

Summary
Here, we present a simple and highly efficient method for generating and detecting mutations of any gene in Drosophila melanogaster through the use of the CRISPR/Cas9 system (clustered regularly interspaced palindromic repeats/CRISPR-associated). We show that injection of RNA into the Drosophila embryo can induce highly efficient mutagenesis of desired target genes in up to 88% of injected flies. These mutations can be transmitted through the germline to make stable lines. Our system provides at least a 10-fold improvement in efficiency over previously published reports, enabling wider application of this technique. We also describe a simple and highly sensitive method of detecting mutations in the target gene by high-resolution melt analysis and discuss how the new technology enables the study of gene function.
Graphical Abstract

Highlights
-
•
Targeted mutagenesis with CRISPR/Cas9 nucleases of Drosophila genes
-
•
Highly efficient mutant generation in up to 88% of injected flies
-
•
Transmission of mutants through the germline, in up to 34.5% of offspring
-
•
Highly sensitive detection of mutations by high-resolution melt analysis
Bassett, Liu, and colleagues present a simple and highly efficient method for generating and detecting mutations of any gene in Drosophila melanogaster through the use of the CRISPR/Cas9 system (clustered regularly interspaced palindromic repeats/CRISPR-associated). They show that injection of RNA into the Drosophila embryo can induce highly efficient mutagenesis of desired target genes in up to 88% of injected flies. These mutations can be transmitted through the germline, and this method offers the opportunity to generate a desired mutant fly line within one month.
Introduction
The fruit fly Drosophila melanogaster is one of the most highly developed genetic model organisms. Nevertheless, despite the large number and power of available techniques, the generation of novel mutant alleles in a chosen gene by homologous recombination remains a relatively time-consuming procedure (Maggert et al., 2008; Rong and Golic, 2000; Venken and Bellen, 2005). Several genome-engineering techniques have recently been developed for guiding nucleases to selected target sites in the genome, making it possible to engineer mutations in a number of model organisms (Cong et al., 2013; DiCarlo et al., 2013; Gaj et al., 2013; Hwang et al., 2013; Mali et al., 2013), including flies (Beumer et al., 2008; Gratz et al., 2013; Liu et al., 2012).
The type II CRISPR/Cas9 system (clustered regularly interspaced short palindromic repeats/CRISPR-associated) is used by bacteria as an RNA-guided defense system against invading viruses and plasmids (Barrangou et al., 2007; Ishino et al., 1987). In Streptococcus pyogenes, the Cas9 endonuclease is guided to its target site by complementary base pairing of CRISPR RNAs (crRNAs) with the target DNA sequence, and this requires a third component, the trans-activating CRISPR RNA (tracrRNA), which recruits the crRNA into the Cas9 complex (Brouns et al., 2008; Gasiunas et al., 2012; Jinek et al., 2012). This system has recently been adapted to target double-strand breaks in the genomes of other organisms, including studies in human cells (Cong et al., 2013; Mali et al., 2013), mice (Wang et al., 2013), and zebrafish (Hwang et al., 2013).
We have modified a two-component system described previously (Cong et al., 2013; Dahlem et al., 2012; Jinek et al., 2012; Mali et al., 2013), in which the crRNA and tracrRNA are fused into a single synthetic guide RNA (sgRNA) (Figure 1A) to efficiently create targeted mutations in the Drosophila yellow and white genes by direct injection of Cas9 mRNA and an sgRNA into the embryo. We show that the induced double-strand breaks can result in small insertions and deletions (indels) at the target sites as a result of inefficient repair by nonhomologous end joining (NHEJ) (Bibikova et al., 2002). This process occurs with extremely high efficiency, with up to 88% of injected flies having detectable mutations. Mutations can also be transmitted through the germline to the following generation at a rate of up to one-third of total offspring. Our method offers a 10- to 100-fold improvement in efficiency over a recently published report (Gratz et al., 2013), enabling its general application to gene knockouts. We also describe a system for simple and effective detection of the indels created in the injected generation (G0) by high-resolution melt analysis (HRMA). This can be applied to any gene and makes it possible to knock out a chosen gene within 1 month. This will allow the rapid mutation of genes that do not appear in the current mutant catalogs (Bellen et al., 2011), enables isogenic lines to be generated more easily, and offers the ability to combine mutations with pre-existing stocks. It also opens up the possibility of a new generation of large-scale genetic screening and mosaic analyses to be performed in Drosophila.
Figure 1.

CRISPR/Cas9 System for Targeting Double-Strand Breaks
(A) The two-component system for inducing double-strand breaks. The synthetic guide RNA (sgRNA) contains a region of complementarity to the target site on the DNA, as well as stem loops from the tracrRNA to mediate binding to the Cas9 protein. Cas9 protein is indicated by a yellow circle, cleavage sites by arrowheads, and the protospacer adjacent motif (PAM, NGG) required for cleavage in red.
(B) The PCR-based system for generation of sgRNAs. Two oligonucleotides are used to generate the sgRNA template for in vitro transcription (black lines). The first includes a T7 promoter (highlighted in blue) and upstream sequence for efficient in vitro transcription, followed by a GGN18 target-site sequence and a portion of the sgRNA stem loops. The second includes the entire sgRNA sequence after the target site. PCR is performed with the two primers but without any other DNA template.
Results
A Simple CRISPR/Cas9 System for Drosophila Mutagenesis
We adapted the CRISPR/Cas9 system to mutagenize genes in Drosophila by injecting a mix of Cas9 mRNA and sgRNA into syncytial blastoderm-stage embryos. The sgRNAs were created by in vitro transcription of a PCR template generated with a unique forward primer containing a T7 polymerase binding site and the gene-specific guide sequence, as well as a common overlapping reverse primer containing the remainder of the sgRNA sequence necessary for incorporation into the Cas9-sgRNA complex (Figure 1B, Figure S1, and Table S1). This process is easily expandable to high-throughput targeting of many genes, because it requires the synthesis of a single oligonucleotide, one PCR, and an in vitro transcription reaction for each new sgRNA.
Figure S1.
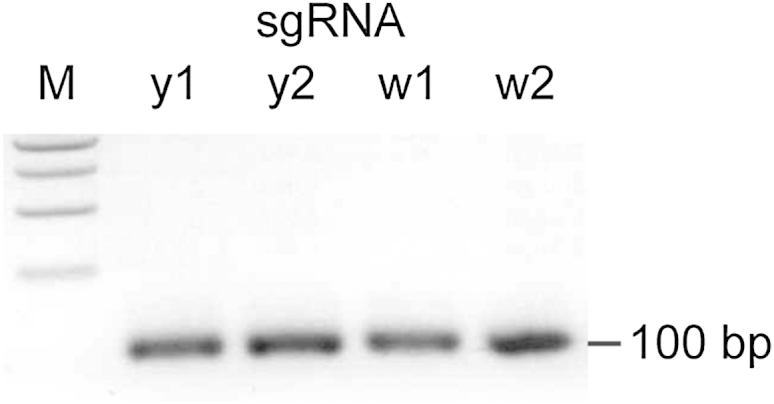
sgRNA Template PCR, Related to Figure 1
Representative gel image of sgRNA template PCR. 5% of the reaction from each sgRNA template PCR was analyzed on a 1.5% agarose gel. A single band of the expected size is observed. M – 1 kb ladder (New England Biolabs).
The sgRNA target sequence (20 nt) must be followed by an NGG protospacer adjacent motif (PAM), which is absolutely required for Cas9 cleavage (Jinek et al., 2012) (Figure 1A). Also, the T7 polymerase requires that the first two bases of the sgRNA target sequence are GG (Figure 1B). Despite these sequence constraints, our analysis shows that there are still more than 5 × 105 potential target sites in the Drosophila melanogaster genome, placed every 300 bases on average. Recent studies have shown that the first seven bases of the 20 nt target sequence are dispensable for CRISPR target recognition and cleavage (Cong et al., 2013), suggesting that the requirement for a GG sequence at the beginning of the target sequence may be relaxed if necessary.
Mutagenesis of the yellow Gene Is Highly Efficient and Concentration Dependent
We initially designed an sgRNA to target a double-strand break at the beginning of the second exon of the yellow gene in flies (y1, Figures 2A and 2B), because its mutation would provide an easy readout of effective mutagenesis by loss of pigmentation in the body of the fly. It is also X-linked, meaning that mutation of a single copy would result in a visible phenotype in males of the injected generation (G0).
Figure 2.

CRISPR/Cas9-Induced Deletions at the yellow and white Loci
(A) A schematic of the yellow and white genes showing the sgRNA target sites. Exons are shown as black boxes, transcriptional start sites as arrows, and sgRNA target sites as black triangles.
(B) Sequence of sgRNA targets. The 20 nt target sequence corresponding to each target site is indicated in orange, along with the neighboring NGG protospacer adjacent motif (PAM) in red.
(C) Mosaic yellow expression in the injected G0 flies. Female flies showing mosaic yellow expression upon injection with Cas9 mRNA and y1 sgRNA are shown. Patches of yellow mutant tissue can be observed (arrowheads) and are outlined by dotted lines.
(D) Mosaic white expression in the injected G0 flies. Female and male flies showing mosaic white expression upon injection with Cas9 mRNA and w2 sgRNA are shown. Patches of white mutant tissue can be observed (arrowheads).
(E) Sequencing of induced mutations in the yellow gene. PCR products spanning the sgRNA target sites were analyzed for indels. The first line in each alignment represents wild-type sequence, and subsequent lines show individual mutant clones. Target sites are indicated in orange, PAM in red, and cleavage sites by a black triangle. Deleted bases are marked with dashes, and inserted or substituted bases are indicated in lower case.
(F) Sequencing of induced mutations in the white gene, as in (E).
We tested a variety of concentrations of Cas9 mRNA:sgRNA and found the efficiency of NHEJ-based deletions to be concentration dependent (Table 1). Remarkably, at the highest concentration (1,000 ng/μl), we found that 86% of the surviving flies from the injected generation (G0) showed mosaic yellow expression. Surprisingly, this included female flies, which require mutation of both alleles of the yellow gene in order to manifest a phenotype, indicating that mutagenesis had been highly efficient (Figure 2C).
Table 1.
y1 Mutagenesis Is Concentration Dependent
| RNA Concentration (ng/μl) | % Survival |
% Mosaic Adults | |
|---|---|---|---|
| Larvae | Adults | ||
| 1000 | 10% | 3% | 86% |
| 500 | 16% | 7% | 50% |
| 250 | 16% | 6% | 17% |
| 125 | 17% | 8% | 10% |
| 0 | 20% | 11% | 0% |
| No injection | 28% | 16% | 0% |
Survival rates of embryos to first instar larvae and adults as well as percentage of mosaic adults are shown for a variety of different concentrations of injected RNA targeting the y1 target site. The highest mutagenesis efficiency was observed with the highest concentration of RNA, although survival rates were reduced slightly. “No injection” indicates embryos that were treated in the same way as the other samples but were not injected.
As the concentration of RNA was reduced, the adult survival rates following injection increased from 3% to 11%, but the proportion of mosaic adults decreased from 86% to 10% (Table 1). We therefore decided to use the highest concentration of RNA for subsequent experiments, because in most situations it is preferable to produce a higher proportion of mutant flies to reduce the number of offspring that need to be screened in order to identify a mutant.
Different sgRNAs Show Different Efficiencies
To compare mutagenesis efficiencies of different sgRNAs, we designed a second sgRNA targeting the yellow gene (y2) and an additional two sgRNAs targeting the second (w1) or third (w2) exons of the white gene (Figures 2A and 2B). The white gene is again X-linked, and its disruption would be expected to result in a visible eye-color phenotype in adult flies. These target sites showed substantially different efficiencies of cleavage, resulting in 88%, 75%, 4%, or 25% mosaic adults for y1, y2, w1, and w2 respectively (Table 2). It seems unlikely that this is due to differences in the chromatin environment around the target sites, given that the pairs of sites are very close; rather, the sgRNA sequence is likely to have an impact on mutagenesis efficiency. The w2 sgRNA efficiently induced large patches of mosaic tissue in the G0 generation, even in female flies, which require two separate targeting events (Figure 2D). However, injection of this construct resulted in low survival rates of <3% (Table 2). In contrast, the w1 sgRNA failed to show any visible mosaic white expression in the injected G0 generation but had an improved survival rate of 12% (Table 2).
Table 2.
Mutagenesis Efficiency Depends on Target Site
| sgRNA | % (#) Survival |
% (#) Mosaic Adults |
||
|---|---|---|---|---|
| Larvae | Adults | Visible | HRMA | |
| y1 | 9% (36) | 8% (33) | 88% (29) | 88% (29) |
| y2 | 11% (24) | 7% (16) | 75% (12) | 75% (12) |
| w1 | 16% (32) | 12% (25) | 0% (0) | 4% (2) |
| w2 | 4% (10) | 3% (8) | 25% (2) | 25% (2) |
Comparison of survival rates and mutagenesis efficiency between different target sites. All RNA was injected at 1000 ng/μl RNA concentration. Different target sites show different survival rates and mutagenesis efficiencies as measured by the proportion of mosaic adults. Mosaic mutations were determined visually (visible) or by high-resolution melt analysis (HRMA). Absolute numbers are indicated in brackets.
The presence of mutations was confirmed by the sequencing of PCR products spanning the cleavage site and showed a selection of indels consistent with previous studies in Drosophila (Liu et al., 2012), zebrafish (Hwang et al., 2013), and human cells (Cong et al., 2013; Mali et al., 2013) (Figure 2E, F).
Transmission of Mutations to Subsequent Generations
We were interested in investigating whether the transmission of mutations to subsequent generations is efficient, because this is a prerequisite for analysis of any mutations generated. All of the flies were backcrossed to flies carrying mutations in the yellow and white genes, and numbers of yellow or white mutant progeny in the subsequent generation were scored (Figure 3). This showed that mutations could be generated within the germline and transmitted to subsequent generations at a very high efficiency. With the use of the y1 sgRNA, 58% of the flies (79% of the fertile flies) were able to give rise to at least one mutant offspring, and a total of 34.5% of all the offspring contained a mutation in the yellow gene (Figure 3A). The proportion of flies obtained from those crosses giving rise to mutant progeny ranged from 5.3% to 88.5% (Figure 3B).
Figure 3.

Germline Transmission of Mutations
(A) Efficiency of germline transmission of mutations induced at each target site. Number of embryos injected, hatched L1 larvae, and total and visibly mosaic G0 adults produced are noted. Each G0 adult was crossed to flies with yellow and white gene mutations (y1w1), and the number of flies producing offspring was noted (fertile), along with the number of crosses that produced at least one mutant offspring (germline mutants). This is also expressed as a percentage of the total G0 adults (brackets). The percentage of mutant progeny shows the total number of mutant offspring compared to the total number of offspring from all crosses. The range shown in brackets shows the percentage of mutant flies produced from each individual positive cross. The bottom row of the table shows the equivalent numbers from Gratz et al. (2013) for comparison.
(B) Percentage of mutant offspring from each injected G0 fly. Offspring of each injected G0 fly were backcrossed to flies with mutations in yellow and white genes (y1w1) and analyzed for the percentage of yellow or white mutant female offspring. Each individual cross is represented by a single point. Distributions for each target site are shown, and they ranged from 0% to 88.5%.
The efficiency of germline transmission largely followed the proportion of mosaic flies in the G0 generation, but it was possible to generate mutant offspring with no visible mosaic tissue (w1, Figure 2F and Figure 3). This is likely to reflect germline mutant tissue not being detectable, because yellow mutants give a cuticular phenotype and white mutants an eye-color phenotype, neither of which overlaps germline tissue.
We also observed that although the w2 sgRNA was able to efficiently target mutagenesis, resulting in mosaic white expression in the injected generation, no mutant offspring were produced (Figure 3). The failure to generate mutant offspring may be due to high efficiency of double-strand break induction, which may result in toxicity or sterility, as indicated by the low survival and fertility rates (Figure 3 and Table 2). This suggests that the concentration of injected RNA may need to be optimized on the basis of the cleavage efficiency of a particular sgRNA.
Detection of Mutations by HRMA
Although the yellow and white genes provide visible readouts in the injected, G0 generation, this is not true of the majority of genes that one would wish to target. A number of techniques have been developed for the detection of indels at the target sites, such as Surveyor assays (Miller et al., 2007), T7 endonuclease assays (Kim et al., 2009), lacZ disruption assays (Hisano et al., 2013), heteroduplex mobility (Ota et al., 2013), HRMA (Dahlem et al., 2012), and loss of restriction sites (Bedell et al., 2012). We found that HRMA was optimal in terms of speed and sensitivity. This technique utilizes differences in the melting temperature of heteroduplexes containing insertions or deletions to differentiate them from wild-type homoduplexes. We therefore designed an HRMA assay for the sgRNA target sites in the yellow and white genes, and we used it to detect mutations in the mosaic G0 flies. This was extremely effective in detecting mosaic mutations (Figures 4A–4D), and it recognized 100% of the flies that had a visible mosaic yellow or white expression in our analysis. Additionally, it identified one fly that did not have visible mosaic white expression but was able to produce mutant offspring (Figure 3). One can apply it to any desired target gene by designing a 100–200 bp PCR amplicon spanning the CRISPR target site.
Figure 4.

High-Resolution Melt Analysis
(A) High resolution melt analysis (HRMA) of G0 mosaic yellow mutant flies. Mosaic mutant flies (red) can be easily differentiated from wild-type Oregon R (green) and nonmosaic (gray) flies by a change in the shape of the melt curve due to heteroduplex formation.
(B) Change in fluorescence relative to control Oregon R and nonmosaic flies highlight the changes in curve shape shown in (A).
(C) HRMA performed as in (A) but for white mosaic flies.
(D) Change in fluorescence as in (B) but for white mosaic flies.
(E) Sequencing of the y1a mutant shows a single point deletion compared to wild-type Oregon R flies at the y1 cleavage site. Target sites are indicated in orange, the protospacer adjacent motif (PAM) in red, and the cleavage site by a black triangle.
(F) HRMA shows a reproducible shift in the melting curve in a heterozygous point deletion in the yellow gene (y1a, red) in comparison to wild-type Oregon R flies (gray).
(G) Change in fluorescence highlights the difference in curve shape shown in (F).
This technique can also be used to detect heterozygous mutations in the following generation in order to screen for those flies that inherit the mutation of interest. We used a fly line containing a single base deletion generated at the y1 target site (Figure 4E) to test the sensitivity of this assay, and we found that even with such a subtle change, the mutation was readily detectable in heterozygous flies (Figures 4F and 4G).
Analysis of Off-Target Cleavage
We also applied the HRMA technique to address potential concerns with off-target cleavage. Other studies have suggested that the final 12 nt of the target sequence within the sgRNA (the “seed”) and the PAM sequence (NGG) are sufficient for efficient cleavage and that substitutions at any position within these sequences abolish target recognition (Cong et al., 2013). When designing the sgRNAs, we avoided any sequences that contained a perfect match to the “seed” sequence of the sgRNA and the adjacent PAM sequence. However, for each sgRNA we designed, at least one other region in the Drosophila genome contained a match of 10 or 11 nt followed by the PAM sequence. We therefore tested these seven sites for off-target mutations by HRMA, but no evidence for any mutagenesis was observed at any of these sites (Figure 5). This information about off-target effects will help to guide target choice in the future.
Figure 5.

Analysis of Off-Target Effects by High-Resolution Melt Analysis
(A) High-resolution melt analysis (HRMA) of the putative off-target site in the CG14073 gene in y1 sgRNA-injected mosaic flies. The upper panel shows the alignment of the off-target site with the y1 sgRNA sequence, indicating the target site in orange, PAM sequence in red, and “seed” sequence previously shown to be required for target recognition (Cong et al., 2013) with a black box. The lower panel shows HRMA of the putative off-target site, showing no difference between wild-type Oregon R flies (gray) and mosaic yellow mutant flies (colored).
(B) HRMA performed as in (A) but for y2 sgRNA off-target sites in CG43672 and H4r.
(C) HRMA performed as in (A) but for w1 sgRNA off-target sites in CG13397 and Myo10A.
(D) HRMA performed as in (A) but for w2 sgRNA off-target sites in CG34422 and Fim.
Discussion
Here, we describe a simple, efficient, and rapid technique for the creation of novel targeted mutations in a chosen gene through adaptation of the CRISPR/Cas9 system to Drosophila melanogaster. This allows the production of a novel mutation in virtually any genetic background within a month, thereby permitting future high-throughput analyses of gene function to be performed.
The ability to rapidly generate new mutations in a chosen gene with our technique will permit simpler compounding of mutations to generate double or triple mutants, particularly with genetically linked genes. Recent studies of CRISPR/Cas9 injection into mouse zygotes or embryonic stem cells (Wang et al., 2013), human cells (Cong et al., 2013), and Drosophila (Gratz et al., 2013) have suggested that several sgRNAs can be used simultaneously to target multiple independent genes, opening up the possibility of removing whole gene families in a single step.
A recent report of the CRISPR/Cas9 system for targeted mutagenesis in Drosophila has demonstrated that mutations and deletions can be generated in the yellow and rosy genes (Gratz et al., 2013). However, the efficiency of mutagenesis in their study is considerably lower than that described here in terms of both the number of flies with germline mutations (5.9% compared to our rate of 58%) and the proportion of mutant progeny (0.25% compared to our 34.5%) (Figure 3A). These differences may be explained by their injection of plasmid DNA encoding the Cas9 and sgRNA components of the system rather than RNA. Expression of RNA from a plasmid template is likely to be at levels lower than those seen with direct RNA injection and perhaps to occur later, once the germline has been established, resulting in fewer germline mutations. Our use of RNA rather than DNA also removes any possibility of integration of the injected DNA into the genome, especially given that the Cas9 endonuclease can induce double-strand breaks, which may result in the insertion of exogenous DNA fragments.
Given the high efficiency of mutant generation, our new system will allow clonal analysis of mutations within a few days of injection, so long as an antibody is available to the targeted protein for detection of the mutant tissue. The observation of mosaic yellow and white females suggests that this can be achieved for genes on all chromosomes, but it is especially straightforward for X-linked genes. It will also be possible to perform semitargeted genetic screening, whereby modifier screens are performed with a library of sgRNAs targeting a desired subset of genes rather than with random mutagens, or with RNAi knockdowns of the genes of interest. Although the system described is able to mutate protein-coding genes through the generation of frameshifts, the deletions generated could also be used to remove small functional sites in the genome, such as transcription factor binding sites, in order to investigate their role in the regulation of gene expression. This may be particularly useful for delineation of the functional consequences of changes observed during evolution or disease, because the system is not limited to Drosophila melanogaster and can be applied to nonmodel Drosophilids and indeed to other animals (Hwang et al., 2013; Wang et al., 2013).
The double-strand breaks generated are predominantly repaired by NHEJ but can also be repaired by homologous recombination (HR) (Chapman et al., 2012). Double-strand breaks have been shown to enhance the rate of homologous gene targeting at a particular locus (Gloor et al., 1991), increasing efficiency as compared to current methods (Maggert et al., 2008; Rong and Golic, 2000). This enables precisely defined changes to be achieved by coinjection with a homologous template (Bibikova et al., 2003; Porteus and Baltimore, 2003). Recent studies have shown that ssDNA oligos can induce small insertions or deletions when combined with CRISPR-induced double-strand breaks (Gratz et al., 2013; Wang et al., 2013) and that homology constructs with larger arms can be used to integrate longer selection cassettes in Drosophila (Beumer et al., 2013). When combined with mutations in the DNA ligase 4 (lig4) gene, this was shown to result in high efficiency of gene targeting in Drosophila (Beumer et al., 2008; Beumer et al., 2013).
The simplicity, efficiency, and power of the CRISPR-based genome engineering system described here will allow novel mutations in a chosen gene to be generated within a few weeks and will undoubtedly facilitate and provide new opportunities for the study of gene function in Drosophila.
Experimental Procedures
sgRNA Design
We designed sgRNA target sites by seeking sequences corresponding to GGN18NGG on the sense or antisense strand of the DNA. Off-target effects were checked through the use of the basic local alignment search tool (BLAST) applied to the Drosophila genome and visual analysis of the results. Sequences that perfectly matched the final 12 nt of the target sequence and NGG PAM sequence were discarded (Cong et al., 2013).
sgRNA Production
PCR was performed with Phusion polymerase (New England Biolabs) in HF buffer with no template; a unique oligonucleotide encoding the T7 polymerase binding site and the sgRNA target sequence, GGN18 (CRISPRF = GAAATTAATACGACTCACTATAGGN18GTTTTA GAGCTAGAAATAGC); and a common oligonucleotide encoding the remainder of the sgRNA sequence (sgRNAR = AAAAGCACCGACTCGGTGCCACTTTTTCAAGTTG ATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAAC) in 100 μl reaction volumes. Reactions were cycled on a GStorm thermal cycler (98°C 30 s, 35 cycles of [98°C 10 s, 60°C 30 s, 72°C 15 s], 72°C 10 min, 10°C ∞) and then purified with a PCR purification kit (QIAGEN).
In vitro transcription was performed with the Megascript T7 Kit (Ambion), with the use of 300 ng purified DNA template for 4 hr at 37°C, and sgRNA was purified by phenol chloroform extraction and isopropanol precipitation. sgRNAs were diluted to 1 μg/μl in water and stored in aliquots at −80°C.
Cas9 mRNA Production
Plasmid MLM3613 (Addgene plasmid 42251; Dahlem et al., 2012) was linearized with Pme I (New England Biolabs) and purified by ethanol precipitation. Cas9 mRNA was produced by in vitro transcription of 1 μg linearized template DNA with the use of the mMESSAGEmMACHINE T7 kit (Ambion) and polyadenylated with the Poly(A) Tailing Kit (Ambion) before purification with the RNeasy Mini Kit (QIAGEN).
Embryo Injection
We mixed 0.5 μg of sgRNA and 10 μg Cas9 mRNA (approximately 2:1 molar ratio) in a 30 μl volume with 3 μl 3 M sodium acetate (pH 5.2) and precipitated it with 3 volumes of absolute ethanol for purification and concentration. After centrifugation, the pellet was washed twice in 70% ethanol and resuspended in 11 μl of water prior to injection. Oregon-R embryos were collected for 30 min at 25°C, washed in water, lined up on coverslips, and left to dry and adhere to the surface. They were injected under a 1:1 mix of halocarbon 700 and 27 oils (Sigma) through the chorion in the posterior end, off center for avoidance of the thickest part of the chorion. A Femtojet Express (Eppendorf) was used at an injection pressure of 1,100 hPa with an InjectMan NI 2 micromanipulator and Femtotip II needles (Eppendorf). Excess oil was drained off, and embryos were incubated at 25°C in food vials for the remainder of development.
Genomic DNA Extraction
Genomic DNA was extracted from single flies by homogenization in 50 μl of squishing buffer (10 mM Tris-HCl [pH 8.2], 1 mM EDTA, 25 mM NaCl, 200 μg/ml proteinase K; Invitrogen), and heating to 37°C for 30 min, followed by inactivation at 95°C for 2 min.
HRMA
Oligonucleotides were designed to give 100–200 nt products spanning the presumed CRISPR cleavage site with the use of Vector NTI (Invitrogen). PCR was performed with Hotshot Diamond PCR MasterMix (Clent Lifescience) in 10 μl reactions with 1 μl gDNA, 5 μl Hotshot diamond mastermix, 200 nM each oligonucleotide, and 1 μl LC Green Plus dye (Idaho Technology). Reactions were cycled on a GStorm thermal cycler (95°C 5 min, 45 cycles of [95°C 20 s, 60°C 30 s, 72°C 30 s], 95°C 30 s, 25°C 30 s, 10°C ∞). Thermal melt profiles were collected on a LightScanner (Idaho Technology) (70°C–98°C, hold 67°C) and analyzed with the LightScanner Call-IT software.
Acknowledgments
More up-to-date information about experimental methods and other links are provided at http://groups.mrcfgu.ox.ac.uk/liu-group. The authors would like to thank Dr. Keith Joung for plasmid MLM3613 (Addgene plasmid 42251; Hwang et al., 2013); the Bloomington Drosophila Stock Center for fly stocks; Sarah Cooper for PCR purifications; and Tamara Sirey, Ana Marques, Stuart Grice, Jenna Schwarz, and Gabriel Aughey for critical reading of the manuscript.
We would also like to thank the European Research Council (DARCGENs, project number 249869) and the Medical Research Council for financial support.
Published: July 1, 2013
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
Supplemental Information includes one figure and one table and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2013.06.020.
Contributor Information
Andrew R. Bassett, Email: andrew.bassett@dpag.ox.ac.uk.
Ji-Long Liu, Email: jilong.liu@dpag.ox.ac.uk.
Supplemental Information
References
- Barrangou R., Fremaux C., Deveau H., Richards M., Boyaval P., Moineau S., Romero D.A., Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bedell V.M., Wang Y., Campbell J.M., Poshusta T.L., Starker C.G., Krug R.G., 2nd, Tan W., Penheiter S.G., Ma A.C., Leung A.Y. In vivo genome editing using a high-efficiency TALEN system. Nature. 2012;491:114–118. doi: 10.1038/nature11537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen H.J., Levis R.W., He Y., Carlson J.W., Evans-Holm M., Bae E., Kim J., Metaxakis A., Savakis C., Schulze K.L. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011;188:731–743. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer K.J., Trautman J.K., Bozas A., Liu J.L., Rutter J., Gall J.G., Carroll D. Efficient gene targeting in Drosophila by direct embryo injection with zinc-finger nucleases. Proc. Natl. Acad. Sci. USA. 2008;105:19821–19826. doi: 10.1073/pnas.0810475105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beumer K.J., Trautman J.K., Mukherjee K., Carroll D. Donor DNA utilization during gene targeting with zinc-finger nucleases. G3 (Bethesda) 2013 doi: 10.1534/g3.112.005439. Published online March 22, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M., Golic M., Golic K.G., Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–1175. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibikova M., Beumer K., Trautman J.K., Carroll D. Enhancing gene targeting with designed zinc finger nucleases. Science. 2003;300:764. doi: 10.1126/science.1079512. [DOI] [PubMed] [Google Scholar]
- Brouns S.J., Jore M.M., Lundgren M., Westra E.R., Slijkhuis R.J., Snijders A.P., Dickman M.J., Makarova K.S., Koonin E.V., van der Oost J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–964. doi: 10.1126/science.1159689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J.R., Taylor M.R., Boulton S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012;47:497–510. doi: 10.1016/j.molcel.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem T.J., Hoshijima K., Jurynec M.J., Gunther D., Starker C.G., Locke A.S., Weis A.M., Voytas D.F., Grunwald D.J. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8:e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo J.E., Norville J.E., Mali P., Rios X., Aach J., Church G.M. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res. 2013;41:4336–4343. doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013 doi: 10.1016/j.tibtech.2013.04.004. Published online May 8, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasiunas G., Barrangou R., Horvath P., Siksnys V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA. 2012;109:E2579–E2586. doi: 10.1073/pnas.1208507109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloor G.B., Nassif N.A., Johnson-Schlitz D.M., Preston C.R., Engels W.R. Targeted gene replacement in Drosophila via P element-induced gap repair. Science. 1991;253:1110–1117. doi: 10.1126/science.1653452. [DOI] [PubMed] [Google Scholar]
- Gratz S.J., Cummings A.M., Nguyen J.N., Hamm D.C., Donohue L.K., Harrison M.M., Wildonger J., O’Connor-Giles K.M. Genome engineering of Drosophila with the CRISPR RNA-guided Cas9 nuclease. Genetics. 2013 doi: 10.1534/genetics.113.152710. Published online May 24, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano Y., Ota S., Arakawa K., Muraki M., Kono N., Oshita K., Sakuma T., Tomita M., Yamamoto T., Okada Y., Kawahara A. Quantitative assay for TALEN activity at endogenous genomic loci. Biol Open. 2013;2:363–367. doi: 10.1242/bio.20133871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang W.Y., Fu Y., Reyon D., Maeder M.L., Tsai S.Q., Sander J.D., Peterson R.T., Yeh J.R., Joung J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino Y., Shinagawa H., Makino K., Amemura M., Nakata A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987;169:5429–5433. doi: 10.1128/jb.169.12.5429-5433.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.J., Lee H.J., Kim H., Cho S.W., Kim J.S. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19:1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Li C., Yu Z., Huang P., Wu H., Wei C., Zhu N., Shen Y., Chen Y., Zhang B. Efficient and specific modifications of the Drosophila genome by means of an easy TALEN strategy. J. Genet. Genomics. 2012;39:209–215. doi: 10.1016/j.jgg.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Maggert K.A., Gong W.J., Golic K.G. Methods for homologous recombination in Drosophila. Methods Mol. Biol. 2008;420:155–174. doi: 10.1007/978-1-59745-583-1_9. [DOI] [PubMed] [Google Scholar]
- Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller J.C., Holmes M.C., Wang J., Guschin D.Y., Lee Y.L., Rupniewski I., Beausejour C.M., Waite A.J., Wang N.S., Kim K.A. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Ota S., Hisano Y., Muraki M., Hoshijima K., Dahlem T.J., Grunwald D.J., Okada Y., Kawahara A. Efficient identification of TALEN-mediated genome modifications using heteroduplex mobility assays. Genes Cells. 2013;18:450–458. doi: 10.1111/gtc.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteus M.H., Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- Rong Y.S., Golic K.G. Gene targeting by homologous recombination in Drosophila. Science. 2000;288:2013–2018. doi: 10.1126/science.288.5473.2013. [DOI] [PubMed] [Google Scholar]
- Venken K.J., Bellen H.J. Emerging technologies for gene manipulation in Drosophila melanogaster. Nat. Rev. Genet. 2005;6:167–178. doi: 10.1038/nrg1553. [DOI] [PubMed] [Google Scholar]
- Wang H., Yang H., Shivalila C.S., Dawlaty M.M., Cheng A.W., Zhang F., Jaenisch R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.