

Abstract
Endothelial adhesion is necessary for the hematogenous dissemination of tumor cells. However, the metastatic breast tumor cell MDA-MB-231 does not bind to the endothelium under physiological flow conditions, suggesting alternate mechanisms of adhesion. Since monocytes are highly represented in the tumor microenvironment, and also bind to endothelium during inflammation, we hypothesized that the monocytes assist in the arrest of MDA-MB-231 on the endothelium. Using in vitro models of the dynamic shear environment of the vasculature, we show that TNF-α-activated THP1/primary human monocytes and MDA-MB-231 cells form stable aggregates, and that the monocytes in these aggregates mediate the adhesion of otherwise nonadherent MDA-MB-231 cells to inflamed endothelium under flow (55±2.4 vs. 1.7±0.82 at a shear stress of 0.5 dyn/cm2, P<0.01). We also show that the hydrodynamic forces determine the size and orientation of aggregates adhered to the endothelium, and strongly favor the attachment of small aggregates with tumor cells downstream of flow (74–86% doublets at 0.5–2 dyn/cm2, P<0.01). The 5-fold up-regulation of ICAM-1 on TNF-α-activated MDA-MB-231 cells through the Nf-κB pathway was found to be critical in MDA-MB-231–monocyte aggregation and endothelial adhesion. Our results demonstrate that, under inflammatory conditions, monocytes may serve to disseminate tumor cells through circulation, and the tumor–monocyte–endothelial axis may represent a new therapeutic target to reduce cancer metastasis.—Evani, S. J., Prabhu, R. G., Gnanaruban, V., Finol, E. A., Ramasubramanian, A. K. Monocytes mediate metastatic breast tumor cell adhesion to endothelium under flow.
Keywords: MDA-MB-231, shear, aggregation
Metastasis is a major cause of cancer mortality. In the continuum of metastatic process, extravasation from circulation is one of the key steps by which circulating tumor cells escape from the bloodstream by binding to the endothelial cells that line the blood vessels and migrate into the extracellular matrix at distant sites (1). An important aspect of this process is that the cells moving with the flow should attain proximity to the endothelium, interact sufficiently strongly with the endothelium against the detaching forces due to flow to get captured, adhere firmly, and eventually extravasate (2, 3). Such interactions are typically mediated by the glycoprotein receptors, such as selectins, integrins, and their ligands, that are expressed at various levels by some of the tumor cells, as these molecules bind to the corresponding counterreceptors on the endothelium. The selectin and integrin receptors are also abundantly expressed on leukocytes and platelets, which allow them to arrest, roll, and firmly adhere on the endothelium during inflammation and hemostasis (3). Recent evidence suggest that several metastatic tumor cells interact with neutrophils and platelets, which, in turn, assist in the arrest of the tumor cells, especially those that express lower levels of endothelial counterreceptors, including colon adenocarcinoma (4), retinoblastoma (5), and melanoma (6, 7), on the endothelium, and eventual extravasation from the vasculature. These interactions suggest a mechanistic link between cancer and inflammation, particularly in the light of strong correlational evidence between abnormalities in coagulation and systemic inflammation normally observed in patients with cancer (8).
Several clinical and experimental studies have shown that monocytes/macrophages facilitate tumor metastasis. Although conventional wisdom suggests that macrophages should be involved in antitumor immunity, clinical studies have made strong associations between poor survival and prognosis and increased macrophage density in several cancers (9–11). Macrophages that are found in the primary tumor site, known as tumor-associated-macrophages (TAMs) promote tumor initiation and progression in several ways (12): producing a mutagenic environment rich in reactive nitrogen and oxygen species (13) and inflammatory cytokines (14), producing epidermal growth factor (EGF), which activates migration and intravasation of tumor cells (15), vascular remodeling, and angiogenesis by releasing matrix metalloproteases and vascular endothelial growth factor (VEGF (16). While most studies have focused on the role of macrophages at the primary tumor site in the initial steps of the metastatic cascade, their contribution to the transit of tumor cells through circulation or lymphatics and extravasation to distant sites is poorly characterized. Only recently, it has been shown in mouse models of breast cancer that at the target site, monocytes/macrophages are abundantly present in close apposition to the tumor cells, and may aid in tumor extravasation (17, 18).
In the light of the aforementioned observations, we hypothesize that monocytes coopt in metastasis by assisting the adhesion of tumor cells to the endothelium under flow. Monocyte rolling on endothelium is mediated by the β1-integrin very late antigen 4 (VLA-4), which on activation results in cell arrest (19). The MDA-MB-231 line is a highly metastatic breast tumor cell line that exhibits genetic and molecular characteristics that support cell proliferation, tissue invasion, and migration (20). However, the MDA-MB-231 cells do not appear to adequately express the appropriate sialyl Lewis-x or -a (sLex/a) or other sialyofucosylated ligands for selectins that are considered essential for tethering to the endothelium under flow conditions (21, 22). We demonstrate that MDA-MB-231 cells, under physiological flow conditions, do not bind to the endothelium directly but utilize interactions with monocytes (THP1 or primary monocytes) to arrest onto the endothelium. We also demonstrate that an inflammatory environment is decisive for stable binding of monocytes and MDA-MB-231 cells, and that the fluid forces dictate the dynamics of interaction between the MDA-MB-231/monocyte aggregates and the endothelial cells. Our results suggest a novel role for monocytes in an inflammatory microenvironment in breast cancer metastasis.
MATERIALS AND METHODS
Cells
Primary human monocytes were isolated from buffy coat using monocyte isolation kit II by indirect magnetic labeling (Miltenyi Biotech, Auburn, CA, USA), and the purity of the monocytes was determined using FITC-conjugated anti-CD14 antibody and a suitable isotype control (Miltenyi Biotech). Human monocyte cell line THP1 [American Type Culture Collection (ATCC), Manassas, VA, USA] was cultured in RPMI 1640 (ATCC) supplemented with 10% FBS and .05 mM mercaptoethanol (Sigma-Aldrich, St. Louis, MO, USA), at 37°C and 5% CO2. The cells were passaged into fresh medium when the cells reached a density of 106/ml. Human metastatic breast cancer cell line, MDA-MB-231 (ATCC), was cultured in L-15 medium (ATCC) supplemented with 10% FBS (Life Technologies, Carlsbad, CA, USA), at 37°C in a non-CO2 incubator. The cells were passaged into fresh medium when the cells reached subconfluence. Human aortic endothelial cells (HAECs) were originally extracted from the aorta of a cadaver of an individual with no known cardiovascular abnormalities, and human dermal microvascular endothelial cells (MVECs) were obtained from Lifeline Cell Technology (Frederick, MD, USA). The cells (up to generation P5) were cultured in medium supplemented with 5% FBS as per the manufacturer's instructions. The cell viability was measured by trypan blue exclusion assay using an automated cell counter (Countess; Invitrogen, Carlsbad, CA, USA).
Aggregation of tumor cells and THP1 or primary monocytes
THP1 cells or primary monocytes and MDA-MB-231 cells were stained in suspension with PKH-26 (λem 567 nm) and PKH-67 (λem 502 nm), respectively, as per the manufacturer's instructions (Sigma-Aldrich). The MDA-MB-231 cells were seeded separately in 6-well plates (106 cells/well) until they reached 80% confluence. All the cells were activated with either 10 ng/ml of recombinant tumor necrosis factor α (TNF-α; R&D Systems, Minneapolis, MN, USA) or medium alone (mock treatment) for 6 h. MDA-MB-231 cells were detached with gentle scraping, washed twice in HBSS, and resuspended in complete L-15 medium with 2% BSA (Research Organics, Cleveland, OH, USA). Monocytes in suspension were washed twice in HBSS and resuspended in complete RPMI medium with 2% BSA. A total of 2 × 106 cells with 106 monocytes and 106 MDA-MB-231 cells in 1 ml were incubated for 20 min at 37°C with gentle rocking. The numbers of THP1 and MDA-MB-231 cells varied, with ratios of 5:1, 1:1, or 1:5. Cell suspension (80 μl) was then sheared in a cone-and-plate rheometer with 0.5° cone (MCR 302; Anton Paar, Scharnhausen Ostfildern, Germany) at 500 s−1 for 2 min, and was immediately fixed with 2% formaldehyde. The samples were analyzed by flow cytometry (LSR II; BD Biosciences, San Jose, CA, USA) and by fluorescence microscopy at ×400 (Leica Microsystems, Wetzlar, Germany). Controls consisted of monocytes and MDA-MB-231 cells alone. In certain experiments, monocytes were incubated with either monoclonal antibody against β2 integrin (MEM-48 clone, AbCam, Cambridge, MA, USA) or the corresponding isotype control (AbCam), or MDA-MB-231 cells were treated with monoclonal antibody against intercellular adhesion molecule 1 (ICAM-1; HA58 clone, eBioscience, San Diego, CA, USA) or corresponding control antibody (eBioscience) at a concentration of 10 μg/106 cells in 100 μl volume for 20 min at 37°C. The cells were washed 2 times with PBS to remove any excess antibody. In certain other experiments, MDA-MB-231 cells were preincubated with 20 μM of calpain inhibitor IV MG-132 (American Peptide Co., Sunnyvale, CA, USA) for 30 min before and through the duration of TNF-α activation.
The aggregates were characterized based on the fluorescence intensities from dual-color flow cytometry (4). The monocytes and MDA-MB-231 cells were identified based on their characteristic forward-scatter (FSC)/side-scatter (SSC) profiles and fluorescence from the FL-1 (530 nm, green) and FL-2 (585 nm, red) channels in unactivated samples. The aggregates in TNF-α-activated samples were distinguished from the increase in the size in the FSC/SSC plots. Homoaggregates consisting of pure population of either monocytes or MDA-MB-231 cells were quantified from an increase in red or green fluorescence intensities over single-cell levels with only background levels of green or red fluorescence intensity, respectively. Heteroaggregates consisting of both monocytes and MDA-MB-231 cells were quantified from an increase in both green and red fluorescence intensities over the background noise.
Cell adhesion assay
For adhesion assays, HAECs or MVECs were seeded into ibiTreat Vl0.1 slides (Ibidi GmbH, Munich, Germany) at a density of 2 × 105 cells/slide and cultured 24 h to achieve confluent monolayers. The nonadherent cells were washed off with medium after 30 min of seeding, and the slide was supplemented with fresh medium and cultured for 24 h with constant rocking to achieve a confluent monolayer. The cells were activated by treatment with 10 ng/ml of recombinant TNF-α for 6 h before the experiment. The flow chamber was assembled on top of an inverted microscope attached to a time-lapse digital camera (FX-360; Leica Microsystems). A suspension consisting of equal concentrations of PKH-26-labeled monocytes and PKH-67-labeled MDA-MB-231 cells that were treated only with medium or activated with 10 ng/ml TNF-α for 6 h were mixed together to form stable aggregates, as described above. The cell suspension was vigorously mixed intermittently throughout the duration of the experiment to dissociate larger pseudoaggregates.
For dynamic adhesion assays, the cell suspension was then drawn through the perfusion chamber using a syringe pump (Harvard Apparatus, Holliston, MA, USA) at a constant flow rate corresponding to a wall shear stress of 0.5, 1, or 2 dyn/cm2 for HAECs. The experiment was repeated at 1 dyn/cm2 for MVECs. After ∼1 min of perfusion, the cellular interactions were captured by bright-field and fluorescence microscopy (λem 527 and 600 nm) at ×10 for 4 min in 5 different fields of view. The field of view was 0.2 × 0.2 mm2. In some cases, the cellular interactions were recorded at 16 frames/s using the time-lapse digital camera. The images were analyzed offline using ImageJ (U.S. National Institutes of Health, Bethesda, MD, USA) and Image Pro software (Media Cybernetics, Bethesda, MD, USA). In certain experiments, THP1 or MDA-MB-231 cells were preincubated with 10 μg/106 cells of unconjugated anti-CD18 or anti-ICAM-1 antibodies, respectively, in 100 μl of medium with 2% BSA for 20 min and washed 2 times with PBS before preparing the cell suspension mixture. The results are reported as the total number of cells adhered from 5 fields of view of each experiment.
For secondary capture assays; i.e., the capture of MDA-MB-231 cells from flow to THP1 cells already attached to endothelium, the THP1 cells were perfused using a syringe pump (Harvard Apparatus) on top of TNF-α-activated endothelium for 4 min at 0.5 dyn/cm2, followed by perfusion of MDA-MB-231 cells for 4 min at 0.5 dyn/cm2. The MDA-MB-231 cells attached to the surface were counted from 5 fields of view for each experiment.
For static attachment assays, the MDA-MB-231 cell suspension was drawn through the perfusion chamber using a syringe pump (Harvard Apparatus) at a constant flow rate, corresponding to a wall shear stress of 2 dyn/cm2, for 2 min. The flow was stopped, and the cells were allowed to adhere onto the HAECs for 4 min. Nonadherent or nonspecifically adhered cells were washed off by perfusing HBSS buffer at 2 dyn/cm2 for 5 min. The firmly adhered cells were counted from 5 fields of view for each experiment.
Surface receptor expression
MDA-MB-231 cells were grown to confluence in 24-well plates until they reached 80% confluence and were then activated with 20 ng/ml TNF-α for 6 h or only with medium (mock treatment). Then, the cells were gently scraped off the plate and were labeled with PE-conjugated antibody against ICAM-1 (eBioscience) or its corresponding isotype at a concentration of 10 μM.
Computational fluid dynamics (CFD) simulations
A 2-dimensional (2D CFD model was developed using the solver ADINA 8.8.0 (ADINA Inc., Watertown, MA, USA) to simulate the interactions between MDA-MB-231, monocytes, and endothelial cells under flow. The diameters of MDA-MB-231 cells and monocytes were experimentally determined to be 12.6 ± 1.1 and 8.6 ± 0.8 μm, respectively. Cell-cell contact was modeled as a common contact interface with contact length and number of tether bonds estimated from published data at the shear stress of 1 dyn/cm2 (4). We used a cell-cell contact length of 2.94 μm between MDA-MB-231 and monocytes, 3.56 μm between 2 MDA-MB-231 cells, and 4 μm between endothelial cells and the monocytes. CFD simulations were performed to estimate the force experienced at the endothelial cell–monocyte interface for different configurations of MDA-MB-231/monocyte aggregates attached to the endothelial cells in steady flow. The monocytes are bound to the endothelial surface by tethers.
We modeled an entry length of 200 μm, sufficient to reach fully developed flow near the cells, and an exit length of 200 μm, adequate to avoid the effects due to the outflow boundary condition. The height of the microchannel was 100 μm. The top surface of the fluid domain was modeled as a free surface with total slip. A no-slip boundary condition was imposed on the bottom surface of fluid domain and the fluid-cell boundaries. A uniform velocity of 0.15 cm/s was prescribed at the inlet to develop a wall shear stress similar to that observed during the experiment. A zero-traction outflow boundary condition was applied. The fluid domains were meshed with ∼2000 triangular elements for the first generation of simulations. Steered adaptive meshing based on pressure and vorticity with same relative variable gradient at each element was used to generate the second generation of meshes (12,000–20,000 triangular elements).
We simulated 3 configurations of MDA-MB-231/monocyte aggregates attached to the endothelial cells: case 1, only monocyte; case 2, a doublet of MDA-MB-231/monocyte; and case 3, a triplet of 2 MDA-MB-231 cells attached to 1 monocyte. For cases 2 and 3, we simulated the various orientations of attachment with respect to the direction of flow while maintaining the intercellular contact lengths constant. For case 2, we simulated 7 different orientations of MDA-MB-231 cells with respect to the direction of flow; for case 3, the centers of the monocyte and 2 MDA-MB-231 cells were aligned on a straight line, and simulations were carried out for 7 orientations of the aggregate with respect to the direction of flow. The reaction forces on the nodes along the fluid-cell boundary were used to calculate the forces experienced by the tether bonds that hold the cell aggregates to the endothelial surface. Each tether is considered to be a cluster of 9 individual bonds (23), and the maximum force on the tether at the trailing edge of the monocyte was reported. The off-rate constant (koff) is estimated from the Bell model for bond dissociation given by koff = koff0 exp (σF/kbT), where koff0 is the off-rate constant in the absence of an external force on the tether F at a given temperature T, σ is the reactive compliance, and kb is the Boltzmann constant (kbT=4.1 pN·nm at 298K) (24).
Statistics
All the experiments were performed in triplicate, and each experiment was repeated ≥2 times under independent conditions. The results are represented as means ± sd in the plots for a representative experiment. The differences between treatments were considered statistically significant at values of P < 0.01 by 2-tailed Student's t test.
RESULTS
Adhesion of MDA-MB-231 cells to endothelium
The metastatic MDA-MB-231 cells escape circulation to reach distant sites by first adhering to the vascular wall endothelium. Consistent with previous reports, we observed that MDA-MB-231 cells attached to TNF-α-activated endothelium under static conditions and integrated with the endothelial monolayer within 1 h (25). The cells needed only a few seconds to attach to the endothelium under static conditions, and remained stably attached even as we perfused buffer at a physiological shear stress of 0.5–2 dyn/cm2 for several minutes (Fig. 1). However, the MDA-MB-231 failed to interact with the endothelium when the cells were perfused continuously at shear rates ranging from 0.5 to 2 dyn/cm2. The MDA-MB-231 cells that were treated with the proinflammatory cytokine TNF-α also showed a similar behavior. This suggests that the MDA-MB-231 may use alternate mechanisms to bind to the endothelium under flow conditions and escape from circulation.
Figure 1.

Adhesion of MDA-MB-231 cells to endothelium. Untreated (mock) or TNF-α-treated (10 ng/ml for 6 h, TNF-α) MDA-MB-231 cells at a concentration of 106 cells/ml were perfused at a constant flow rate, corresponding to a wall shear stress of 0.5 dyn/cm2 for 2 min over a confluent monolayer of HAECs activated with TNF-α (10 ng/ml for 6 h). The flow was stopped, and the cells were allowed to adhere onto the HAECs for 4 min (static group). Nonspecifically adhered cells were washed off by perfusing HBSS buffer at 2 dyn/cm2 for 5 min. In other experiments, the flow was not stopped, and the cells were perfused continuously (perfusion group). The firmly adhered cells were counted from five different fields of view for each experiment. Results are expressed as means ± sd of a representative experiment performed in triplicate; experiments were performed 3 times.
Formation of tumor cell–monocyte aggregates
Since MDA-MB-231 cells did not attach to endothelium under flow, we hypothesized that monocytes may bridge MDA-MB-231 cells to the endothelium. To test the hypothesis that tumor cells and monocytes form tumor-monocyte heteroaggregates that may assist in MDA-MB-231 extravasation, we first analyzed for heteroaggregate formation in a suspension of monocytes and MDA-MB-231 cells. The experiments were initially performed with the monocytic cell line THP1, and the key results were confirmed with freshly isolated primary human monocytes. THP1 cells have been widely used as a reliable, fairly accurate approximation for human monocytes in numerous studies (26). The MDA MB-231 and THP1 cells were treated with exogenous recombinant TNF-α to simulate an inflammatory microenvironment typical of primary tumor. The tumor cells and THP1 were distinguished by labeling with membrane dyes conjugated with fluorophores that emit at 502 (FITC) and 567 (PE), respectively. To simulate the fluid mechanical environment in the vasculature, the tumor cell/monocyte suspension was sheared at 500 s−1 for 2 min. Shearing the suspension disaggregated pseudoaggregates, and the remaining stable aggregates were analyzed by dual-color flow cytometry (Fig. 2A). Activation with TNF-α increased the number of MDA-MB-231/THP1 heteroaggregates by 2-fold compared to unactivated cells. Analysis by fluorescence microscopy revealed that the heteroaggregates consisted of varying numbers of MDA-MB-231 and THP1 cells (Fig. 2B). We noticed that activation of MDA-MB-231 with TNF-α is essential for aggregation, since activation of THP1 cells alone did not enhance compared to unactivated controls (Fig. 2C).
Figure 2.
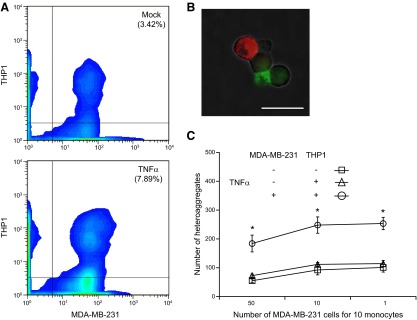
Aggregation of MDA-MB-231 and THP1 cells. Prestained THP1 cells (PKH-26, FITC channel) and MDA-MB-231 cells (PKH-67, PE channel) were either untreated (mock) or treated with 10 ng/ml TNF-α (TNF-α) for 6 h, and resuspended to 2 × 106 cells/ml in corresponding complete medium with 2% BSA. Equal volumes of the cell suspensions were incubated for 20 min with rocking at 37°C and gently mixed with pipette to disaggregate any nonspecific aggregates. Suspensions (80 μl) were subjected to a shear stress of 5 dyn/cm2 for 2 min in a cone-and-plate viscometer at 37°C. Samples (20 μl) were immediately fixed and analyzed by flow cytometry. A) MDA-MB-231/THP1 heteroaggregates were quantified as the number of events that were positive for fluorescence in both FITC and PE channels. B) Fluorescence microscopic images of aggregates in mock- and TNF-α-treated samples. Scale bar = 20 μm. C) MDA-MB-231/THP1 heteroaggregates in suspension containing different initial ratios of MDA-MB-231:THP1 cells. Results are expressed as means ± sd of a representative experiment performed in triplicate; experiments were performed 3 times. *P < 0.01 vs. untreated controls.
Further, we observed that the increase in heteroaggregates due to TNF-α activation was independent of the initial ratio of THP1 to MDA-MB-231 cells (Fig. 2C). At high initial concentration of MDA-MB-231, there was a modest decrease in the number of heteroaggregates, because the MDA MB-231 cells form large numbers of homoaggregates (Supplemental Fig. S1). Thus, these results suggest that the activation of tumor cells by TNF-α significantly enhances the aggregation of MDA-MB-231 to THP1 cells, and this enhancement is independent of the initial ratio of the two types of cells.
Monocyte-mediated adhesion of tumor cells to endothelium
We hypothesized that since MDA-MB-231 cells form heteroaggregates with THP1 cells, the THP1 cells may act as an anchor to arrest the tumor cells to the endothelium. To test this hypothesis, the MDA-MB-231/THP1 heteroaggregates were perfused over a monolayer of TNF-α-activated endothelium at shear rates of 0.5-2 dyn/cm2. The THP1 and MDA-MB-231 cells were fluorescently labeled with green and red membrane dyes, respectively, and the number of MDA MB231 cells attached to the endothelium was enumerated by microscopy. As expected, a large number of single and homoaggregated THP1 cells adhered on the endothelium, as seen by red fluorescent cells adhered to the endothelial monolayer (Fig. 3A). When the cells were not activated, neither single nor homoaggregated MDA-MB-231 cells attached to the endothelial surface, as seen by the absence of green fluorescent cells. However, on activation with TNF-α, significantly large numbers of MDA-MB-231 were seen bound to the endothelium, as seen by the presence of both red and green fluorescent cells. Interestingly, the red and green cells overlapped exclusively, indicating that the MDA-MB-231 cells that adhered to the endothelium were only seen in close juxtaposition to the THP1 cells. The number of bound heteroaggregates decreased linearly with an increase in shear stress but was significantly more at all shear rates when activated with TNF-α (Fig. 3B). We also observed that proximity of MDA-MB-231 and THP1 cells is by far a result of the binding of MDA-MB-231/THP1 heteroaggregates than the secondary capture of individual MDA-MB-231 cells from flow by THP1 cells that already bound to the endothelium. At 0.5 dyn/cm2, <10% of MDA-MB-231 cells were bound to endothelium by secondary capture, and at higher shear stresses, we did not observe any secondary capture at all (Fig. 3B vs. Supplemental Fig. S2). To confirm that these results were not specific to HAECs, we studied the adhesion of heteroaggregates at 1 dyn/cm2 to MVECs. Similar to HAECs, the MDA-MB-231 cells by themselves did not bind to MVECs but were bound only as MDA-MB-231/THP1 heteroaggregates (Supplemental Fig. S3). Further, the MDA-MB-231 cells were always found downstream of THP1 cells. These results demonstrate that monocytes assist in the binding of MDA-MB-231 as heteroaggregates to the endothelium under flow conditions.
Figure 3.
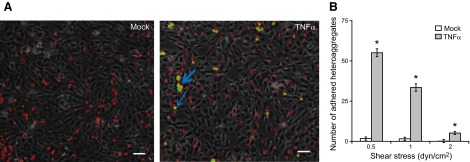
Adhesion of MDA-MB-231/THP1 heteroaggregates to endothelium. A suspension of MDA-MB-231/THP1 aggregates was prepared as described in Fig. 2. The suspension was perfused at 0.5, 1 and 2 dyn/cm2 on top of TNF-α-activated HAECs, and the number of adherent MDA-MB-231 cells was surveyed by fluorescence and bright-field microscopy. A) In cell suspension not treated with TNF-α, only THP1 cells (red) bind to the endothelium, but in cell suspension treated with TNF-α, MDA-MB-231/THP1 heteroaggregates (green-red) bind to the endothelium. Arrows represent aggregates attached to endothelium. Scale bars = 20 μm. B) Number of adherent MDA-MB-231 cells (in heteroaggregates) was calculated from 5 fields of view. Results are expressed as means ± sd of a representative experiment performed in triplicate; experiments were performed 3 times. *P < 0.01 vs. untreated controls.
Biophysical mechanisms of adhesion of tumor cells to endothelium
We characterized the biophysics of adhesion of MDA-MB231/THP1 heteroaggregates to the endothelium by analyzing the adhesion events by time-lapse fluorescence microscopy under flow. First, we surveyed the number and types of cells in the heteroaggregates captured on the endothelium at wall shear stresses of 0.5, 1, and 2 dyn/cm2. We observed that most of the heteroaggregates that stably bound to the endothelium were small aggregates consisting of 1 or 2 MDA-MB231 cells attached to a THP1 cell (Fig. 4A). The analysis of the distribution showed that a vast majority of the cells (75–90%) were doublets consisting of 1 MDA-MB-231 cell bound to 1 THP1 cell, and the rest (10–25%) were triplets or larger aggregates consisting of mostly of 2 or 3 MDA-MB-231 cells bound 1 or 2 THP1 cells. The larger macroaggregates did not bind to the endothelium at all. Next, we analyzed the temporal evolution of the adhesion of MDA-MB-231/THP1 heteroaggregates to the endothelium by quantifying the adhesion process in every frame. We measured the velocities of single THP1 cells and MDA-MB-231/THP1 doublets and triplets interacting with the endothelium (Fig. 4B). Single monocytes rolled for a very short distance, lasting less than a couple of seconds before adhering firmly to the endothelium, while the MDA MB-231 cells by themselves did not adhere to the endothelium at all. The MDA-MB-231/THP1 doublet heteroaggregates rolled on the endothelium for much longer duration and a greater distance than a single monocyte before arresting firmly. Triplets rolled much faster than doublets, and typically rejoined the flow after short, transient interactions with the endothelium (Fig. 4B).
Figure 4.

Size and orientation of MDA-MB-231/THP1 heteroaggregates attached to the endothelium. Untreated or TNF-α-treated MDA-MB-231/THP1 heteroaggregates were perfused over activated HAECs as described in Fig. 3. A) Numbers of doublets, containing 1 MDA-MB-231 cell and 1 THP1 cell, or triplets and larger aggregates that attached stably to the endothelium were counted. B) Representative instantaneous velocity profiles of THP1 cells, MDA-MB-231/THP1 doublets, and triplet heteroaggregates of 2 MDA-MB-231 cells and a THP1 cell rolling on the endothelium at a wall shear rate of 1 dyn/cm2. C) Orientation of MDA-MB-231 cells with respect to the flow direction in a stably attached aggregate at a wall shear rate of 1 dyn/cm2. Fluorescence microscopic images of THP1 (red) and MDA-MB-231 (green) were merged, with phase-contrast image of endothelial cells in the background. Scale bar = 20 μm. Center red circle represents a THP1 cell; inner green circles and outer blue circles represent the locations of MDA MB-231 cells during initial tethering and final stable attachment, respectively. Angle between direction of flow and the location of MDA-MB-231 cells with respect to THP1 in heteroaggregates was calculated from 5 fields of view. Results are expressed as means ± sd of a representative experiment performed in triplicate; experiments were performed 3 times.
To further characterize the dynamics of interaction between heteroaggregates and the endothelium, we analyzed the temporal evolution of the relative positions of MDA-MB-231 and THP1 as the heteroaggregates eventually are arrested on the surface. We measured the orientation of MDA-MB-231 cells in the heteroaggregates with respect to the direction of flow, as shown in Fig. 4C. As the heteroaggregates are initially captured from flow due to the interactions between monocytes and endothelium, the orientation of MDA-MB-231 cells with respect to the endothelium is random. However, as the aggregates are firmly arrested, and equilibrate to a stable position, the MDA-MB-231 cells align almost exclusively downstream of the bound THP1 cells. The distribution of final orientation of the MDA-MB-231 cells with respect to the THP1 cells in the aggregates show that the majority of the MDA-MB-231 cells (75%) are located within 45° downstream of flow from the center of the monocyte (Fig. 4C). Taken together, this analysis show that only MDA-MB-231/monocyte aggregates firmly adhere to the endothelium and that the monocyte acts as an anchor in recruiting MDA-MB-231 cells to the endothelium under flow.
Molecular mechanisms of heteroaggregate formation and adhesion to endothelium
We characterized the adhesion molecules that are responsible for the formation of MDA-MB-231/THP1 heteroaggregates. β2-Integrins (CD18) on leukocytes mediate firm adhesion of leukocytes with other leukocytes, platelets, endothelial cells (27, 28), and also certain tumor cells (5). Since an important counterreceptor for leukocyte CD18 is ICAM-1, we measured the ICAM-1 expression levels on MDA-MB-231 cells activated with TNF-α. We noticed that there was a modest increase in the ICAM-1+ population of cells and a drastic increase in the expression levels in MDA-MB-231 cells on activation from low basal levels (Fig. 5A). We then evaluated the role of ICAM-1 on MDA-MB-231 cells and CD18 on THP1 on the formation of stable heteroaggregates (Fig. 5B). Compared to isotype control, blocking with monoclonal antibodies against either ICAM-1 or CD18 essentially abrogated TNF-α-induced aggregation down to unactivated levels.
Figure 5.
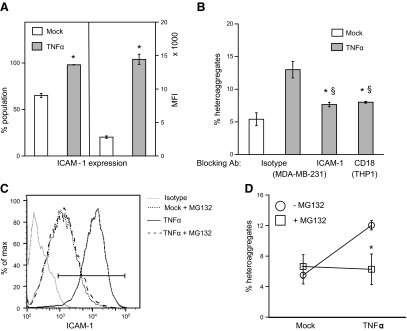
Mechanism of MDA-MB-231/THP1 aggregation. A) Untreated or TNF-α-treated MDA-MB-231 cells were fixed, stained with PE-conjugated ICAM-1 antibody or the corresponding isotype control, and analyzed by flow cytometry. Results are averages of a representative experiment run in triplicate; experiment was repeated 2 times. *P < 0.01 vs. mock treatment. B) TNF-α-activated MDA-MB-231 and THP1 cells were blocked with anti-ICAM-1 or anti-CD18 antibody, respectively, or with the corresponding isotype controls, before preparing and quantifying heteroaggregates as described in Fig. 2. *P < 0.01 vs. TNF-α-treated cells without blocking; §P < 0.01 vs. mock-treated control. C, D) MDA-MB-231 cells were preincubated with 0 or 20 μM of MG132 for 30 min before TNF-α treatment. ICAM-1 expression levels were analyzed by flow cytometry (C), and the aggregation experiments (D) were performed as described above. Results are expressed as means ± sd of a representative experiment performed in triplicate; experiments were performed 3 times. *P < 0.01 vs. no MG132.
One of the well-established pathways of TNF-α signaling is the activation of nuclear receptor Nf-κB, which, in turn, up-regulates a number of genes, including ICAM-1. We examined whether TNF-α activates MDA-MB-231 through this pathway by using MG132, a cell-permeable tripeptide derivative that blocks Nf-κB activity, to inhibit the effect of TNF-α on MDA-MB-231 cells. We observed that MG132 comprehensively nullified the effect of TNF-α on ICAM-1 expression (Fig. 5C), and also on MDA-MB-231/THP1 heteroaggregation, thus bringing these values to baseline levels (Fig. 5D).
Next, we examined the role of ICAM-1 on the adhesion of heteroaggregates to the endothelium under flow. The MDA-MB-231 cells were blocked with either F(ab′)2 fragment (isotype control) or the whole monoclonal antibody against ICAM-1. These blocked MDA-MB-231 cells were mixed with THP1 cells to form aggregates, and the aggregates were perfused on top of endothelial cells at 1 dyn/cm2. We observed that blocking ICAM-1 on MDA-MB-231 cells significantly reduced the number of heteroaggregates adhered to the endothelium compared to the suspension containing untreated or isotype-treated MDA-MB-231 cells and THP1 cells (Fig. 6A). We also observed that the binding of heteroaggregates to the endothelium was completely eliminated when MDA-MB-231 cells were pretreated with MG132 (Fig. 6B). In all cases, similar numbers of single or homoaggregated monocytes adhered to the endothelium. These results clearly indicate that TNF-α-induced ICAM-1 expression in MDA-MB-231 plays a critical role in the stabilizing MDA-MB-231/THP1 interactions, and in mediating adhesion to the endothelium.
Figure 6.

Mechanism of MDA-MB-231/THP1 adhesion to endothelium. A) MDA-MB-231 cells were blocked with anti-ICAM-1 antibody, and a suspension of MDA-MB-231/THP1 aggregates was prepared as described in Fig. 2. Aggregates were perfused on top of endothelium at 1 dyn/cm2, and the number of aggregates attaching stably to the endothelium was quantified as described in Fig. 3. B) MDA-MB-231 cells were preincubated with 0 or 20 μM of MG132 for 30 min before TNF-α treatment. A suspension of MG132-treated MDA-MB-231 and THP1 aggregates was perfused on top of endothelium, and stable aggregates attached to the endothelium were quantified. *P < 0.01 vs. untreated controls.
To confirm that our results are not specific to the monocytic cell line THP1, we examined the role of freshly isolated primary human monocytes in forming stable aggregates with MDA-MB-231 and in mediating the adhesion to the endothelium. After isolation, 95% of monocytes were CD14+ (data not shown) and were allowed to aggregate with MDA-MB-231 cells, as described in Fig. 2. We quantified the aggregation of MDA-MB-231 cells with human monocytes using flow cytometry (Fig. 7A). On activation with TNF-α, we observed a 4-fold increase in the number of stable MDA-MB231/monocyte heteroaggregates. This aggregation process was blocked by antibodies against ICAM-1 on MDA-MB-231 cell surface (Fig. 7B). When we perfused the MDA-MB-231/monocyte heteroaggregates over TNF-α-activated endothelium under flow at 1 dyn/cm2, the TNF-α-activated heteroaggregates adhered stably to the endothelial cells (Fig. 7C). In addition, we also found that the number and orientation of MDA-MB-231 cells in stably attached MDA-MB-231/monocyte heteroaggregates were similar to those observed with THP1 cells (data not shown). In essence, the results obtained with freshly isolated human monocytes are identical to those obtained with the THP1 model system, and together they clearly demonstrate that monocytes play an important role in the recruitment of MDA-MB-231 cells to the endothelium under flow.
Figure 7.
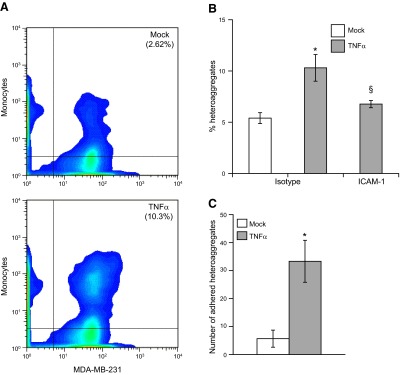
MDA-MB-231/primary monocyte heteroaggregation and endothelial adhesion. A) Aggregation experiments and quantification were identical to those described in Fig. 2, except that human monocytes were used. Formaldehyde-fixed MDA-MB-231/primary monocyte heteroaggregates were quantified as the percentage of events that were positive for fluorescence in both FITC and PE channels. B) TNF-α-activated MDA-MB-231 cells were blocked with anti-ICAM-1 or the corresponding isotype controls, before heteroaggregates were prepared with human monocytes and quantified by flow cytometry. *,§P < 0.05 vs. untreated controls. C) Suspension containing MDA-MB-231/monocyte aggregates was perfused at 1 dyn/cm2 on top of TNF-α-activated HAECs, and the number of adherent MDA-MB-231 cells was surveyed by fluorescence microscopy in 5 fields of view. Results are means ± sd of a representative experiment performed in triplicate; experiments were repeated 3 times. *P < 0.01 vs. mock treatment.
Role of hydrodynamic flow in heteroaggregate adhesion to the endothelium
We have seen that the fluid flow is a determinant of the adhesion of MDA-MB-231/monocyte heteroaggregates to the endothelium. We used a CFD model to understand the role of biophysical forces on MDA-MB-231–monocyte–endothelial interactions. Using this numerical technique, we simulated the adhesion of a single monocyte, a doublet consisting of 1 monocyte and 1 MDA-MB-231 cell, and a triplet consisting of 1 monocyte and 2 MDA-MB-231 cells (Fig. 8A). The monocytes act as a bridge by holding the MDA-MB-231 cells while being attached to the endothelium. The entire channel was divided into small domains over which the equations describing the fluid flow were solved to evaluate the forces acting on the aggregates bound to the endothelium (Supplemental Fig. S4). At a shear stress of 1 dyn/cm2, we estimated the reaction force at the endothelial cell-aggregate interface for various orientations of the aggregates with respect to the direction of fluid flow. A vertical orientation of an MDA-MB-231 cell on top of a monocyte is 0°, and it increases to 90° when the MDA-MB-231 cell is directly downstream of the monocyte. The reaction force is the net detachment force due to fluid flow, which the cells have to overcome for a stable adhesion. The aggregates are assumed to be attached to the surface by tethers, which are clusters of bonds (tether bonds) that break simultaneously under flow (24). Based on published estimates of contact area of an adherent cell and receptor density (23, 29), we calculated the reaction force per tether bond. The maximum reaction force experienced at the trailing edge is reported in Fig. 8B for various orientations of single cells and doublet and triplet aggregates. Our calculations reveal that larger the aggregate, greater the detachment force at the monocyte-endothelial interface. Further, this force decreases as the MDA-MB-231 cells align downstream of monocytes, and is minimum when the MDA-MB-231 cells are in the wake of adhered monocyte.
Figure 8.
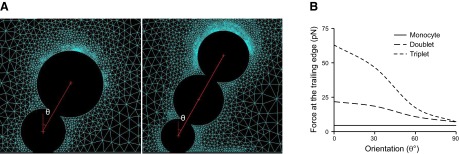
Estimation of detachment force at the monocyte-endothelial interface of heteroaggregates due to fluid flow. A) Geometric configurations of MDA-MB-231/monocyte heteroaggregates attached to endothelial surface under laminar shear flow. Configurations include a MDA-MB-231/THP1 doublet and a triplet consisting of 2 MDA-MB-231 cells and a monocyte. Angle θ represents the orientation of the MDA-MB-231 cell that is attached to the monocyte with respect to the vertical axis. B) Reaction force experienced at the trailing edge of the monocyte–endothelial contact area.
To evaluate whether the reaction forces acting at the trailing edge of the monocyte can detach the cells, we estimated the off-rate due to fluid force-induced bond dissociation. Based on our simulations, the maximum forces acting at the trailing edge of a single cell, a doublet, and a triplet are 6.35, 21.77, and 62.93 pN, respectively. Using experimental estimates of koff0 and σ in the Bell model for activated VLA4–VCAM-1 bonds as 75 s−1 and 0.1 nm, respectively (30), koff at the trailing edge of a single monocyte, doublet, and triplet is 87.6, 127.5, and 348 s−1, respectively. This comparison reveals that as the aggregate size increases, the dissociation rates increase considerably, resulting in faster rolling, detachment, and poor adhesion to endothelium. These results support our experimental observations in Fig. 4, and illustrate that fluid mechanical forces stipulate the size and orientation of aggregates that adhere to the endothelium.
DISCUSSION
In this work, we have demonstrated that monocytes and the metastatic breast tumor cell MDA-MB-231 form stable aggregates in suspension, which play a critical role in mediating the adhesion of otherwise nonadherent tumor cells to the vascular endothelium under flow. We also show that an inflammatory microenvironment is vital for the formation of stable MDA-MB-231/monocyte aggregates, and that smaller aggregates are most efficient in adhering to the endothelium. The aggregation process is likely mediated by the interaction between β2-integrin on monocytes and ICAM-1 on MDA-MB-231 cells. Cumulatively, our data provide a compelling evidence for a novel role of monocyte in cancer metastasis.
MDA-MB-231 cells were initially isolated from the pleural effusion of a patient with breast cancer in widespread metastasis (20), and have been extensively studied as a model for tumor with high metastatic potential. A key event in tumor metastasis is the escape of MDA-MB-231 cells from circulation by adhesion and transmigration through the endothelium. Previous studies have focused on the molecular determinants of this process as the MDA-MB-231 cells adhere to the endothelium (31), invade through a layer of endothelial cells (32), and migrate through the matrix (33, 34). Consistent with these studies, we observed that MDA-MB-231 cells adhered to the endothelial monolayer when the cells were incubated together even for a few minutes (Fig. 1), and migrated through the endothelial layer in a transwell chamber (data not shown). However, MDA-MB-231 cells failed to adhere to the endothelium under microvascular flow conditions, leading us to hypothesize alternate mechanisms for the adhesion of MDA-MB-231 cells to the endothelium. Primary tumor lesions contain innate immune cells that were initially considered tumoricidal. However, recent clinical and experimental evidence strongly suggests that monocyte-derived macrophages that are closely associated with tumor cells, the TAMs, promote tumor progression to malignancy directly or indirectly by participating in several stages, including mutagenesis, tumor cell proliferation, migration and intravasation (35, 36), angiogenesis (37), and immune regulation (38). The monocytes/macrophages at the primary tumor site establish a low-grade smoldering inflammation that renders the microenvironment rich in reactive free radicals, growth factors, and cytokines (10).
TNF-α is a proinflammatory pleiotropic cytokine that is found abundantly in a number of solid tumors (39). Though TNF-α is cytotoxic at high doses, chronic lower doses favor tumor progression. Serum concentrations of TNF-α correlate with breast cancer progression (40), and clinical trials of TNF-α inhibition have shown at least partial responses (41). The dysregulated production of TNF-α results in chronic inflammation that manifests in all 6 hallmarks of cancer, namely, angiogenesis, autocrine growth, limitless growth potential, insensitivity of growth-inhibiting signals, survival factors, and invasion and metastasis. It has been shown that the incubation of monocytes with MDA-MB-231 cells induces monocytes to secrete TNF-α, which, in turn, activates the MDA-MB-231 cells instead of inducing apoptosis (42). Our results show that, in addition to the effects localized to the primary tumor site listed above, TNF-α has downstream systemic consequences. TNF-α activation is necessary for the stable aggregation of monocytes with MDA-MB-231 cells that can survive the disaggregating shear forces of blood flow. Unactivated monocytes and tumor cells associate only weakly by nonspecific adhesion and detach under physiological ranges of shear stresses within a short duration.
The aggregation due to TNF-α activation is largely dependent on the binding of ICAM-1 (CD54) on the tumor cell surface to its counterreceptor, the β2 integrins (CD18), on the monocyte surface. Previous studies have shown that ICAM-1 expression correlates with the metastatic potential of 5 breast tumor cells, possibly due to an increase in cell migration and tissue invasion (43). Here, we show that a significant increase in ICAM-1 expression on the MDA-MB-231 cells can also contribute to metastasis by fostering tumor-monocyte aggregation, and hence endothelial adhesion. The up-regulation of ICAM-1 in MDA-MB-231 cells by TNF-α activation is mediated by a key transcriptional regulator, Nf-κB. Nf-κB appears to be a critical link between inflammation and cancer, since Nf-κB activation promotes apoptosis, and is tumor protective only in models without an inflammatory component, such as liver and skin cancer, but promotes tumorigenesis and metastasis in models with an inflammatory component, such as breast cancer (44). Blockade of Nf-κB completely abolished tumor-monocyte aggregation, suggesting that Nf-κB activation may be solely responsible for stable aggregation. Thus, our model assigns another role to Nf-κB signaling, namely, the formation of tumor-monocyte aggregates. Interestingly, while blocking Nf-κB completely abrogated aggregate formation, blocking ICAM-1 decreased aggregation by only 75%, suggesting that there may be receptors other than ICAM-1 that contribute to a lesser extent to the aggregation process.
In our experiments, the MDA-MB-231/monocyte heteroaggregates are essential in transporting the MDA-MB-231 cells from flow to the endothelium, since we did not observe any unassisted interactions between MDA-MB-231 cells and the endothelium at the 0.5–2 dyn/cm2 shear stress evaluated in this work. Cell adhesion under flow involves the initial capture of free-flowing cells (tethering), slowing down on the surface (rolling), and complete arrest (stable adhesion). However, MDA-MB-231 cells do not express selectin receptors or hitherto known selectin ligands necessary for capture on to endothelium under flow. On the other hand, monocytes roll and arrest on the endothelium, mediated by β1 integrin VLA-4 interaction with endothelium VCAM-1 (19). Hence, MDA-MB-231 cells utilize monocytes as a carrier for arresting on the endothelium. We show compelling evidence for the involvement of monocyte and MDA-MB-231 cell aggregates in this process rather than the capture of single MDA-MB231 cells to monocyte previously attached to the endothelium (Supplemental Fig. S3). It is significant to note that under the experimental conditions tested in this study, the fundamental role of monocyte/MDA-MB-231 heteroaggregates in the extravasation process is similar on both aortic and microvascular endothelium. Previous studies have shown that neutrophil-tumor cell aggregates serve to facilitate adhesion to endothelium and extravasation of metastatic tumor cells, including colon carcinoma (4) and melanoma (45), under flow.
Fluid forces play a primary role in MDA-MB-231/monocyte heteroaggregate adhesion to the endothelium. As the cells roll, the bonds in the trailing edge of the contact zone are subjected to stress due to fluid flow, and the time scale of bond rupture will delineate the difference between arrest and detachment (46). According to the Bell model for force-induced bond dissociation, the off-rate constant koff increases exponentially with the detachment force on the tether bond. The CFD simulations reveal that the effective force at the trailing edge of an adherent cell is dependent on aggregate size and orientation with respect to flow. A large aggregate perpendicular to the direction of flow will experience the greatest drag, while a small aggregate parallel to direction of flow will experience the lowest drag. Consequently, a large koff for a larger aggregate or a triplet may result in spontaneous detachment from the endothelium, while a lower koff for smaller aggregates or doublets may sustain steady rolling. As doublets roll, they align in the direction of flow that has minimum drag, and hence even lower koff, resulting in a firm arrest. It should be noted that the current 2D CFD study is limited to the movement of aggregates on a vertical plane parallel to the flow, while the actual flow is in 3D. However, 2D models are effective in outlining the limiting cases and providing insights into experimental observations. This is evident by the same order of magnitude of single-cell detachment force found in our study and previously reported in a 3D model of Jadhav et al. (25).
The arrest of MDA-MB-231/monocyte heteroaggregates brings the MDA-MB-231 cells within molecular proximity of the endothelium. This process is akin to static incubation of MDA-MB-231 cells to the endothelium, which has been shown by us (Fig. 1), and also by others to support stable adhesion (21). In addition, we observed that even a few seconds of static incubation is sufficient for the stable adhesion of MDA-MB-231 cells. Based on these results, we speculate that the rolling of doublets anchored by the monocytes even for only a couple of seconds may be a legitimate substitute for the lack of selectin-like molecules on MDA-MB-231 surface needed for the initial capture from flow. After their arrest, the MDA-MB-231 cells firmly adhere to the endothelium, modulate endothelial properties, and actively extravasate by processes mediated by ICAM-1 and β1 integrins (25, 47). In addition, since monocytes can rapidly extravasate, the MDA-MB-231 cells in the heteroaggregates may also be passively transported across the endothelium.
In summary, we have addressed what alternate strategy may be employed by the nonadherent MDA-MB-231 cells to adhere to the endothelium during hematogenous dissemination. We show that, under inflammatory conditions such as those commonly found in primary tumor sites, MDA-MB-231 cells bound to monocytes can withstand the disaggregating forces in circulation, and use monocytes as a vehicle to attach to the endothelium. We also show that the process is efficient, since forces due to blood flow favor the attachment of smaller aggregates. While our in vitro observations outline an important and novel possibility, it needs to be tested in suitable in vivo mouse models. There is sufficient clinical and experimental evidence that monocytes play an important role in cancer metastasis. Current work presents yet another facet of their role in tumor metastasis in addition to their well-established contribution at the primary tumor site, and more recently at the secondary metastatic sites. Therefore, the tumor–monocyte–endothelial axis may represent a new therapeutic target to reduce cancer metastasis and mortality.
Supplementary Material
Acknowledgments
This work was funded by a grant from the Breast Cancer Research Program, U.S. Department of Defense (W81XWH-10-1-0754), and partially by a grant from the U.S. National Institutes of Health (HL112629).
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
- 2D
- 2-dimensional
- 3D
- 3-dimensional
- CFD
- computational fluid dynamics
- EGF
- epidermal growth factor
- HAEC
- human aortic endothelial cell
- ICAM-1
- intercellular adhesion molecule 1
- MVEC
- microvascular endothelial cell
- PE
- phycoerythrin
- sLex/a
- sialyl Lewis-x or -a
- TAM
- tumor-associated-macrophage
- TNF-α
- tumor necrosis factor α
- VEGF
- vascular endothelial growth factor
- VLA-4
- very late antigen 4
REFERENCES
- 1. Valastyan S., Weinberg R. A. (2011) Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liang S., Slattery M. J., Wagner D., Simon S. I., Dong C. (2008) Hydrodynamic shear rate regulates melanoma-leukocyte aggregation, melanoma adhesion to the endothelium, and subsequent extravasation. Ann. Biomed. Eng. 36, 661–671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Konstantopoulos K., Thomas S. N. (2009) Cancer cells in transit: the vascular interactions of tumor cells. Annu. Rev. Biomed. Eng. 11, 177–202 [DOI] [PubMed] [Google Scholar]
- 4. Jadhav S., Bochner B., Konstantopoulos K. (2001) Hydrodynamic shear regulates the kinetics and receptor specificity of polymorphonuclear leukocyte-colon carcinoma cell adhesive interactions. J. Immunol. 167, 5986–5993 [DOI] [PubMed] [Google Scholar]
- 5. Geng Y., Narasipura S., Seigel G. M., King M. R. (2010) Vascular recruitment of human retinoblastoma cells by multi-cellular adhesive interactions with circulating leukocytes. Cell. Mol. Bioeng. 3, 361–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Peng H. H., Liang S., Henderson A. J., Dong C. (2007) Regulation of interleukin-8 expression in melanoma-stimulated neutrophil inflammatory response. Exp. Cell Res. 313, 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huh S. J., Liang S., Sharma A., Dong C., Robertson G. P. (2010) Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 70, 6071–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gay L. J., Felding-Habermann B. (2011) Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 11, 123–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leek R. D., Lewis C. E., Whitehouse R., Greenall M., Clarke J., Harris A. L. (1996) Association of macrophage infiltration with angiogenesis and prognosis in invasive breast carcinoma. Cancer Res. 56, 4625–4629 [PubMed] [Google Scholar]
- 10. Qian B. Z., Pollard J. W. (2010) Macrophage diversity enhances tumor progression and metastasis. Cell 141, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Joyce J. A., Pollard J. W. (2009) Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 9, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mantovani A., Sica A. (2010) Macrophages, innate immunity, and cancer: balance, tolerance, and diversity. Curr. Opin. Immunol. 22, 231–237 [DOI] [PubMed] [Google Scholar]
- 13. Meira L. B., Bugni J. M., Green S. L., Lee C. W., Pang B., Borenshtein D., Rickman B. H., Rogers A. B., Moroski-Erkul C. A., McFaline J. L., Schauer D. B., Dedon P. C., Fox J. G., Samson L. D. (2008) DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Invest. 118, 2516–2525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Robinson B. D., Sica G. L., Liu Y. F., Rohan T. E., Gertler F. B., Condeelis J. S., Jones J. G. (2009) Tumor microenvironment of metastasis in human breast carcinoma: a potential prognostic marker linked to hematogenous dissemination. Clin. Cancer Res. 15, 2433–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wyckoff J., Wang W., Lin E. Y., Wang Y., Pixley F., Stanley E. R., Graf T., Pollard J. W., Segall J., Condeelis J. (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022–7029 [DOI] [PubMed] [Google Scholar]
- 16. Lin E. Y., Li J. F., Bricard G., Wang W., Deng Y., Sellers R., Porcelli S. A., Pollard J. W. (2007) Vascular endothelial growth factor restores delayed tumor progression in tumors depleted of macrophages. Mol. Oncol. 1, 288–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qian B. Z., Li J., Zhang H., Kitamura T., Zhang J., Campion L. R., Kaiser E. A., Snyder L. A., Pollard J. W. (2011) CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 475, 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qian B., Deng Y., Im J. H., Muschel R. J., Zou Y., Li J., Lang R. A., Pollard J. W. (2009) A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS One 4, e6562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ley K., Laudanna C., Cybulsky M. I., Nourshargh S. (2007) Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat. Rev. Immunol. 7, 678–689 [DOI] [PubMed] [Google Scholar]
- 20. Cailleau R., Olive M., Cruciger Q. V. (1978) Long-term human breast carcinoma cell lines of metastatic origin: preliminary characterization. In Vitro 14, 911–915 [DOI] [PubMed] [Google Scholar]
- 21. Julien S., Ivetic A., Grigoriadis A., QiZe D., Burford B., Sproviero D., Picco G., Gillett C., Papp S. L., Schaffer L., Tutt A., Taylor-Papadimitriou J., Pinder S. E., Burchell J. M. (2011) Selectin ligand sialyl-Lewis x antigen drives metastasis of hormone-dependent breast cancers. Cancer Res. 71, 7683–7693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burdick M. M., Henson K. A., Delgadillo L. F., Choi Y. E., Goetz D. J., Tees D. F. J., Benencia F. (2012) Expression of E-selectin ligands on circulating tumor cells: cross-regulation with cancer stem cell regulatory pathways? Front Oncol. 2, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jadhav S., Eggleton C. D., Konstantopoulos K. (2005) A 3-D computational model predicts that cell deformation affects selectin-mediated leukocyte rolling. Biophys. J. 88, 96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar R. A., Dong J.-f., Thaggard J. A., Cruz M. A., Lopez J. A., McIntire L. V. (2003) Kinetics of GPIbα-vWF-A1 tether bond under flow: Effect of GPIba mutations on the association and dissociation rates. Biophys. J. 85, 4099–4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haidari M., Zhang W., Caivano A., Chen Z., Ganjehei L., Mortazavi A., Stroud C., Woodside D. G., Willerson J. T., Dixon R. A. (2012) Integrin α2β1 mediates tyrosine phosphorylation of vascular endothelial cadherin induced by invasive breast cancer cells. J. Biol. Chem. 287, 32981–32992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Auwerx J. (1991) The human leukemia cell line, THP-1: a multifacetted model for the study of monocyte-macrophage differentiation. Experientia 47, 22–31 [DOI] [PubMed] [Google Scholar]
- 27. Langer H. F., Chavakis T. (2009) Leukocyte-endothelial interactions in inflammation. J. Cell. Mol. Med. 13, 1211–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang P., Ozdemir T., Chung C. Y., Robertson G. P., Dong C. (2011) Sequential binding of αVβ3 and ICAM-1 determines fibrin-mediated melanoma capture and stable adhesion to CD11b/CD18 on neutrophils. J. Immunol. 186, 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dong C., Cao J., Struble E. J., Lipowsky H. H. (1999) Mechanics of leukocyte deformation and adhesion to endothelium in shear flow. Ann. Biomed. Eng. 27, 298–312 [DOI] [PubMed] [Google Scholar]
- 30. Zhang X., Craig S. E., Kirby H., Humphries M. J., Moy V. T. (2004) Molecular basis for the dynamic strength of the integrin α4β1/VCAM-1 interaction. Biophys. J. 87, 3470–3478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Draffin J. E., McFarlane S., Hill A., Johnston P. G., Waugh D. J. (2004) CD44 potentiates the adherence of metastatic prostate and breast cancer cells to bone marrow endothelial cells. Cancer Res. 64, 5702–5711 [DOI] [PubMed] [Google Scholar]
- 32. Gramling M. W., Beaulieu L. M., Church F. C. (2010) Activated protein C enhances cell motility of endothelial cells and MDA-MB231 breast cancer cells by intracellular signal transduction. Exp. Cell Res. 316, 314–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Peyri N., Berard M., Fauvel-Lafeve F., Trochon V., Arbeille B., Lu H., Legrand C., Crepin M. (2009) Breast tumor cells transendothelial migration induces endothelial cell anoikis through extracellular matrix degradation. Anticancer Res. 29, 2347–2355 [PubMed] [Google Scholar]
- 34. Narasimhan M., Ammanamanchi S. (2008) Curcumin blocks RON tyrosine kinase-mediated invasion of breast carcinoma cells. Cancer Res. 68, 5185–5192 [DOI] [PubMed] [Google Scholar]
- 35. Beck A. H., Espinosa I., Edris B., Li R., Montgomery K., Zhu S., Varma S., Marinelli R. J., van de Rijn M., West R. B. (2009) The macrophage colony-stimulating factor 1 response signature in breast carcinoma. Clin. Cancer Res. 15, 778–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Condeelis J. S., Pollard J. W. (2006) Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124, 263–266 [DOI] [PubMed] [Google Scholar]
- 37. Ahn G.-O., Tseng D., Liao C.-H., Dorie M. J., Czechowicz A., Brown J. M. (2010) Inhibition of Mac-1 (CD11b/CD18) enhances tumor response to radiation by reducing myeloid cell recruitment. Proc. Natl. Acad. Sci. U. S. A. 107, 8363–8368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bailey C., Negus R., Morris A., Ziprin P., Goldin R., Allavena P., Peck D., Darzi A. (2007) Chemokine expression is associated with the accumulation of tumour associated macrophages (TAMs) and progression in human colorectal cancer. Clin. Exp. Metastasis 24, 121–130 [DOI] [PubMed] [Google Scholar]
- 39. Balkwill F. (2006) TNFα in promotion and progression of cancer. Cancer Metastasis Rev. 25, 409–416 [DOI] [PubMed] [Google Scholar]
- 40. Sheen-Chen S. M., Chen W. J., Eng H. L., Chou F. F. (1997) Serum concentration of tumor necrosis factor in patients with breast cancer. Breast Cancer Res. Treat. 43, 211–215 [DOI] [PubMed] [Google Scholar]
- 41. Brown E. R., Charles K. A., Hoare S. A., Rye R. L., Jodrell D. I., Aird R. E., Vora R., Prabhakar U., Nakada M., Corringham R. E., DeWitte M., Sturgeon C., Propper D., Balkwill F. R., Smyth J. F. (2008) A clinical study assessing the tolerability and biological effects of infliximab, a TNFα inhibitor, in patients with advanced cancer. Ann. Oncol. 19, 1340–1346 [DOI] [PubMed] [Google Scholar]
- 42. Blot E., Chen W., Vasse M., Paysant J., Denoyelle C., Pillé J.-Y., Vincent L., Vannier J.-P., Soria J., Soria C. (2003) Cooperation between monocytes and breast cancer cells promotes factors involved in cancer aggressiveness. Br. J. Cancer. 88, 1207–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosette C., Roth R. B., Oeth P., Braun A., Kammerer S., Ekblom J., Denissenko M. F. (2005) Role of ICAM1 in invasion of human breast cancer cells. Carcinogenesis 26, 943–950 [DOI] [PubMed] [Google Scholar]
- 44. Karin M., Greten F. R. (2005) Nf-κB: Linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5, 749–759 [DOI] [PubMed] [Google Scholar]
- 45. Liang S., Dong C. (2008) Integrin VLA-4 enhances sialyl-Lewisx/a-negative melanoma adhesion to and extravasation through the endothelium under low flow conditions. Am. J. Physiol. Cell. Physiol. 295, C701–C707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chang K.-C., Tees D. F. J., Hammer D. A. (2000) The state diagram for cell adhesion under flow: leukocyte rolling and firm adhesion. Proc. Natl. Acad. Sci. (U. S. A.). 97, 11262–11267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mierke C. T. (2011) Cancer cells regulate biomechanical properties of human microvascular endothelial cells. J. Biol. Chem. 286, 40025–40037 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.