

Abstract
Selenoprotein P (Sepp1) is taken up by receptor-mediated endocytosis for its selenium. The other extracellular selenoprotein, glutathione peroxidase-3 (Gpx3), has not been shown to transport selenium. Mice with genetic alterations of Sepp1, the Sepp1 receptors apolipoprotein E receptor-2 (apoER2) and megalin, and Gpx3 were used to investigate maternal-fetal selenium transfer. Immunocytochemistry (ICC) showed receptor-independent uptake of Sepp1 and Gpx3 in the same vesicles of d-13 visceral yolk sac cells, suggesting uptake by pinocytosis. ICC also showed apoER2-mediated uptake of maternal Sepp1 in the d-18 placenta. Thus, two selenoprotein-dependent maternal-fetal selenium transfer mechanisms were identified. Selenium was quantified in d-18 fetuses with the mechanisms disrupted. Maternal Sepp1 deletion, which lowers maternal whole-body selenium, decreased fetal selenium under selenium-adequate conditions but deletion of fetal apoER2 did not. Fetal apoER2 deletion did decrease fetal selenium, by 51%, under selenium-deficient conditions, verifying function of the placental Sepp1-apoER2 mechanism. Maternal Gpx3 deletion decreased fetal selenium, by 13%, but only under selenium-deficient conditions. These findings indicate that the selenoprotein uptake mechanisms ensure selenium transfer to the fetus under selenium-deficient conditions. The failure of their disruptions (apoER2 deletion, Gpx3 deletion) to affect fetal selenium under selenium-adequate conditions indicates the existence of an additional maternal-fetal selenium transfer mechanism.—Burk, R. F., Olson, G. E., Hill, K. E., Winfrey, V. P., Motley, A. K., and Kurokawa, S. Maternal-fetal transfer of selenium in the mouse.
Keywords: selenoprotein P, apolipoprotein E receptor-2, glutathione peroxidase-3, visceral yolk sac, placenta
Selenium is an essential micronutrient that exerts its functions through selenoproteins. Inability of the embryo to synthesize selenoproteins leads to its death (1). Therefore, the pregnant female must supply the embryo and the fetus with selenium to support selenoprotein synthesis.
Two selenoproteins contain >97% of the selenium in mouse plasma (2). Selenoprotein P (Sepp1), produced mostly in the liver, transports selenium to extrahepatic tissues (3). The other extracellular selenoprotein, glutathione peroxidase-3 (Gpx3), has not yet been shown to have a selenium transport role. Sepp1 is made up of an N-terminal domain, containing 1 selenocysteine residue in a redox motif, and a C-terminal domain, containing multiple selenocysteine residues (3). The total number of Sepp1 selenocysteine residues is 10 in humans, mice, and rats. Full-length Sepp1 and isoforms that terminate at the second, third, and seventh selenocysteine residues have been isolated from rat plasma (4).
Apolipoprotein E receptor-2 (apoER2) binds Sepp1 and mediates its endocytosis from the systemic circulation (5). Megalin, another member of the low-density lipoprotein receptor family, is present on the urinary aspect of renal proximal convoluted tubule (PCT) cells and mediates endocytosis of Sepp1 forms from the glomerular filtrate (6). Evidence indicates that apoER2 binds only Sepp1 forms that contain all or part of the selenium-rich C-terminal domain (5), while megalin binds selenium-poor Sepp1 forms that lack the C-terminal domain (unpublished results). The placenta expresses apoER2 (7), and megalin is expressed by the visceral yolk sac (8).
We postulated that Sepp1 and its receptors are involved in maternal to fetal transfer of selenium via the visceral yolk sac and the placenta. We investigated this possibility using mice with genetic alterations of Sepp1 and its receptors. In addition, we investigated the possibility that Gpx3 contributes to maternal-fetal selenium transfer. Our results indicate that both extracellular selenoproteins contribute to maternal-fetal selenium transfer and provide outlines of two mechanisms by which the transfer is accomplished. In addition, they demonstrate the existence of at least one more selenium transfer mechanism that does not appear to depend directly on the extracellular selenoproteins.
MATERIALS AND METHODS
Animals
Weanling mice were fed Torula yeast-based diet supplemented with 0, 0.25, 1.0, or 2.0 mg Se/kg diet in the form of sodium selenite unless otherwise indicated. The diet was formulated and pelleted to our specifications by Harlan-Teklad (Madison, WI, USA; ref. 9). The mice had free access to food and tap water.
Sepp1+/−/ breeding pairs (10) that had been backcrossed 7–10 times with C57BL/6 mice, except when otherwise indicated, were used to produce Sepp1−/− and Sepp1+/+ fetuses. Sepp1Δ240–361/+ breeding pairs (11) were used to produce Sepp1Δ240–361/Δ240–361 females for mating with Sepp1+/+ sires. Gpx3−/− and Gpx3+/+ dams and sires that had not been backcrossed were used to produce fetuses for experiments (2). ApoER2+/− breeding pairs congenic with C57BL/6 mice were used to produce fetuses. ApoER2−/− mice, strain name: B6;129S6-Lp8tm1Her/J, were originally purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Megalin+/− breeding pairs that had not been backcrossed were used to produce fetuses for experiments (12).
Blood was removed from the inferior vena cava under isoflurane anesthesia using a syringe and needle. Coagulation of the blood was prevented by the addition of 1 mg disodium EDTA/ml, and plasma was separated from formed elements by centrifugation. The Vanderbilt University Institutional Animal Care and Use Committee approved the studies described herein.
Genotyping
PCR primers for genotyping Sepp1 and apoER2 are given in a previous publication (13). Megalin genotypes were determined by PCR using primers G20 (5′-GCACATTTGGCCAGCCAAGG-3′), G21 (5′-CATATCTTGGAAATAAAGCGAC-3′), and SI-75 (5′-GATTGGGAAGACAATAGCAGGCATGC-3′. G20 and G21 gave a 275-bp product for megalin+/+ mice, while G21 and SI-75 gave a 325-bp product for megalin−/− mice. Megalin+/− mice had both PCR products.
Fetal experiments
Timed-mating experiments to produce fetuses of a specific gestational age were carried out by overnight housing of a single male mouse with 3 female mice. The male was removed in the morning, and the females were examined for vaginal plugs. The presence of a plug indicated that the female had mated, and the female's record was marked as d 1 of gestation.
When the selenium content of fetuses was determined, pregnant dams were killed by isoflurane inhalation on d 18. The fetuses were removed from the uterus, separated from membranes and placenta, and weighed. A 0.5-cm portion of the tail was taken from each fetus for isolation of DNA. Fetuses were frozen in liquid N2 and stored at −80°C. The genotype of each fetus was determined by PCR.
For immunocytochemistry (ICC) experiments, pregnant dams were killed by isoflurane inhalation. The uterus was removed and placed in ice-cold PBS. The placenta and the visceral yolk sac were dissected from each fetus. Tissues were then processed for ICC.
Biochemical assays
Plasma Sepp1 concentration and Gpx activity were measured in samples that had been stored at −80°C. Sepp1 concentration was determined using an ELISA method described previously (11). Gpx activity was measured as described previously using the coupled assay method with 0.25 mM H2O2 as a substrate (14). For selenium analysis, tissues were predigested in concentrated nitric acid. Then selenium was determined by the method of Koh and Benson (15), as modified by Sheehan and Gao (16).
ICC
Tissues were fixed at 4°C for 1 h with 4% formaldehyde in 0.1 M sodium phosphate (pH 7.4). They were then rinsed in phosphate buffer and infiltrated overnight at 4°C in phosphate buffer containing 20% sucrose. Tissue was then immersed in optimal cutting temperature compound (OCT; Fisher Scientific; Atlanta, GA, USA), frozen in liquid nitrogen, and stored at −80°C until sectioning at 5 μm was performed. Cryosections were rinsed with TBST2 (20 mM Tris·HCl, pH 8.0, 150 mM sodium chloride, 0.05% Tween 20, and 0.025% sodium azide) and blocked for 1 h with TBST2 containing 1% BSA and 0.1% glycine. Sections were then incubated with the primary antibody at 0.7 μg/ml in blocking solution for 1 h at room temperature. Primary antibodies were 9S4 (monoclonal antibody to N-terminal domain of Sepp1), 8096 (polyclonal antibody preparation to Gpx3), 2561 (polyclonal antibody preparation to apoER2), and polyclonal anti-megalin (2, 6, 17, 18). Sections were then washed 3 times for 5 min each in TBST2 and incubated for 1 h in affinity-purified Cy3-F(ab′)2 donkey anti-rabbit IgG, Cy3-F(ab′)2 mouse anti-rat IgG, Dylight 488-F(ab′)2 donkey anti-rat IgG (Jackson ImmunoResearch Laboratories, West Grove, PA, USA), or Alexa Fluor 488-Phalloidin (Molecular Probes/Invitrogen, Carlsbad, CA, USA) diluted in blocking solution. Hoechst 33258 was added to the secondary antibody solution to stain DNA. Slides were washed 3 times for 5 min each with TBST2 and mounted in Fluoromount G (Fisher Scientific, Pittsburgh, PA, USA). Specimens were examined by phase-contrast and epifluorescence microscopy, and digital images of experimental and control samples were made using identical photographic settings. Sections from knockout tissues stained with the same primary antibody served as negative controls. Each experiment was carried out with ≥3 mice, and the images shown are representative of all replicates.
Statistics
Statistical comparisons between groups were made on an iMac (Apple, Cupertino, CA, USA) using Prism 4 for Macintosh 4.0c software (GraphPad Software, Inc., La Jolla, CA, USA). Tukey's multiple-comparison test was applied after analysis by 1-way ANOVA. Where appropriate, Student's t test was used to compare groups. Groups were considered to be significantly different at values of P < 0.05.
RESULTS
Sepp1 and Gpx3 trafficking in the visceral yolk sac
During early to midgestation, the visceral yolk sac transfers nutrients from uterine fluid to the fetus. We studied Sepp1 and Gpx3 in the d-13 visceral yolk sac using ICC. Sepp1 was present in apical vesicles of the visceral yolk sac epithelial cells (Fig. 1A), as was Gpx3 (Fig. 1B). Gpx3 was also present beneath the epithelial cells at the position of the basement membrane, as has been reported in the intestine and several other tissues (19). Merging the figures of Sepp1 and Gpx3 staining shows that these selenoproteins colocalized in apical vesicles of the epithelial cells (Fig. 1C). These observations indicate that both Sepp1 and Gpx3 were taken up in the same vesicles.
Figure 1.
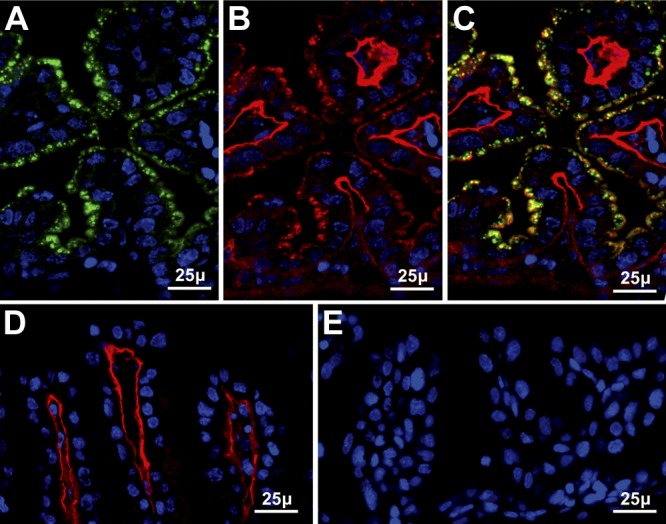
Uptake of maternal Sepp1 (green) and Gpx3 (red) by d-13 visceral yolk sac. Nuclei are stained blue. A, B) Sepp1 (A) and Gpx3 (B) staining of the same tissue section from a wild-type mouse. C) Merged image of panels A and B. D) Gpx3 staining of the visceral yolk sac from a Gpx3+/− fetus in a Gpx3−/− dam. E) Sepp1 staining of the visceral yolk sac from a Sepp1+/− fetus in a Sepp1−/− dam.
When a Gpx3−/− dam and her d-13 Gpx3+/− fetus were studied, no Gpx3 was detected in the visceral yolk sac epithelial cell vesicles, but Gpx3 was detected at the basement membrane (Fig. 1D). This indicates that the Gpx3 in the vesicles had a maternal origin and the Gpx3 at the basement membrane had a fetal origin. When a Sepp1−/− dam and her d-13 Sepp1+/− fetus were studied, no Sepp1 was detected in the visceral yolk sac epithelial cell vesicles (Fig. 1E). Thus, both Gpx3 and Sepp1 in the visceral yolk sac epithelial cell vesicles originated in the dam and were, therefore, transported to the fetus via the uterine fluid. Endocytosis of both Sepp1 and Gpx3 in the same vesicles could have been accomplished by bulk pinocytosis of uterine fluid or by endocytosis after the selenoproteins had bound to receptors on the epithelial cell membrane.
Because Sepp1 forms are taken up by receptor-mediated endocytosis in many tissues, we studied the known Sepp1 receptors in the visceral yolk sac. Megalin was detected at the apical surface of visceral yolk sac epithelial cells (Fig. 2A) but ICC did not detect apoER2 in the visceral yolk sac (Fig. 2B). The presence in the visceral yolk sac of vesicles containing both Sepp1 and Gpx3 was not eliminated by deletion of fetal megalin (Fig. 2C) or of fetal apoER2 (Fig. 2D). These findings indicate that d-13 visceral yolk sac cells take up maternal Sepp1 and Gpx3 from the uterine fluid by a mechanism that does not require megalin or apoER2. Thus, uptake of those selenoproteins by the visceral yolk sac can likely be attributed to pinocytosis.
Figure 2.
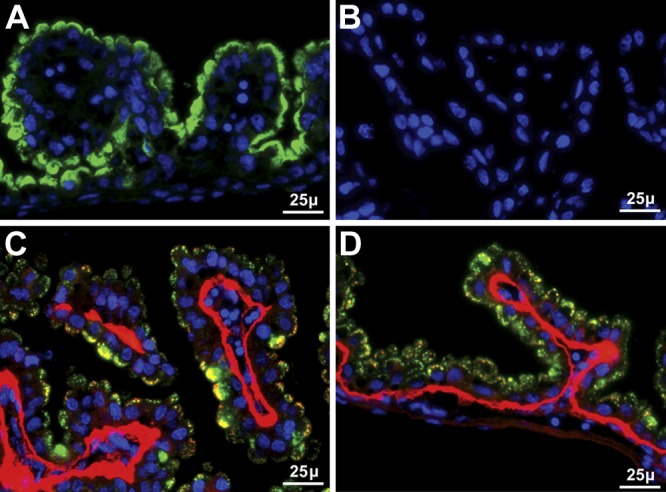
Uptake of Sepp1 and Gpx3 by d-13 visceral yolk sac independent of apoER2 and megalin. Nuclei are stained blue. A) Presence of megalin (green) on the apical aspect of visceral yolk sac epithelial cells. B) Absence of apoER2 staining (red). C) Merged image of Sepp1 (green) and Gpx3 (red) staining of the visceral yolk sac of a megalin−/− fetus in a megalin+/− dam. D) Merged image of Sepp1 (green) and Gpx3 (red) staining of the visceral yolk sac of an apoER2−/− fetus in an apoER2+/− dam.
The visceral yolk sac of the d-18 fetus resulting from mating of wild-type dams and sires was studied (not shown). No uptake of maternal Sepp1 or Gpx3 by the epithelial cells was detected, but Gpx3 was present at the basement membrane, as had been seen in the d-13 visceral yolk sac (Fig. 1B). Thus, uptake of maternal Sepp1 and Gpx3 by the visceral yolk sac epithelial cells ceased between d 13 and 18 of gestation.
Sepp1 trafficking in the placenta
The placenta provides nutrients to the mouse fetus during the last half of gestation. ICC of the d-18 placenta (late pregnancy) demonstrated the presence of Sepp1 in vesicles within trophoblast cells of the placental labyrinth zone (Fig. 3A), but no Gpx3 was detected in the placenta (not shown). ApoER2 was present in the placental labyrinth (Fig. 3B), but megalin was not detected there (not shown).
Figure 3.
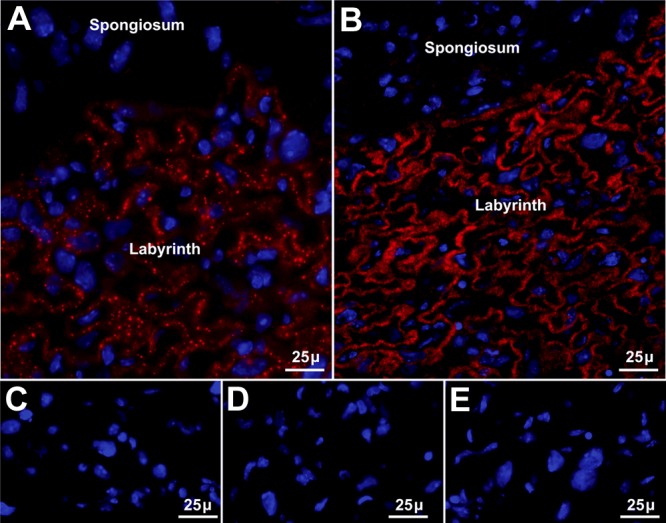
ApoER2-mediated uptake of maternal long-isoform Sepp1 by d-18 placenta. Panels show Sepp1 or apoER2 immunolocalization in red and nuclei in blue. A) Placenta of a Sepp1+/+ d-18 fetus from a Sepp1+/+ dam stained with antibody to Sepp1. B) Placenta of a Sepp1+/+ d-18 fetus from a Sepp1+/+ dam stained with antibody to apoER2. C) Placenta of a d-18 Sepp1+/− fetus from a Sepp1−/− dam stained for Sepp1. D) Placenta of a d-18 Sepp1Δ240–361/+ fetus whose Sepp1Δ240–361/Δ240–361 mother expresses only the N-terminal domain of Sepp1 stained for Sepp1. E) Placenta of a d-18 apoER2−/− fetus from an apoER2+/− dam stained with antibody to Sepp1.
Sepp1 was not detected in the placental labyrinth when Sepp1 was absent from the maternal circulation (Fig. 3C) or when the form of Sepp1 in the maternal circulation consisted only of its N-terminal domain without the selenium-rich C-terminal domain (Fig. 3D). Sepp1 was also not detected in the placental labyrinth when the fetal genotype was apoER2−/− (Fig. 3E). These findings indicate that fetal apoER2 binds maternal long-isoform Sepp1 in the placental labyrinth. Thus, apoER2-mediated endocytosis of maternal long-isoform Sepp1 in the placental labyrinth appears to be a mechanism of maternal-fetal selenium transfer.
The mechanism by which selenium delivered to the placenta by maternal Sepp1 is subsequently transferred to the fetus is not known. We considered the possibility that transcytosis of maternal Sepp1 occurs in the placental labyrinth (or elsewhere). If transcytosis from the maternal circulation to the fetal circulation does occur, maternal Sepp1 should be detectable in vesicles of d-18 fetal kidney PCT cells (6). We stained fetal kidneys for Sepp1 when the dam was Sepp1−/− and the fetus was Sepp1+/− (Fig. 4A) and when the dam was Sepp1+/− and the fetus was Sepp1−/− (Fig. 4B). Sepp1 appeared in the fetal kidney only when the fetus was able to express Sepp1 (Fig. 4A). We conclude that transcytosis of maternal Sepp1 in the placental labyrinth with transport of it to the fetal circulation does not occur.
Figure 4.
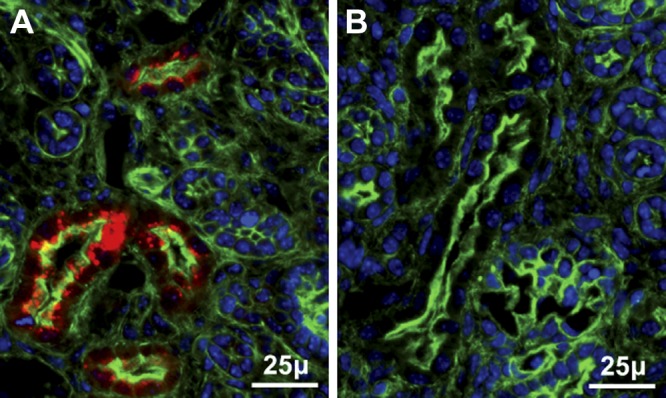
Maternal Sepp1 is not transferred to the d-18 fetal kidney. Sections are from d-18 fetus kidney cortex stained red for Sepp1, blue for nuclei, and green (phalloidin) for actin. A) Sepp1+/− fetus from a Sepp1−/− dam. B) Sepp1−/− fetus from a Sepp1+/− dam.
Both Sepp1 and Gpx3 affect fetal selenium concentration
Because Sepp1 and Gpx3 mechanisms of maternal-fetal selenium transfer had been demonstrated in Figs. 1–4, we examined the effect of pregnancy on their levels in maternal plasma. Fig. 5 confirms that maternal plasma Sepp1 concentration falls as pregnancy progresses (20) and shows that it rebounded after parturition. Plasma Gpx activity, over 90% of which is due to Gpx3 (2), varied little during pregnancy. These results are consistent with the fetus taking up Sepp1 from the maternal circulation, especially late in pregnancy.
Figure 5.

Maternal plasma selenoproteins during pregnancy. Plasma from pregnant C57BL/6 dams fed rodent chow was assayed for Sepp1 concentration and Gpx activity. Values are expressed as means with 1 sd shown by a bracket; n = 3. *P < 0.05 vs. preceding value; 1-way ANOVA with Tukey's multiple-comparison test.
The effects of deletion of Sepp1, Sepp1 receptors, and Gpx3 on fetal selenium concentration were determined. Female mice with deletion of genes for the plasma selenoproteins were mated and, on d 18 of gestation, their fetuses were taken for selenium assay. Maternal Sepp1 deletion sharply reduced fetal selenium concentration when dams were fed selenium-adequate diet, but Gpx3 deletion did not affect fetal selenium concentration under selenium-adequate conditions (Fig. 6). It was not possible to feed selenium-deficient diet to Sepp1−/− dams because that would have caused severe neurological injury and death (9, 10). Inducing selenium deficiency in Gpx3−/− dams, however, led to a small, but statistically significant, decrease (13%) in d-18 fetal selenium concentration (Fig. 6). These results are consistent with the known role of Sepp1 in selenium transport and regulation. The salutary effect of Gpx3 on fetal selenium in selenium-deficient dams, while small, challenges the general opinion that Gpx3 does not have a selenium transport role. However, it is consistent with the observation that the d-13 visceral yolk sac takes up Gpx3 from uterine fluid and indicates that Gpx3 does have a maternal-fetal selenium transfer role (Fig. 1B).
Figure 6.

Effect of Sepp1 and Gpx3 maternal genotypes on selenium concentrations of their d-18 fetuses. In the Sepp1 experiment, the mice had not been backcrossed. Sepp1−/− dams were mated with Sepp1+/+ sires, resulting in Sepp1+/− fetuses (n=48). Sepp1+/+ dams were mated with Sepp1+/− sires, and the resulting fetuses (n=63) were genotyped. Fetal Sepp1 genotype had no statistically significant effect on fetal selenium, so values of all fetuses were combined. In the Gpx3 experiment with the dams receiving 0.25 mg Se/kg diet, fetuses resulted from Gpx3−/− by Gpx3−/− or Gpx3+/+ by Gpx3+/+ matings, and thus were Gpx3−/− (n=36) or Gpx3+/+ (n=22). In the Gpx3 experiment with the dams receiving selenium-deficient diet, Gpx3+/− fetuses (n=10 fetuses for each maternal genotype) resulted from mating Gpx3−/− dams with Gpx3+/+ sires and Gpx3+/+ dams with Gpx3−/− sires. Values are expressed as means with a bracket indicating 1 sd. Paired values that were different by Student's t test are indicated by the percentages.
Sepp1 receptors and fetal selenium concentration
Figure 7A shows that deletion of apoER2 in the fetus did not affect d-18 fetal selenium when the maternal diet was selenium adequate, but lack of fetal apoER2 lowered fetal selenium concentration to 49% of that present in apoER2+/+ fetuses when the pregnant dams were selenium deficient. Deletion of fetal megalin did not affect d-18 fetal selenium concentration regardless of the maternal selenium status (Fig. 7B). These results show that apoER2 has a role in fetal selenium supply that becomes obvious only in maternal selenium deficiency, implying that the depression of fetal selenium to 40% in selenium-replete Sepp1−/− dams (Fig. 6) was not caused solely by disruption of apoER2-mediated Sepp1 endocytosis. No evidence was found that megalin is involved in maintaining selenium in d-18 fetuses.
Figure 7.

Effects of apoER2 (A) and megalin (B) genotypes of d-18 fetuses on their selenium concentrations under conditions of selenium adequacy and selenium deficiency. Fetuses resulted from heterozygous matings, and the dams had been fed the respective diets for ≥4 wk before mating. Values are expressed as means with a bracket indicating 1 sd; n = 4–13. The value marked 49% in panel A was significantly different from the corresponding wild-type value by Student's t test. Other values were not different from the wild-type values within dietary groups.
Maternal selenium status affects fetal selenium concentration
Deletion of Sepp1 in the mouse decreases its whole-body selenium (21). We performed an experiment to assess the effect of maternal whole-body selenium concentration on d-18 fetus selenium concentration in pregnant Sepp1−/− mice. Figure 8A confirms that deletion of Sepp1 reduced whole-body selenium concentration in nonpregnant mice fed selenium-adequate diet (supplemented with 0.25 mg Se/kg) and high-selenium diet (supplemented with 2 mg Se/kg).
Figure 8.

Effects of dietary selenium on whole-body selenium concentrations of nonpregnant female Sepp1−/− mice (A) and pregnant Sepp1−/− dams and their d-18 fetuses (B). The mice were fed the respective diets for ≥8 wk beginning at weaning. Values are means with a bracket indicating 1 sd; n = 4–7 adult female mice and 13–34 fetuses. Pairs were all significantly different (P<0.05) by Student's t test, and differences are indicated by the percentage values.
A similar experiment was carried out using pregnant Sepp1−/− mice carrying Sepp1−/− and Sepp1+/− fetuses. The high-selenium diet raised maternal and fetal whole-body selenium in approximately the same proportions (Fig. 8B). This result indicates that transfer of selenium from the pregnant mouse to its fetuses can occur by a pathway that does not require the presence of maternal Sepp1. This transfer mechanism appears to be related to the concentration of selenium in the pregnant mouse and/or its dietary selenium intake. Therefore, some of the decrease in fetal selenium caused by maternal Sepp1 deletion (Fig. 6) was likely due to this indirect effect of Sepp1 deletion.
DISCUSSION
The results presented here indicate that both extracellular selenoproteins, Sepp1 and Gpx3, are involved in the transfer of selenium from the pregnant dam to the fetus (Fig. 6). We identified visceral yolk sac and placental mechanisms by which these maternal selenoproteins are taken up. In the d-13 visceral yolk sac, apical vesicles in the epithelial cells contained maternal Sepp1 and Gpx3 (Fig. 1). That uptake from uterine fluid was not abolished by deletion of apoER2 or megalin. In the d-18 placenta, trophoblasts in the labyrinth zone took up maternal long-isoform Sepp1 by apoER2-mediated endocytosis (Fig. 3). A third maternal-fetal selenium transfer process was suggested by experiments in Sepp1−/− dams. It was sensitive to maternal whole-body selenium status (Fig. 8).
The visceral yolk sac supplies nutrients to the developing conceptus beginning early in gestation. It takes up nutrients from the uterine fluid by receptor- or transporter-mediated processes and by bulk pinocytosis. To assess the potential roles of Sepp1 and Gpx3 in its selenium uptake mechanism(s), we studied the midpregnancy, d-13 visceral yolk sac. We observed epithelial cell uptake of maternal Sepp1 and Gpx3, with both selenoproteins being present in the same apical vesicles (Fig. 1C). Furthermore, we showed that uptake of the selenoproteins was not abolished by deletion of megalin (Fig. 2C), which was present in the epithelial cells, or by deletion of apoER2 (Fig. 2D), which we did not detect in the epithelial cells. Coupled with our observation that this type of uptake does not occur in late pregnancy, d-18 visceral yolk sac (not shown), these findings imply that bulk pinocytosis of Sepp1 and Gpx3 by the visceral yolk sac supplies the midpregnancy—and possibly early pregnancy—fetus with selenium, although we cannot rule out a partial contribution by receptors. To our knowledge, bulk pinocytosis of selenoproteins has not previously been suggested to be a selenium transfer mechanism. Moreover, we are unaware of other instances in which Gpx3 serves as a selenium transport protein.
Because pinocytosis is a nonspecific uptake mechanism, the amount of selenium taken up by it should depend on the selenium content of the uterine fluid. Thus, we speculate that the selenium acquired by the conceptus through this mechanism is regulated by secretion of Sepp1, Gpx3, and other selenium forms into the uterine fluid. Assessment of the selenium in uterine fluid was not carried out in this study, but it will be needed for further characterization of this mechanism.
The placenta develops later in gestation than the visceral yolk sac, and it supplies the fetus with its increasing nutrient needs from midgestation until parturition. Our study of the late-pregnancy, d-18 placenta (Fig. 3) delineated an apoER2-dependent pathway of maternal long-isoform Sepp1 uptake that has properties similar to those of the apoER2-mediated Sepp1 endocytosis mechanism characterized in other tissues (3, 5). No uptake of Gpx3 was detected in the placenta. Thus, the placenta appears to take up Sepp1 from the maternal circulation in a manner similar to Sepp1 uptake by other tissues. The decline in maternal plasma Sepp1 concentration toward the end of pregnancy (Fig. 5) is compatible with increasing uptake of maternal Sepp1 by the placenta as the fetal growth rate increases.
We did not determine how the fetus assimilated the selenium taken up as selenoproteins by the visceral yolk sac and the placenta. However, we eliminated the possibility that maternal Sepp1 taken up by the placenta at d 18 of gestation was transferred to the fetal blood (Fig. 4). This finding implies that the placenta catabolizes maternal Sepp1 and releases the Sepp1 selenium into the fetal circulation. We did not determine the nature of the selenium released by the placenta into the fetal blood.
At least one more mechanism of maternal-fetal selenium transfer was detected in these studies. Sepp1−/− dams transferred selenium to their d-18 fetuses in direct relation to maternal dietary selenium intake and whole-body selenium concentration (Fig. 8B). The mechanism of this selenium transfer to the fetus is unknown, but possibilities include transfer by nonselenoprotein forms and/or by Gpx3. Pinocytosis of Gpx3 in the visceral yolk sac, the apparent mechanism of its uptake, ceases before d 18 of gestation. Nevertheless, we cannot rule out a role for Gpx3 in these results because Gpx3−/− mice were not studied in this experiment. Others have reported the presence of a sulfate transporter in the rat placental labyrinth that is inhibited by selenite and selenate, suggesting that it might also transport selenium forms (22). Further research will be necessary to characterize the additional selenium transport mechanism.
This third mechanism would appear to explain why fetal selenium concentration was decreased under selenium-adequate conditions by maternal deletion of Sepp1 (Fig. 6) but not by fetal deletion of apoER2 (Fig. 7A). Deletion of Sepp1 causes loss of selenium from the body, resulting in lower whole-body selenium content (21), but deletion of apoER2 does not significantly affect whole-body selenium (23). Thus, lowering of maternal whole-body selenium is likely to be responsible for the decrease in fetal selenium seen in Fig. 6. That deletion of apoER2 decreased fetal selenium only under conditions of selenium deficiency (Fig. 7A) indicates that the Sepp1-apoER2 uptake mechanism becomes most important under selenium-deficient conditions.
There is precedent for transport of selenium that does not depend on the extracellular selenoproteins. Mice with knockout of both Sepp1 and Gpx3 are viable if fed high-selenium diet, demonstrating that selenium can reach tissues by other routes (2). Thus, there appear to be two tiers of specific selenium transport. The lower-tier mechanism does not depend directly on selenoproteins and, therefore, likely involves one or more small-molecule forms of selenium. Function of this transport mechanism is highly dependent on the selenium intake of the animal and does not protect critical tissues, such as the brain and the testis under selenium-deficient conditions (10). One upper tier mechanism is the Sepp1-apoER2 mechanism (5). This mechanism ensures transport of selenium to tissues according to their need for it—even when dietary selenium is limiting. Pinocytosis by the visceral yolk sac, as newly described in this report, has characteristics of an upper-tier mechanism. Although it does not depend on receptors for selenoproteins, it does depend on uptake of selenoproteins by cells and appears to function under selenium-deficient conditions. Thus, both the visceral yolk sac and the placenta have upper-tier, selenoprotein-dependent mechanisms for maternal-fetal selenium transfer that should protect the fetus when the pregnant dam becomes selenium-deficient.
Some of the observations we made on visceral yolk sac and placenta Sepp1 uptake systems provide insight into Sepp1 interactions with its receptors. Placental apoER2 appears to have Sepp1-uptake properties similar to apoER2 in other tissues. This observation supports our suggestion that apoER2 is the principal receptor for long-isoform Sepp1 in the systemic circulation (5, 24).
Megalin mediates endocytosis of Sepp1 forms in the proximal renal tubule (1, 6). It is also present in the visceral yolk sac but does not appear to mediate endocytosis of the Sepp1 presented to that tissue (Figs. 2C and 7). It is possible that the megalin environment, or megalin itself, is different between visceral yolk sac and kidney PCT, causing this discrepancy. Another possibility is that the Sepp1 forms in glomerular filtrate and in uterine fluid are different. Sepp1 forms present in the urine of megalin−/− mice are N-terminal fragments that appear to have been generated by proteolytic cleavage of Sepp1 (unpublished results). The nature of Sepp1 forms in the uterine fluid has not been determined beyond the finding that they contain the N-terminal domain epitope for the monoclonal antibody 9S4, so further experiments will be necessary to assess binding of Sepp1 forms by megalin. In any case, it appears that megalin and apoER2 bind different forms of Sepp1.
The experiments presented in this report were designed to identify mechanisms of maternal-fetal selenium transfer that are specific for selenium. A nonselenium-specific pathway of selenium transfer also exists, and it was not evaluated here. That pathway is the transfer of selenium in the form of selenomethionine. Dietary selenium of free-living animals and humans is present in several forms, the most abundant one of which is selenomethionine. We eliminated selenomethionine from the diets used in these experiments except for the one depicted in Fig. 5.
Ingested selenomethionine enters the methionine pool and is incorporated randomly into proteins at methionine positions. Its selenium is recognized as such only after the selenomethionine is catabolized. Thus, transfer of selenomethionine-containing proteins or of selenomethionine itself (via methionine transporters) to the fetus will supply it with selenium. This transfer depends on selenomethionine intake by the dam and appears to be unregulated. Thus, superimposed on the maternal-fetal selenium transfer mechanisms described in this report, the selenomethionine transfer of selenium must be considered to occur in free-living animals and in humans. It is important to note that selenium transfer by selenomethionine will be negligible under conditions of dietary selenium deficiency. The selenium transfer mechanisms characterized in this work allow animals to survive in just such low-selenium environments.
Our observations may have implications for maternal-fetal selenium transfer in humans. Human selenium nutritional status is borderline or low in many areas of the world. Under those conditions, it seems possible that Sepp1 could be depleted in maternal plasma because of fetal demands (Fig. 5). If this occurs, transfer of selenium to the fetus might be suboptimal and/or selenium supply to maternal tissues might be compromised. These possibilities need further investigation in animals and in human populations with marginal or low-selenium nutritional status.
In summary, several mechanisms ensure the transfer of selenium from the pregnant dam to its conceptus. The visceral yolk sac imbibes uterine fluid that contains Sepp1 and Gpx3, and the placenta takes up Sepp1 from maternal blood via apoER2-mediated endocytosis. Both of these mechanisms depend on selenoproteins and function under conditions of selenium deficiency. A third mechanism that does not appear to depend directly on selenoproteins was detected. It is highly dependent on the selenium status of the pregnant female and, therefore, becomes less effective under conditions of selenium deficiency.
Acknowledgments
The authors are grateful to Lori M. Austin, Teri D. Stevenson, Kristin V. Bradshaw (deceased), and Michelle L. Chatterton for animal husbandry. Dr. Takeshi Naruse (Kaketsuken, The Chemo-Sero-Therapeutic Research Institute, Kumamoto, Japan) kindly provided the hybridoma that produced 9S4, the monoclonal antibody to Sepp1. Dr. Daniel Biemesderfer (Yale University, New Haven, CT, USA) provided the antibody to megalin, and Dr. Joachim Herz (University of Texas Southwestern Medical School, Dallas, TX, USA) provided the antibody to apoER2 and the megalin+/− mouse strain.
These studies were supported by U.S. National Institutes of Health grants R37 ES02497, R01 DK82813, P30 ES00267, and P30 DK58404.
The authors declare no conflicts of interest.
Footnotes
- apoER2
- apolipoprotein E receptor-2
- Gpx3
- glutathione peroxidase-3
- ICC
- immunocytochemistry
- PCT
- proximal convoluted tubule
- Sepp1
- selenoprotein P
REFERENCES
- 1. Bösl M. R., Takaku K., Oshima M., Nishimura S., Taketo M. M. (1997) Early embryonic lethality caused by targeted disruption of the mouse selenocysteine tRNA gene (Trsp). Proc. Natl. Acad. Sci. U. S. A. 94, 5531–5534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Olson G. E., Whitin J. C., Hill K. E., Winfrey V. P., Motley A. K., Austin L. M., Deal J., Cohen H. J., Burk R. F. (2010) Extracellular glutathione peroxidase (Gpx3) binds specifically to basement membranes of mouse renal cortex tubule cells. Am. J. Physiol. Renal Physiol. 298, F1244–F1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Burk R. F., Hill K. E. (2009) Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 1790, 1441–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma S., Hill K. E., Caprioli R. M., Burk R. F. (2002) Mass spectrometric characterization of full-length rat selenoprotein P and three isoforms shortened at the C terminus. Evidence that three UGA codons in the mRNA open reading frame have alternative functions of specifying selenocysteine insertion or translation termination. J. Biol. Chem. 277, 12749–12754 [DOI] [PubMed] [Google Scholar]
- 5. Kurokawa S., Hill K. E., McDonald W. H., Burk R. F. (2012) Long isoform mouse selenoprotein P (Sepp1) supplies rat myoblast L8 cells with selenium via endocytosis mediated by heparin binding properties and apolipoprotein E receptor-2 (ApoER2). J. Biol. Chem. 287, 28717–28726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olson G. E., Winfrey V. P., Hill K. E., Burk R. F. (2008) Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 283, 6854–6860 [DOI] [PubMed] [Google Scholar]
- 7. Kim D. H., Iijima H., Goto K., Sakai J., Ishii H., Kim H. J., Suzuki H., Kondo H., Saeki S., Yamamoto T. (1996) Human apolipoprotein E receptor 2. A novel lipoprotein receptor of the low density lipoprotein receptor family predominantly expressed in brain. J. Biol. Chem. 271, 8373–8380 [DOI] [PubMed] [Google Scholar]
- 8. Sahali D., Mulliez N., Chatelet F., Laurent-Winter C., Citadelle D., Roux C., Ronco P., Verroust P. (1992) Coexpression in humans by kidney and fetal envelopes of a 280 kDa-coated pit-restricted protein. Similarity with the murine target of teratogenic antibodies. Am. J. Pathol. 140, 33–44 [PMC free article] [PubMed] [Google Scholar]
- 9. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Burk R. F. (2004) Neurological dysfunction occurs in mice with targeted deletion of selenoprotein P gene. J. Nutr. 134, 157–161 [DOI] [PubMed] [Google Scholar]
- 10. Hill K. E., Zhou J., McMahan W. J., Motley A. K., Atkins J. F., Gesteland R. F., Burk R. F. (2003) Deletion of selenoprotein P alters distribution of selenium in the mouse. J. Biol. Chem. 278, 13640–13646 [DOI] [PubMed] [Google Scholar]
- 11. Hill K. E., Zhou J., Austin L. M., Motley A. K., Ham A. J., Olson G. E., Atkins J. F., Gesteland R. F., Burk R. F. (2007) The selenium-rich C-terminal domain of mouse selenoprotein P is necessary for supply of selenium to brain and testis but not for maintenance of whole-body selenium. J. Biol. Chem. 282, 10972–10980 [DOI] [PubMed] [Google Scholar]
- 12. Willnow T. E., Hilpert J., Armstrong S. A., Rohlmann A., Hammer R. E., Burns D. K., Herz J. (1996) Defective forebrain development in mice lacking gp330/megalin. Proc. Natl. Acad. Sci. U. S. A. 93, 8460–8464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Olson G. E., Winfrey V. P., Nagdas S. K., Hill K. E., Burk R. F. (2007) Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 282, 12290–12297 [DOI] [PubMed] [Google Scholar]
- 14. Lawrence R. A., Burk R. F. (1976) Glutathione peroxidase activity in selenium-deficient rat liver. Biochem. Biophys. Res. Commun. 71, 952–958 [DOI] [PubMed] [Google Scholar]
- 15. Koh T. S., Benson T. H. (1983) Critical re-appraisal of fluorometric method for determination of selenium in biological materials. J. Assoc. Off. Anal. Chem. 66, 918–926 [PubMed] [Google Scholar]
- 16. Sheehan T. M. T., Gao M. (1990) Simplified fluorometric assay of total selenium in plasma and urine. Clin. Chem. 36, 2124–2126 [PubMed] [Google Scholar]
- 17. Trotter J. H., Klein M., Jinwal U. K., Abisambra J. F., Dickey C. A., Tharkur J., Masiulis I., Ding J., Locke K. G., Rickman C. B., Birch D. G., Weeber E. J., Herz J. (2011) ApoER2 function in the establishment and maintenance of retinal synaptic connectivity. J. Neurosci. 31, 14413–14423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zou Z., Chung B., Nguyen T., Mentone S., Thomson B., Biemesderfer D. (2004) Linking receptor-mediated endocytosis and cell signaling: evidence for regulated intramembrane proteolysis of megalin in proximal tubule. J. Biol. Chem. 279, 34302–34310 [DOI] [PubMed] [Google Scholar]
- 19. Burk R. F., Olson G. E., Winfrey V. P., Hill K. E., Yin D. (2011) Glutathione peroxidase-3 produced by the kidney binds to a population of basement membranes in the gastrointestinal tract and in other tissues. Am. J. Physiol. Gastrointest. Liver Physiol. 301, G32–G38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Anan Y., Ogra Y., Somekawa L., Suzuki K. T. (2009) Effects of chemical species of selenium on maternal transfer during pregnancy and lactation. Life Sci. 84, 888–893 [DOI] [PubMed] [Google Scholar]
- 21. Burk R. F., Hill K. E., Motley A. K., Austin L. M., Norsworthy B. K. (2006) Deletion of selenoprotein P upregulates urinary selenium excretion and depresses whole-body selenium content. Biochim. Biophys. Acta 1760, 1789–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miyauchi S., Srinivas S. R., Fei Y. J., Gopal E., Umapathy N. S., Wang H., Conway S. J., Ganapathy V., Prasad P. D. (2006) Functional characteristics of NaS2, a placenta-specific Na+-coupled transporter for sulfate and oxyanions of the micronutrients selenium and chromium. Placenta 27, 550–559 [DOI] [PubMed] [Google Scholar]
- 23. Burk R. F., Hill K. E., Olson G. E., Weeber E. J., Motley A. K., Winfrey V. P., Austin L. M. (2007) Deletion of apolipoprotein E receptor-2 in mice lowers brain selenium and causes severe neurological dysfunction and death when a low-selenium diet is fed. J. Neurosci. 27, 6207–6211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hill K. E., Wu S., Motley A. K., Stevenson T. D., Winfrey V. P., Capecchi M. R., Atkins J. F., Burk R. F. (2012) Production of selenoprotein P (Sepp1) by hepatocytes is central to selenium homeostasis. J. Biol. Chem. 287, 40414–40424 [DOI] [PMC free article] [PubMed] [Google Scholar]