lesions resulting in a faster response as compared to iiia. CSB remodeling helps to efficiently remove oxidative lesions likely via the BER pathway or to pass stalled complexes over the lesion (lesion bypass). However, due to the faster response, protection of CSB from ubiquitination-dependent degradation by UVSSA/USP7 is not necessary. After repair is completed (iv), transcription is resumed (v)
Cockayne syndrome versus UV-sensitive syndrome
In humans, defective TC-NER is associated with two autosomal recessive DNA repair deficiency disorders: Cockayne syndrome (CS) and UV-sensitive syndrome (UVsS) (Nance and Berry 1992; Spivak 2005). Although patient-derived cells are equally deficient in UV-induced TC-NER in vitro, the patients exhibit strikingly distinct clinical symptoms: CS individuals display severe developmental, neurological, and premature aging features, whereas UVSS patients express much milder features, mostly restricted to UV hypersensitivity. CS is caused by mutations in two genes indispensable for TC-NER, CSA and CSB. UVsS comprises three complementation groups, which are defined by specific mutations in CSA, CSB, and in the recently identified gene encoding for UV-stimulated scaffold protein A (UVSSA) (Table 1). Intriguingly, neither the site nor the nature of the mutations in CSA or CSB seems to correlate with the clinical differences observed among patients with CS and UVSS (Nance and Berry 1992; Laugel et al. 2010). Thus, an important question remains: how do molecular defects within the same TC-NER pathway and even different mutations within the same genes lead to such diverse pathologies?
Table 1.
Overview of UVSS mutations in CSA, CSB, and UVSSA
| Patient/cell line | Gene affected | Mutation | References |
|---|---|---|---|
| KPS2 | UVSSA | p.Lys123* | 1, 3, 4, 5 |
| KPS3 | UVSSA | p.Lys123* | 1, 2, 3, 4, 5 |
| UVS24TA | UVSSA | p.Ile31Phefs*9 | 1, 2, 6 |
| XP24KO | UVSSA | p.Lys123* | 1, 2, 7, 8 |
| XP70TO | UVSSA | p.Cys32Arg | 1, 7, 9 |
| UVSS1VI | CSA | p.Trp361Cys | 10 |
| UVS1KO | CSB | p.Arg77* | 3, 4, 6, 11, 12 |
| CS3AM | CSB | p.Arg77* | 13 |
References: 1. Nakazawa et al. (2012), 2. Zhang et al. (2012), 3. Itoh et al. (1996), 4. Itoh et al. (1995), 5. Itoh et al. (1994), 6. Cleaver and Thomas (1993), 7. Itoh et al. (2000), 8. Fujiwara et al. (1985), 9. Kawada et al. (1986), 10. Nardo et al. (2009), 11. Horibata et al. (2004), 12. Fujiwara et al. (1981), 13. Miyauchi-Hashimoto et al. (1998)
Several models are proposed that try to explain the underlying molecular reason for the wide variety in TC-NER-deficient phenotype (Sarasin 2012; Cleaver 2012). Most of these hypotheses are based on additional functions of the CS proteins outside of their role in TC-NER while assuming UVSSA is not implicated in these processes, hence explaining the milder phenotype in UVSS (Spivak and Hanawalt 2006) (Fig. 2).
Fig. 2.
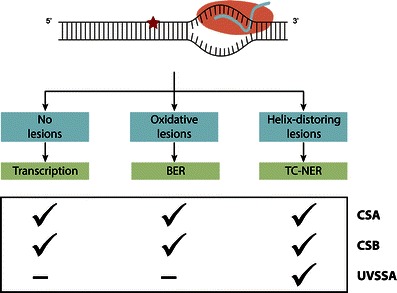
Involvement of CSA, CSB, and UVSSA in transcription, BER, and TC-NER. While the CS proteins have functions outside of TC-NER, a role for UVSSA in these other processes has presently not been shown
One of these models, the transcription defect model, is mainly based on data obtained for the CSB protein. CSB is a member of the SNF2/SWI2 family of DNA-dependent ATPases (Troelstra et al. 1990) and is suggested to have chromatin remodeling abilities, possibly through recruitment of the p300 histone acetyltransferase (Citterio et al. 2000; Newman et al. 2006; Frontini and Proietti-De-Santis 2009). CSB transiently interacts with elongating RNA Pol II and stimulates transcription (Balajee et al. 1997; Dianov et al. 1997; Selby and Sancar 1997; Tantin et al. 1997; van Gool et al. 1997; van den Boom et al. 2004). This suggests that when CSB is absent or mutated, transcription would be affected even in the absence of DNA damage. There is a growing list of human diseases which are associated with defects in transcription, and many of these diseases are characterized by congenital defects (Villard 2004), as also observed in CS patients. This suggests that the severe developmental and premature aging features of CS could partially be caused by a defect in transcription, while the sun sensitivity in these patients is most likely caused by the DNA repair deficiency of the TC-NER pathway.
Another hypothesis involves a role of the CS proteins in repairing endogenous DNA damage from reactive oxygen species (ROS), which is usually repaired by the base excision repair (BER) machinery (Hegde et al. 2008). Both human and mouse cell lines deficient in the CS proteins indeed display enhanced sensitivity to agents that produce oxidative DNA damage (Dianov et al. 1999; Tuo et al. 2003; de Waard et al. 2004; Gorgels et al. 2007; D’Errico et al. 2007; Stevnsner et al. 2008). Furthermore, Menoni et al. (2012) recently showed recruitment of CSB to local oxidative DNA lesions in living cells without an accumulation of downstream NER factors, indicating the involvement of CSB in the response to oxidative DNA lesions independent of the downstream NER reaction. Importantly, CSB binding to oxidative damage was reduced upon transcription inhibition which suggests a role of CSB in transcription-associated repair of oxidative lesions. In CS patients, the accumulation of unrepaired oxidative DNA damage and the subsequent prolonged transcription arrest at these lesions could result in apoptosis and consequentially tissue degeneration. Since active metabolism during development and a high level of oxidative metabolism in neural tissues may generate a considerable amount of oxidative damage, the consequences of the transcription arrest are mainly disclosed by developmental and neurological symptoms which are a hallmark of CS (Hoeijmakers 2007). This model is further supported by transcriptomic analysis of CSB-deficient mice which revealed systematic suppression of growth and oxidative metabolism, an altered glycolysis and an upregulated antioxidant defense, collectively referred to as survival response (van der Pluijm et al. 2007). Moreover, Pascucci et al. (2012) recently observed a disturbed redox balance in patient-derived CS cells. They showed increased intracellular ROS levels and oxidative DNA damage. In addition, alterations in cellular metabolism including the glycolysis pathway and oxidative metabolism were observed. Addition of antioxidants to the cells reduced ROS levels and at least partially reverted the alterations in cellular metabolism, suggesting that oxidative stress plays a causative role in CS pathology. Although there is compelling evidence supporting this hypothesis, no direct proof is currently present and it needs further exploration.
Within a third model, a role for the CS proteins in repair of oxidative DNA damage specifically in mitochondrial DNA (mtDNA) has been reported (Osenbroch et al. 2009; Kamenisch et al. 2010; Scheibye-Knudsen et al. 2012). Mitochondria possess an independent repair machinery for oxidative lesions, which is thought to play a crucial role in protecting the integrity of mtDNA from the relatively high levels of ROS generated in this organelle. Certain clinical symptoms associated with mitochondrial dysfunction are also observed in CS patients, including the severe neurological and premature aging features (Stevnsner et al. 2002). This would suggest that the cause of these symptoms in CS results in part from the loss of mitochondrial function as a consequence of unrepaired oxidative mtDNA damage due to mutations in the CS proteins.
The abovementioned hypotheses to explain the severe phenotypical consequences of CS do not uphold, however, for the intriguing case of two unrelated patients with null mutations in CSB associated with mild UVSS symptoms (Miyauchi-Hashimoto et al. 1998; Horibata et al. 2004). In contrast, two other patients with a deletion of the promoter sequence of CSB (resulting in no detectable CSB protein) have been found, which showed the most severe CS symptoms (Laugel et al. 2008). Thus, the absence of CSB alone cannot explain the striking variability in phenotype between CS and UVSS. A factor possibly involved in this variability might be the expression of an evolutionary conserved CSB-PGBD3 fusion protein, containing exons 1–5 of CSB joined in frame with the PiggyBac transposase. Reports have indicated that this fusion protein might play a role in the CS phenotype (Bailey et al. 2012).
The UVSSA factor
Recently, three laboratories identified UVSSA as the causative gene for the UVsS-A complementation group. Microcell-mediated chromosome transfer (Zhang et al. 2012), whole-exome sequencing (Nakazawa et al. 2012), and quantitative proteomics (Schwertman et al. 2012) were used to identify KIAA1530 as a new TC-NER factor, which was subsequently renamed UVSSA. Sequencing of this gene in five UVSS-A patient cell lines revealed three different inactivating mutations in the UVSSA gene (Table 1).
Within UVSSA, two conserved, though poorly characterized, domains are identified with homology to the Vps27-Hrs-STAM (VHS) domain and the DUF2043 domain. UVSS-A cells expressing UVSSA mutants without either of these domains fail to complement UVSS-A deficiency (Nakazawa et al. 2012), indicating that both domains are important for TC-NER activity. The UVSSA protein was shown to interact with the TC-NER factors RNA Pol II, CSA, CSB, and TFIIH (Schwertman et al. 2012; Nakazawa et al. 2012; Zhang et al. 2012). A specialized chromatin immunoprecipitation (ChIP) procedure showed that UVSSA resides in active, chromatin-bound TC-NER complexes upon UV damage. Furthermore, GFP-tagged UVSSA accumulated at local UV damage in living cells with similar recruitment kinetics as the TC-NER factor CSB (Schwertman et al. 2012). Together, these results showed that UVSSA is a novel TC-NER factor.
UVSSA recruitment
There are two, though not mutually exclusive, models explaining how UVSSA is recruited to UV lesions: (1) as an RNA Pol II interaction partner (Schwertman et al. 2012) and (2) as a CSA interaction partner (Zhang et al. 2012; Fei and Chen 2012).
We (Schwertman et al. 2012) observed a UV-independent interaction between RNA Pol II and UVSSA in ChIP experiments. This interaction was also observed in CSB-deficient patient cells, indicative of a CSB-independent binding of UVSSA to TC-NER complexes. In line with this, a CSA- and CSB-independent accumulation of GFP-UVSSA was observed at sites of local UV damage using live cell imaging. In contrast, Zhang et al. (2012) observed UV dependency for the UVSSA–RNA Pol II interaction and showed a CSA-dependent interaction with UVSSA in the chromatin fraction using non-cross-linking IPs; the latter was also observed by Fei and Chen (2012). An explanation for this apparent discrepancy in UVSSA recruitment could be that the CS proteins may be required for stable integration into a functional TC-NER complex rather than for the recruitment of UVSSA. Transient or low-affinity interactions could appear CSA/CSB independent if interactions are fixed by cross-linking as in ChIP experiments (Schwertman et al. 2012) and CSA/CSB dependent as observed in non-cross-linking IPs (Zhang et al. 2012; Fei and Chen 2012).
The observed CSA-dependent stable association of UVSSA into TC-NER complexes provides the basis for a possible molecular explanation of the UVSS/CS phenotypic difference. The CSA protein is a subunit of an E3 ubiquitin ligase complex (Groisman et al. 2003) that is recruited to the site of damage by CSB (Fousteri et al. 2006). Although essential for TC-NER, the precise role of CSA remains largely unknown. Nardo et al. (2009) described a patient with a specific missense CSA mutation, which causes the mild UV-sensitive phenotype in the absence of the severe premature aging features common in CS. Cells of this UVSS patient were hypersensitive to UV light, but not to oxidative damaging agents. This finding thus implies a separable role for CSA in response to UV and oxidative DNA damage. Interestingly, Fei and Chen (2012) showed that this same CSA mutant disrupts the interaction of CSA with UVSSA. This might suggest that an impaired interaction of UVSSA with mutated CSA at sites of DNA damage results in UVSS in this patient, while an unaffected role of CSA in other processes—such as in oxidative DNA damage repair—prevents the additional CS symptoms.
UVSSA function: a role in CSB stability
In addition to its interaction with CSA and RNA Pol II, UVSSA forms a UV-independent protein complex with the deubiquitinating enzyme (DUB) ubiquitin-specific protease 7 (USP7). USP7 (also known as HAUSP) has multiple roles in the DNA damage response, as illustrated by the wide variety of substrates including Mdm2, p53, claspin, Chfr, and histone H2B (Li et al. 2002; Meulmeester et al. 2005; Faustrup et al. 2009; Khoronenkova et al. 2011; Khoronenkova et al. 2012). Additionally, its diverse activity also includes targeting tumor suppressors, immune responders, viral proteins, and epigenetic modulators (Nicholson and Suresh Kumar 2011). Through its interaction with UVSSA, USP7 is recruited to active TC-NER complexes upon UV damage (Schwertman et al. 2012). Depletion of USP7 leads to a similar TC-NER deficiency as seen with UVSSA depletion, such as a decrease in UV survival and RNA synthesis recovery after UV (Schwertman et al. 2012; Zhang et al. 2012). From previous studies, it is known that CSB is ubiquitinated and degraded by the proteasome in response to UV (Groisman et al. 2006; Wei et al. 2011). In the absence of either UVSSA or USP7, the degradation of CSB by the 26S proteasome is faster upon UV damage. It is therefore suggested that UVSSA stabilizes CSB after UV by targeting the pleiotropic DUB USP7 to TC-NER complexes, which subsequently removes UV-induced ubiquitin chains. The purpose of this could be to provide an increased time frame for CSB to orchestrate TC-NER complex formation or to efficiently complete repair before CSB is degraded.
The stabilizing function of UVSSA/USP7 on CSB might be restricted to UV-induced TC-NER, since the reduced CSB levels in UVSS-A cells do not result in developmental and neurological symptoms in UVSS patients. Interestingly, overexpression of CSB in UVSS-A cells did not correct the TC-NER defect (Schwertman et al. 2012). This implies that the reduced level of CSB in UVSS-A cells alone is not sufficient to explain the UVSS phenotype. Therefore, the ubiquitination state of CSB itself might have an additional function for proper TC-NER, for example to mediate a ubiquitin-mediated functional change in CSB, or UVSSA/USP7 might have additional substrates that can cause the UVSS phenotype.
UVSSA function: a role in the processing of stalled RNA Pol II
Several transcription-associated factors recognize the C-terminal domain (CTD) of RNA Pol II by means of a conserved CTD-interacting domain (CID). The CID fold closely resembles that of VHS domains (Misra et al. 2002; Meinhart and Cramer 2004), suggesting that the observed interaction between UVSSA and RNA Pol II is mediated via the VHS domain of UVSSA. This interaction might be important for an additional function of UVSSA, as Nakazawa et al. (2012) showed that UVSSA is needed for processing stalled RNA Pol II at sites of UV damage. During TC-NER, elongating RNA Pol II can be recycled for a new round of transcription by means of dephosphorylating the elongating form of RNA Pol II into the hypophosphorylated initiating form (Rockx et al. 2000). Without the UVSSA protein, this dephosphorylation step is substantially inhibited (Nakazawa et al. 2012), as previously found in CS patient cells (Rockx et al. 2000), leading to impaired resumption of transcription initiation.
Next to its possible involvement in RNA Pol II binding, the VHS domain has also been implicated in ubiquitin binding (Mizuno et al. 2003), raising the possibility that UVSSA binds ubiquitinated TC-NER proteins. Upon UV irradiation, RNA Pol II is ubiquitinated and, under specific conditions, degraded (Bregman et al. 1996; Anindya et al. 2007; Malik et al. 2008). Nakazawa et al. (2012) identified a UVSSA-dependent ubiquitination of RNA Pol II, which however is not subject to proteasomal degradation. Interestingly, this new ubiquitinated form of stalled elongating RNA Pol II is UV specific, as it was not observed for oxidative DNA damage induced by hydrogen peroxide. Interaction of UVSSA with ubiquitinated RNA Pol II was indeed observed by Nakazawa et al. (2012). Additionally, we (Schwertman et al. 2012) identified UVSSA in a stable isotope labeling with amino acids in cell culture (SILAC)-based proteomic screen for differentially ubiquitinated proteins following UV irradiation. While the ubiquitination status of UVSSA was not changed after UV, a similar SILAC ratio for UVSSA and RNA Pol II was observed. This would suggest that UVSSA was co-purified by virtue of the enhanced ubiquitination of RNA Pol II in response to UV. However, whether this interaction is ubiquitination dependent remains unclear.
The VHS domain of UVSSA is suggested to be also involved in the interaction between UVSSA and CSB, since a UVSS-A patient missense mutation (Cys32Arg) within the VHS domain disrupts the interaction with CSB in response to UV. Interestingly, UVSS-A cells expressing UVSSA with this mutation were able to restore CSB stability to wild-type levels upon UV, while RNA Pol II ubiquitination and dephosphorylation remained absent (Nakazawa et al. 2012). This might indicate that while the VHS domain of UVSSA is dispensable for CSB stabilization, it is important for RNA Pol II processing. Apparently, the action of UVSSA on both CSB and RNA Pol II contributes to the UVSS phenotype. Although the precise involvement of UVSSA in processing these RNA Pol II modifications remains to be elucidated, it is hypothesized that the modifications themselves contribute to the coordination of the removal of stalled RNA Pol II and formation of the TC-NER complex and the subsequent resumption of transcription.
New insights in transcription-coupled DNA repair from a UVSSA perspective
Mutations in UVSSA were recently identified to cause UVSS, a previously unresolved TC-NER deficiency disorder with rather mild clinical manifestations as compared to CS. UVSSA and its interaction partner USP7 were identified as new factors involved in TC-NER repair efficiency and cell survival after UV damage. Additionally, UVSSA is also important for the fate and ubiquitination state of both RNA Pol II and CSB. The identification of these new transcription-coupled DNA repair factors presents us with the opportunity to gain more insight into the molecular mechanisms underlying the clinical differences between UVSS and CS.
The transcription defect model discussed previously is based on a role for the CS proteins in transcription, while assuming UVSSA is not involved in this process. The UV-independent transient interaction between UVSSA and RNA Pol II may suggest a role for UVSSA in transcription. However, no other data are currently available to conclude whether UVSSA is as equally important as CSB for transcription on undamaged DNA. Therefore, further studies are needed to determine the possible involvement of UVSSA in transcription.
One of the models including UVSSA involves the aberrant processing of elongating RNA Pol II when stalled at a lesion (Fig. 3). For efficient repair to take place, the RNA Pol II complex must be remodeled since it will likely shield the DNA lesion and consequently prevent accessibility of repair proteins (Donahue et al. 1994; Tornaletti et al. 2001; Brueckner et al. 2007; Fousteri and Mullenders 2008; Wilson et al. 2013). Additionally, when a lesion cannot be repaired or bypassed, stalled RNA Pol II is thought to be ubiquitinated and degraded in order to prevent persistent transcriptional arrest (Wilson et al. 2013). In the absence of UVSSA, stalled RNA Pol II can still be ubiquitinated in a CSA/CSB-dependent way, which results in proteasomal degradation. In contrast, stalled RNA Pol II cannot be degraded by the proteasome in CS cells (Hanawalt and Spivak 2008). This would suggest that the lack of DNA repair combined with persistent arrest of RNA Pol II at DNA lesions in CS cells, which results in apoptosis or senescence, might be causative for the severe CS phenotype. In UVSS-A cells, prolonged arrest is prevented and the lesion is made accessible for alternative DNA repair by degradation of RNA Pol II, hence resulting in the milder UVSS phenotype (Fig. 3).
Fig. 3.
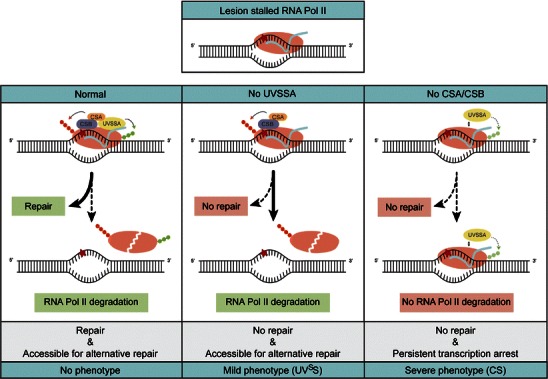
Differential aberrant processing of stalled elongating RNA Pol II is responsible for the differences in UVSS/CS phenotype. In wild-type cells, lesion stalled RNA Pol II initiates TC-NER to remove the transcription-blocking damage. In the infrequent cases that repair cannot take place, RNA Pol II is ubiquitinated and degraded by the proteasome to make the lesion accessible for alternative DNA repair. In UVSS-A cells, TC-NER does not take place. Since the prolonged stalled RNA Pol II can still be ubiquitinated in a CSA/CSB dependent way, RNA Pol II is degraded and the lesion is made accessible for alternative DNA repair. In CS cells, TC-NER is blocked as well. In addition, the ubiquitination and degradation of RNA Pol II does not take place. The resultant persistent transcription arrest is more harmful for the cell, since it leads to apoptosis or senescence and might contribute to the more severe phenotype in CS patients
The CS proteins play important roles in both TC-NER and oxidative DNA damage repair, while the precise function of UVSSA in these processes remains unclear. The previously discussed oxidative damage model assumes that UVSSA does not play a role in oxidative DNA damage repair (Spivak and Hanawalt 2006), hence explaining the milder phenotype of UVSS. We would like to postulate a model including UVSSA and its role in both repair pathways (Fig. 1). We hypothesize that based on the nature of the DNA damage, distinct modulations of lesion stalled RNA Pol II are required and that this involves a remodeling function of CSB. While oxidative lesions only slow down or transiently arrest RNA Pol II, more helix-distorting lesions (such as those induced by UV) would lead to a prolonged transcriptional arrest. In the case of transient transcription arrest, CSB remodeling assists in the efficient removal of a lesion or in bypassing the lesion (Doetsch 2002; Charlet-Berguerand et al. 2006; Damsma and Cramer 2009). In contrast, for a prolonged blocked transcription, more time would be required for CSB to assemble a functional TC-NER complex and to assist in remodeling of the stalled RNA Pol II. The presence of CSB should, in the latter case, be protected by the concerted action of UVSSA/USP7 to allow sufficient time for CSB to perform its remodeling function. Whether the ubiquitination of CSB takes place at all after transient transcription arrest at oxidative DNA lesions or if CSB is ubiquitinated but not degraded before the repair of these lesions is completed remains to be elucidated. In this proposed model, mutations in UVSSA would lead to dysfunctional TC-NER only, since the lack of CSB protection would not affect repair or restart of transcription blocked by oxidative lesions.
The discovery of UVSSA enables us to test the proposed models in greater detail and whether or not they are mutually exclusive. To further substantiate the hypothesis that the phenotypical difference between UVSS and CS is derived from different sensitivities for endogenous levels of oxidative DNA damage, dedicated lesion-specific DNA repair assays are required (Spivak et al. 2009). Furthermore, dissecting the differences in molecular composition between repair complexes stalled at UV-induced lesions or oxidative lesions will help us to better understand the molecular mechanism of transcription-coupled repair for different types of DNA damage. Characterization of repair complexes present on the different lesions using immunoprecipitation combined with mass spectrometry would be a powerful tool to study this. Another interesting area for further investigation is the differential ubiquitination of several DNA repair factors, which is likely an important driver of protein hand over and passing of repair intermediates through successive steps during TC-NER. Especially the actual and/or precise involvement of USP7 in changing the ubiquitination state of CSB and RNA Pol II upon UV is of great interest, as is the establishment of identified or new E3 ubiquitin ligases responsible for the ubiquitin modifications. Finally, with the identification of UVSSA, a UVSS-A mouse model can now be generated for a more extensive study of the UVSS phenotype and molecular defect. Also, an overall comparison of the UVSS-A mouse with the already existing CS mouse models in an isogenic background will be a valuable source of information. Phenotypic consequences and possible differential responses to various genotoxic agents in the whole organism will contribute to our understanding of the clinical differences of CS versus UVSS on a molecular level.
Acknowledgments
This work is funded by the Netherlands Genomics Initiative NPCII and the Dutch Organization for Scientific Research ZonMW: Horizon Zenith (935.11.042), Veni (917.96.120), and TOP grant (912.08.031 and 912.12.132).
References
- Anindya R, Aygun O, Svejstrup JQ. Damage-induced ubiquitylation of human RNA polymerase II by the ubiquitin ligase Nedd4, but not Cockayne syndrome proteins or BRCA1. Mol Cell. 2007;28(3):386–397. doi: 10.1016/j.molcel.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Bailey AD, Gray LT, Pavelitz T, Newman JC, Horibata K, Tanaka K, Weiner AM. The conserved Cockayne syndrome B-piggyBac fusion protein (CSB-PGBD3) affects DNA repair and induces both interferon-like and innate antiviral responses in CSB-null cells. DNA Repair (Amst) 2012;11(5):488–501. doi: 10.1016/j.dnarep.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balajee AS, May A, Dianov GL, Friedberg EC, Bohr VA. Reduced RNA polymerase II transcription in intact and permeabilized Cockayne syndrome group B cells. Proc Natl Acad Sci U S A. 1997;94(9):4306–4311. doi: 10.1073/pnas.94.9.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci U S A. 1996;93(21):11586–11590. doi: 10.1073/pnas.93.21.11586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueckner F, Hennecke U, Carell T, Cramer P. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315(5813):859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- Charlet-Berguerand N, Feuerhahn S, Kong SE, Ziserman H, Conaway JW, Conaway R, Egly JM. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006;25(23):5481–5491. doi: 10.1038/sj.emboj.7601403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citterio E, Van Den Boom V, Schnitzler G, Kanaar R, Bonte E, Kingston RE, Hoeijmakers JH, Vermeulen W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol Cell Biol. 2000;20(20):7643–7653. doi: 10.1128/MCB.20.20.7643-7653.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleaver JE. Photosensitivity syndrome brings to light a new transcription-coupled DNA repair cofactor. Nat Genet. 2012;44(5):477–478. doi: 10.1038/ng.2255. [DOI] [PubMed] [Google Scholar]
- Cleaver JE, Thomas GH. Clinical syndromes associated with DNA repair deficiency and enhanced sun sensitivity. Arch Dermatol. 1993;129(3):348–350. doi: 10.1001/archderm.1993.01680240088014. [DOI] [PubMed] [Google Scholar]
- D’Errico M, Parlanti E, Teson M, Degan P, Lemma T, Calcagnile A, Iavarone I, Jaruga P, Ropolo M, Pedrini AM, Orioli D, Frosina G, Zambruno G, Dizdaroglu M, Stefanini M, Dogliotti E. The role of CSA in the response to oxidative DNA damage in human cells. Oncogene. 2007;26(30):4336–4343. doi: 10.1038/sj.onc.1210232. [DOI] [PubMed] [Google Scholar]
- Damsma GE, Cramer P. Molecular basis of transcriptional mutagenesis at 8-oxoguanine. J Biol Chem. 2009;284(46):31658–31663. doi: 10.1074/jbc.M109.022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard H, de Wit J, Andressoo JO, van Oostrom CT, Riis B, Weimann A, Poulsen HE, van Steeg H, Hoeijmakers JH, van der Horst GT. Different effects of CSA and CSB deficiency on sensitivity to oxidative DNA damage. Mol Cell Biol. 2004;24(18):7941–7948. doi: 10.1128/MCB.24.18.7941-7948.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov G, Bischoff C, Sunesen M, Bohr VA. Repair of 8-oxoguanine in DNA is deficient in Cockayne syndrome group B cells. Nucleic Acids Res. 1999;27(5):1365–1368. doi: 10.1093/nar/27.5.1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dianov GL, Houle JF, Iyer N, Bohr VA, Friedberg EC. Reduced RNA polymerase II transcription in extracts of cockayne syndrome and xeroderma pigmentosum/Cockayne syndrome cells. Nucleic Acids Res. 1997;25(18):3636–3642. doi: 10.1093/nar/25.18.3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetsch PW. Translesion synthesis by RNA polymerases: occurrence and biological implications for transcriptional mutagenesis. Mutat Res. 2002;510(1–2):131–140. doi: 10.1016/s0027-5107(02)00258-0. [DOI] [PubMed] [Google Scholar]
- Donahue BA, Yin S, Taylor JS, Reines D, Hanawalt PC. Transcript cleavage by RNA polymerase II arrested by a cyclobutane pyrimidine dimer in the DNA template. Proc Natl Acad Sci U S A. 1994;91(18):8502–8506. doi: 10.1073/pnas.91.18.8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faustrup H, Bekker-Jensen S, Bartek J, Lukas J, Mailand N. USP7 counteracts SCFbetaTrCP- but not APCCdh1-mediated proteolysis of Claspin. J Cell Biol. 2009;184(1):13–19. doi: 10.1083/jcb.200807137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei J, Chen J. KIAA1530 protein is recruited by Cockayne syndrome complementation group protein A (CSA) to participate in transcription-coupled repair (TCR) J Biol Chem. 2012;287(42):35118–35126. doi: 10.1074/jbc.M112.398131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18(1):73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23(4):471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Frontini M, Proietti-De-Santis L. Cockayne syndrome B protein (CSB): linking p53, HIF-1 and p300 to robustness, lifespan, cancer and cell fate decisions. Cell Cycle. 2009;8(5):693–696. doi: 10.4161/cc.8.5.7754. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Ichihashi M, Kano Y, Goto K, Shimizu K. A new human photosensitive subject with a defect in the recovery of DNA synthesis after ultraviolet-light irradiation. J Invest Dermatol. 1981;77(3):256–263. doi: 10.1111/1523-1747.ep12482447. [DOI] [PubMed] [Google Scholar]
- Fujiwara Y, Uehara Y, Ichihashi M, Yamamoto Y, Nishioka K. Assignment of 2 patients with xeroderma pigmentosum to complementation group E. Mutat Res. 1985;145(1–2):55–61. doi: 10.1016/0167-8817(85)90040-9. [DOI] [PubMed] [Google Scholar]
- Gorgels TG, van der Pluijm I, Brandt RM, Garinis GA, van Steeg H, van den Aardweg G, Jansen GH, Ruijter JM, Bergen AA, van Norren D, Hoeijmakers JH, van der Horst GT. Retinal degeneration and ionizing radiation hypersensitivity in a mouse model for Cockayne syndrome. Mol Cell Biol. 2007;27(4):1433–1441. doi: 10.1128/MCB.01037-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Kuraoka I, Chevallier O, Gaye N, Magnaldo T, Tanaka K, Kisselev AF, Harel-Bellan A, Nakatani Y. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006;20(11):1429–1434. doi: 10.1101/gad.378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groisman R, Polanowska J, Kuraoka I, Sawada J, Saijo M, Drapkin R, Kisselev AF, Tanaka K, Nakatani Y. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113(3):357–367. doi: 10.1016/S0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9(12):958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008;18(1):27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JH. Genome maintenance mechanisms are critical for preventing cancer as well as other aging-associated diseases. Mech Ageing Dev. 2007;128(7–8):460–462. doi: 10.1016/j.mad.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Horibata K, Iwamoto Y, Kuraoka I, Jaspers NG, Kurimasa A, Oshimura M, Ichihashi M, Tanaka K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci U S A. 2004;101(43):15410–15415. doi: 10.1073/pnas.0404587101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Fujiwara Y, Ono T, Yamaizumi M. UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Am J Hum Genet. 1995;56(6):1267–1276. [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Linn S, Ono T, Yamaizumi M. Reinvestigation of the classification of five cell strains of xeroderma pigmentosum group E with reclassification of three of them. J Invest Dermatol. 2000;114(5):1022–1029. doi: 10.1046/j.1523-1747.2000.00952.x. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ono T, Yamaizumi M. A new UV-sensitive syndrome not belonging to any complementation groups of xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat Res. 1994;314(3):233–248. doi: 10.1016/0921-8777(94)90068-X. [DOI] [PubMed] [Google Scholar]
- Itoh T, Yamaizumi M, Ichihashi M, Hiro-Oka M, Matsui T, Matsuno M, Ono T. Clinical characteristics of three patients with UVs syndrome, a photosensitive disorder with defective DNA repair. Br J Dermatol. 1996;134(6):1147–1150. doi: 10.1111/j.1365-2133.1996.tb07963.x. [DOI] [PubMed] [Google Scholar]
- Kamenisch Y, Fousteri M, Knoch J, von Thaler AK, Fehrenbacher B, Kato H, Becker T, Dolle ME, Kuiper R, Majora M, Schaller M, van der Horst GT, van Steeg H, Rocken M, Rapaport D, Krutmann J, Mullenders LH, Berneburg M. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med. 2010;207(2):379–390. doi: 10.1084/jem.20091834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawada A, Satoh Y, Fujiwara Y. Xeroderma pigmentosum complementation group E: a case report. Photodermatol. 1986;3(4):233–238. [PubMed] [Google Scholar]
- Khoronenkova SV, Dianova II, Parsons JL, Dianov GL. USP7/HAUSP stimulates repair of oxidative DNA lesions. Nucleic Acids Res. 2011;39(7):2604–2609. doi: 10.1093/nar/gkq1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoronenkova SV, Dianova II, Ternette N, Kessler BM, Parsons JL, Dianov GL. ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol Cell. 2012;45(6):801–813. doi: 10.1016/j.molcel.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Durand M, Sauvanaud F, Kristensen U, Vincent MC, Pasquier L, Odent S, Cormier-Daire V, Gener B, Tobias ES, Tolmie JL, Martin-Coignard D, Drouin-Garraud V, Heron D, Journel H, Raffo E, Vigneron J, Lyonnet S, Murday V, Gubser-Mercati D, Funalot B, Brueton L, Sanchez Del Pozo J, Munoz E, Gennery AR, Salih M, Noruzinia M, Prescott K, Ramos L, Stark Z, Fieggen K, Chabrol B, Sarda P, Edery P, Bloch-Zupan A, Fawcett H, Pham D, Egly JM, Lehmann AR, Sarasin A, Dollfus H. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat. 2010;31(2):113–126. doi: 10.1002/humu.21154. [DOI] [PubMed] [Google Scholar]
- Laugel V, Dalloz C, Stary A, Cormier-Daire V, Desguerre I, Renouil M, Fourmaintraux A, Velez-Cruz R, Egly JM, Sarasin A, Dollfus H. Deletion of 5' sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur J Hum Genet. 2008;16(3):320–327. doi: 10.1038/sj.ejhg.5201991. [DOI] [PubMed] [Google Scholar]
- Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416(6881):648–653. doi: 10.1038/nature737. [DOI] [PubMed] [Google Scholar]
- Ljungman M, Lane DP. Transcription - guarding the genome by sensing DNA damage. Nat Rev Cancer. 2004;4(9):727–737. doi: 10.1038/nrc1435. [DOI] [PubMed] [Google Scholar]
- Ljungman M, Zhang F. Blockage of RNA polymerase as a possible trigger for u.v. light-induced apoptosis. Oncogene. 1996;13(4):823–831. [PubMed] [Google Scholar]
- Malik S, Bagla S, Chaurasia P, Duan Z, Bhaumik SR. Elongating RNA polymerase II is disassembled through specific degradation of its largest but not other subunits in response to DNA damage in vivo. J Biol Chem. 2008;283(11):6897–6905. doi: 10.1074/jbc.M707649200. [DOI] [PubMed] [Google Scholar]
- Meinhart A, Cramer P. Recognition of RNA polymerase II carboxy-terminal domain by 3'-RNA-processing factors. Nature. 2004;430(6996):223–226. doi: 10.1038/nature02679. [DOI] [PubMed] [Google Scholar]
- Menoni H, Hoeijmakers JH, Vermeulen W. Nucleotide excision repair-initiating proteins bind to oxidative DNA lesions in vivo. J Cell Biol. 2012;199(7):1037–1046. doi: 10.1083/jcb.201205149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meulmeester E, Maurice MM, Boutell C, Teunisse AF, Ovaa H, Abraham TE, Dirks RW, Jochemsen AG. Loss of HAUSP-mediated deubiquitination contributes to DNA damage-induced destabilization of Hdmx and Hdm2. Mol Cell. 2005;18(5):565–576. doi: 10.1016/j.molcel.2005.04.024. [DOI] [PubMed] [Google Scholar]
- Misra S, Puertollano R, Kato Y, Bonifacino JS, Hurley JH. Structural basis for acidic-cluster-dileucine sorting-signal recognition by VHS domains. Nature. 2002;415(6874):933–937. doi: 10.1038/415933a. [DOI] [PubMed] [Google Scholar]
- Miyauchi-Hashimoto H, Akaeda T, Maihara T, Ikenaga M, Horio T. Cockayne syndrome without typical clinical manifestations including neurologic abnormalities. J Am Acad Dermatol. 1998;39(4 Pt 1):565–570. doi: 10.1016/S0190-9622(98)70005-2. [DOI] [PubMed] [Google Scholar]
- Mizuno E, Kawahata K, Kato M, Kitamura N, Komada M. STAM proteins bind ubiquitinated proteins on the early endosome via the VHS domain and ubiquitin-interacting motif. Mol Biol Cell. 2003;14(9):3675–3689. doi: 10.1091/mbc.E02-12-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Sasaki K, Mitsutake N, Matsuse M, Shimada M, Nardo T, Takahashi Y, Ohyama K, Ito K, Mishima H, Nomura M, Kinoshita A, Ono S, Takenaka K, Masuyama R, Kudo T, Slor H, Utani A, Tateishi S, Yamashita S, Stefanini M, Lehmann AR, Yoshiura K, Ogi T. Mutations in UVSSA cause UV-sensitive syndrome and impair RNA polymerase IIo processing in transcription-coupled nucleotide-excision repair. Nat Genet. 2012;44(5):586–592. doi: 10.1038/ng.2229. [DOI] [PubMed] [Google Scholar]
- Nance MA, Berry SA. Cockayne syndrome: review of 140 cases. Am J Med Genet. 1992;42(1):68–84. doi: 10.1002/ajmg.1320420115. [DOI] [PubMed] [Google Scholar]
- Nardo T, Oneda R, Spivak G, Vaz B, Mortier L, Thomas P, Orioli D, Laugel V, Stary A, Hanawalt PC, Sarasin A, Stefanini M. A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci U S A. 2009;106(15):6209–6214. doi: 10.1073/pnas.0902113106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman JC, Bailey AD, Weiner AM. Cockayne syndrome group B protein (CSB) plays a general role in chromatin maintenance and remodeling. Proc Natl Acad Sci U S A. 2006;103(25):9613–9618. doi: 10.1073/pnas.0510909103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60(1–2):61–68. doi: 10.1007/s12013-011-9185-5. [DOI] [PubMed] [Google Scholar]
- Osenbroch PO, Auk-Emblem P, Halsne R, Strand J, Forstrom RJ, van der Pluijm I, Eide L. Accumulation of mitochondrial DNA damage and bioenergetic dysfunction in CSB defective cells. FEBS J. 2009;276(10):2811–2821. doi: 10.1111/j.1742-4658.2009.07004.x. [DOI] [PubMed] [Google Scholar]
- Pascucci B, Lemma T, Iorio E, Giovannini S, Vaz B, Iavarone I, Calcagnile A, Narciso L, Degan P, Podo F, Roginskya V, Janjic BM, Van Houten B, Stefanini M, Dogliotti E, D’Errico M. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell. 2012;11(3):520–529. doi: 10.1111/j.1474-9726.2012.00815.x. [DOI] [PubMed] [Google Scholar]
- Rockx DA, Mason R, van Hoffen A, Barton MC, Citterio E, Bregman DB, van Zeeland AA, Vrieling H, Mullenders LH. UV-induced inhibition of transcription involves repression of transcription initiation and phosphorylation of RNA polymerase II. Proc Natl Acad Sci U S A. 2000;97(19):10503–10508. doi: 10.1073/pnas.180169797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin A. UVSSA and USP7: new players regulating transcription-coupled nucleotide excision repair in human cells. Genome Med. 2012;4(5):44. doi: 10.1186/gm343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DM, 3rd, Cooper M, Spencer R, de Cabo R, Croteau DL, Bohr VA. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. 2012;209(4):855–869. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwertman P, Lagarou A, Dekkers DH, Raams A, van der Hoek AC, Laffeber C, Hoeijmakers JH, Demmers JA, Fousteri M, Vermeulen W, Marteijn JA. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat Genet. 2012;44(5):598–602. doi: 10.1038/ng.2230. [DOI] [PubMed] [Google Scholar]
- Selby CP, Sancar A. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem. 1997;272(3):1885–1890. doi: 10.1074/jbc.272.3.1885. [DOI] [PubMed] [Google Scholar]
- Spivak G. UV-sensitive syndrome. Mutat Res. 2005;577(1–2):162–169. doi: 10.1016/j.mrfmmm.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Spivak G, Cox RA, Hanawalt PC. New applications of the Comet assay: Comet-FISH and transcription-coupled DNA repair. Mutat Res. 2009;681(1):44–50. doi: 10.1016/j.mrrev.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivak G, Hanawalt PC. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006;5(1):13–22. doi: 10.1016/j.dnarep.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Stevnsner T, Muftuoglu M, Aamann MD, Bohr VA. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev. 2008;129(7–8):441–448. doi: 10.1016/j.mad.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevnsner T, Nyaga S, de Souza-Pinto NC, van der Horst GT, Gorgels TG, Hogue BA, Thorslund T, Bohr VA. Mitochondrial repair of 8-oxoguanine is deficient in Cockayne syndrome group B. Oncogene. 2002;21(57):8675–8682. doi: 10.1038/sj.onc.1205994. [DOI] [PubMed] [Google Scholar]
- Tantin D, Kansal A, Carey M. Recruitment of the putative transcription-repair coupling factor CSB/ERCC6 to RNA polymerase II elongation complexes. Mol Cell Biol. 1997;17(12):6803–6814. doi: 10.1128/mcb.17.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornaletti S, Maeda LS, Lloyd DR, Reines D, Hanawalt PC. Effect of thymine glycol on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. J Biol Chem. 2001;276(48):45367–45371. doi: 10.1074/jbc.M105282200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troelstra C, Odijk H, de Wit J, Westerveld A, Thompson LH, Bootsma D, Hoeijmakers JH. Molecular cloning of the human DNA excision repair gene ERCC-6. Mol Cell Biol. 1990;10(11):5806–5813. doi: 10.1128/mcb.10.11.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuo J, Jaruga P, Rodriguez H, Bohr VA, Dizdaroglu M. Primary fibroblasts of Cockayne syndrome patients are defective in cellular repair of 8-hydroxyguanine and 8-hydroxyadenine resulting from oxidative stress. FASEB J. 2003;17(6):668–674. doi: 10.1096/fj.02-0851com. [DOI] [PubMed] [Google Scholar]
- van den Boom V, Citterio E, Hoogstraten D, Zotter A, Egly JM, van Cappellen WA, Hoeijmakers JH, Houtsmuller AB, Vermeulen W. DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J Cell Biol. 2004;166(1):27–36. doi: 10.1083/jcb.200401056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Pluijm I, Garinis GA, Brandt RM, Gorgels TG, Wijnhoven SW, Diderich KE, de Wit J, Mitchell JR, van Oostrom C, Beems R, Niedernhofer LJ, Velasco S, Friedberg EC, Tanaka K, van Steeg H, Hoeijmakers JH, van der Horst GT. Impaired genome maintenance suppresses the growth hormone—insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2007;5(1):e2. doi: 10.1371/journal.pbio.0050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool AJ, Citterio E, Rademakers S, van Os R, Vermeulen W, Constantinou A, Egly JM, Bootsma D, Hoeijmakers JH. The Cockayne syndrome B protein, involved in transcription-coupled DNA repair, resides in an RNA polymerase II-containing complex. EMBO J. 1997;16(19):5955–5965. doi: 10.1093/emboj/16.19.5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard J. Transcription regulation and human diseases. Swiss Med Wkly. 2004;134(39–40):571–579. doi: 10.4414/smw.2004.10191. [DOI] [PubMed] [Google Scholar]
- Wei L, Lan L, Yasui A, Tanaka K, Saijo M, Matsuzawa A, Kashiwagi R, Maseki E, Hu Y, Parvin JD, Ishioka C, Chiba N. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011;102(10):1840–1847. doi: 10.1111/j.1349-7006.2011.02037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MD, Harreman M, Svejstrup JQ. Ubiquitylation and degradation of elongating RNA polymerase II: the last resort. Biochim Biophys Acta. 2013;1829(1):151–157. doi: 10.1016/j.bbagrm.2012.08.002. [DOI] [PubMed] [Google Scholar]
- Zhang X, Horibata K, Saijo M, Ishigami C, Ukai A, Kanno S, Tahara H, Neilan EG, Honma M, Nohmi T, Yasui A, Tanaka K. Mutations in UVSSA cause UV-sensitive syndrome and destabilize ERCC6 in transcription-coupled DNA repair. Nat Genet. 2012;44(5):593–597. doi: 10.1038/ng.2228. [DOI] [PubMed] [Google Scholar]