

Abstract
Heterotrimeric G proteins are conformational switches that turn on intracellular signaling cascades in response to the activation of G-protein-coupled receptors. Receptor activation by extracellular stimuli promotes a cycle of GTP binding and hydrolysis on the G protein α-subunit (Gα). Important conformational transitions occurring during this cycle have been characterized from extensive crystallographic studies of Gα. However, the link between the observed conformations and the mechanisms involved in G-protein activation and effector interaction remain unclear. Here we describe a comprehensive principal component analysis of available Gα crystallographic structures supplemented with extensive unbiased conventional and accelerated molecular dynamics simulations that together characterize the response of Gα to GTP binding and hydrolysis. Our studies reveal details of activating conformational changes as well as the intrinsic flexibility of the α-helical domain that includes a large-scale 60° domain opening under nucleotide-free conditions. This result is consistent with the recently reported open crystal structure of Gs, the stimulatory G protein for adenylyl cyclase, in complex with the α2 adrenergic receptor. Sets of unique interactions potentially important for the conformational transition are also identified. Moreover simulations reveal nucleotide-dependent dynamical couplings of distal regions and residues potentially important for the allosteric link between functional sites.
Heterotrimeric G proteins undergo cycles of GTP-dependent conformational rearrangements and alterations of their oligomeric αβγ form to convey receptor signals to downstream effectors that control diverse cellular processes ranging from movement to division and differentiation. Interaction with activated receptor promotes the exchange of GDP for GTP on the G protein α subunit (Gα) and its separation from its βγ subunit partners (Gβγ). Both isolated Gα and Gβγ then interact with downstream effectors. GTP hydrolysis deactivates Gα, which reassociates with Gβγ, becoming ready to restart the cycle. Each of these stages has been subjected to extensive crystallographic studies with high-resolution structures of Gα in complex with GDP, GTP analog, Gβγ, and, most recently, the G-protein-coupled receptors now available. These studies have provided extensive mechanistic insight. However, a number of important questions remain, including:
How do the distinct conformations evident in the accumulated structures interconvert?
How do disease-associated mutations affect the fidelity of these transitions?
And, critically, how do distal functional sites responsible for nucleotide and protein partner binding allosterically coordinate their activities?
Here we describe a comprehensive analysis of the accumulated Gα crystallographic structures supplemented with extensive conventional (cMD) and accelerated molecular dynamics (aMD) simulations (1) that together map the structural and dynamical features of Gα in different nucleotide states. These enhanced sampling simulations reveal the spontaneous interconversion between GDP and GTP conformations and also characterize large-scale opening motions of the α-helical domain (HD) that were not accessible to previous simulation studies (2–5). Furthermore, the current simulations results reveal a distinctive pattern of collective motions that provide evidence for a nucleotide-dependent network of dynamic communication between the active site and the receptor and effector binding sites.
Principal component analysis of 53 Gα experimental structures homologous to transducin (Gαt) reveals that the major variation in accumulated structures is the concerted association/disassociation of three nucleotide-binding site loops termed the switch regions (SI, SII, and SIII). An additional small-scale (<10°) rotation of the HD relative to the main catalytic Ras-like domain (RasD) is also apparent (see Fig. S1 in the Supporting Material). The distinct conformation of SI–SIII regions gives rise to nucleotide-associated segregation of GDP- and GTP-analog-bound experimental structures along the PC1-PC2 plane. Interestingly, both GDP- and GTP-bound structures display a skewed distribution along the PC1-PC2 plane that arises from HD rotation. In comparison, the distribution of the GTP-bound structures becomes more restricted and the skew decreases when the mapping is based on a principle component analysis that excludes the HD region (see Fig. S1).
Recently, the HD region of Gαs (the α-subunit of the stimulatory G protein for adenyl cyclase) was shown to adopt a dramatically more open conformation in a crystal structure complex with the β2 adrenergic receptor (β2AR) (6). This clam-shell-like 127° opening in the absence of nucleotide and presence of receptor is consistent with electron microscopy (7) and double electron-electron resonance analysis (8). These results, together with recent hydrogen-deuterium exchange mass spectrometry data (9), indicate that there may be additional functional motions and inherent flexibility in the ensemble of native states beyond those apparent in the accumulated crystal structures of Gαt (9). To address this question, we performed multiple 100-ns aMD simulations of nucleotide-free Gαt. These simulations reveal a spontaneous large-scale opening and closing motion of larger magnitude (>60°) than those evident in the distribution of crystallographic structures (Fig. 1 A and see Fig. S2). In addition, the trajectory reveals two dominant modes of HD opening: an out-of-plane shifting (PC1 in Fig. S3) and an in-plane rotation (PC2 in Fig. S3). It is also notable that nucleotide-free aMD simulations sample both active (GTP-like) and inactive (GDP-like) structures (see Fig. S2) in an analogous manner to the spontaneous GDP to GTP interconversion sampled for Ras and Rho small G proteins with similar methods (10–12).
Figure 1.
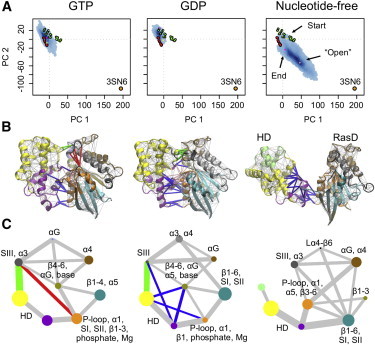
Nucleotide-associated differences in flexibility and dynamic coupling. (A) Mapping aMD simulation trajectories (blue points) onto the principal components obtained from analysis of Gα crystallographic GDP-bound (green) and GTP-analog bound (red) experimental structures. (Orange) Open β2AR-Gαs complex structure. (B) Results of dynamic coupling analysis mapped onto the average structure for each nucleotide state. (Spheres) Nodes for the nucleotide; the protein cartoon is colored by community structure. (C) Community network graph. (Circles) Communities, colored as in panel B. Radius of the circle indicates the number of residues in the community. Thickness of linking lines is determined by the maximum betweenness of the respective intercommunity edges (see the Supporting Material). (Red, blue, and green edges) Major topological difference between states.
The low sequence identity between Gαt and Gαs (44.5%), as well as the absence of the receptor and Gβγ in the simulations, may explain the difference between the predicted ∼60° Gαt-HD rotation and that displayed in the β2AR-Gαs crystallographic structure (see Fig. S3). It is notable that, although the amplitude is much smaller, aMD simulations with bound nucleotide display similar dominant HD motions to those observed in the nucleotide-free simulations (see Fig. S4). This suggests that the interdomain flexibility of RasD and HD is likely an intrinsic feature of Gαt regardless of nucleotide state.
The transition between distinct conformations (structural clusters; see Fig. S5) was observed to correspond to significant dynamical changes in side-chain contacts (see Fig. S6). Specifically, we found sequential contacts breaking during the HD in-plane rotation motion starting from the region between HD helix αD and RasD helix αG toward that between HD helix αE and RasD SIII and the P-loop. In comparison, for the out-of-plane shift, we found simultaneous breaking and formation of contacts in the region containing the loop between helices αB and αC, the N-terminus of αA, αE, and αF of HD; α1, SI, and the loop between strand β6 and helix α5 of RasD. Interactions highlighted in these regions as potentially important for the conformational transitions include D137::K276, S140::K273, S140::D227, Q143::R238, N145::E39, and D146::K266, the effect of which can be further evaluated by mutagenesis experiments and simulations.
Dynamic network analysis methods developed by Sethi et al. (13) were used to examine whether the motions of one residue were correlated to the motions of another (distant) residue. In this approach, a weighted graph is constructed where each residue represents a node and the weight of the connection between nodes represents their respective correlation value. A clustering of edges is then used to define local communities of highly correlated residues that represent substructures that are highly intraconnected, but loosely interconnected. Applying this approach to multiple 40-ns cMD simulations initiated from GTP-, GDP-, and aMD-derived APO conformations revealed a consistent community composition as well as a distinct pattern of intercommunity connection between nucleotide states (Fig. 1, B and C).
The dynamics of the RasD region can be decomposed into two main communities that stem from the nucleotide base and phosphate regions in GDP and GTP states: The first community is composed of residues from the P-loop, helix α1, strands β1–β3, and the phosphates of the nucleotide (orange in Fig. 1, B and C). The second community comprises residues from helix αG, strands β4–β6, and the nucleotide base region (tan in Fig. 1, B and C). This dynamic partitioning of the central β-sheet and central role of the nucleotide is consistent with the bilobal structure and dynamics previously reported for Ras (14). In the presence of GTP, the first community includes or is dynamically coupled to SI, SII, and SIII regions (see the orange node and the red edge in Fig. 1 C). Removal of the γ-phosphate of GTP disrupts this region, leading to decoupling of the switch regions from the nucleotide. Also evident for GDP states is an apparent tighter coupling of RasD and HD regions (blue edges in Fig. 1 C). We note that these findings are robust to the choice of initial simulation conditions and are observed in both cMD and aMD simulations (see Fig. S7 and Fig. S8). Nucleotide-free Gαt simulations display an altered dynamical network with respect to those of nucleotide bound states. In particular, RasD and HD regions lose connecting edges consistent with the large-scale opening of these domains (e.g., SIII-HD green edges in Fig. 1 C).
A number of residues highlighted here as potentially important for mediating the coupling between prominent communities (see Table S1 in the Supporting Material) have been shown by previous mutagenesis studies to affect GDP release. For example, the double mutation A322S/R174M was found to significantly enhance the rate of GDP release (15). The current results indicate that these positions are involved in coupling the nucleotide and RasD. Also, mutations R144A and L232Q caused a faster basal GDP release rate in Gαi1 (16). The current analysis indicates that the equivalent positions in Gαt (S140 and M228) couple the RasD and HD, and suggests that their mutation could promote domain-domain motions. We also note the apparent coupling of α5 with the nucleotide base and Ploop-β1 with the phosphate regions of GDP. These direct connections of the receptor connecting N- and C-terminus to GDP are suggestive of potential routes for receptor-mediated GDP release. We expect further study of these sites and of receptor-bound dynamics to be informative in this regard.
In conclusion, simulations suggest a flexible HD in Gαt similar to that found for Gαs. In particular, in the absence of nucleotide we observed the spontaneous large-scale opening and closing of HD relative to RasD, which was unseen in previous computational studies. Moreover, we found that the functional states of Gαt are associated with the distinct dynamical couplings of functional regions including SI–SIII, P-loop, α5, and the HD region. Finally, our results indicate that nucleotide may not directly induce large-scale conformational changes but, instead, act as a modulator of intrinsically accessible conformations and as a central participant in their associated dynamical couplings.
Acknowledgments
We thank Drs. G. Scarabelli and R. K. Sunahara for valuable discussions and we gratefully acknowledge support from the University of Michigan.
Supporting Material
References and Footnotes
- 1.Hamelberg D., Mongan J., McCammon J.A. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J. Chem. Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- 2.Ceruso M.A., Periole X., Weinstein H. Molecular dynamics simulations of transducin: interdomain and front to back communication in activation and nucleotide exchange. J. Mol. Biol. 2004;338:469–481. doi: 10.1016/j.jmb.2004.02.064. [DOI] [PubMed] [Google Scholar]
- 3.Khafizov K., Lattanzi G., Carloni P. G protein inactive and active forms investigated by simulation methods. Proteins. 2009;75:919–930. doi: 10.1002/prot.22303. [DOI] [PubMed] [Google Scholar]
- 4.Louet M., Perahia D., Floquet N. A concerted mechanism for opening the GDP binding pocket and release of the nucleotide in heterotrimeric G-proteins. J. Mol. Biol. 2011;411:298–312. doi: 10.1016/j.jmb.2011.05.034. [DOI] [PubMed] [Google Scholar]
- 5.Raimondi F., Seeber M., Fanelli F. Mechanisms of inter- and intramolecular communication in GPCRs and G proteins. J. Am. Chem. Soc. 2008;130:4310–4325. doi: 10.1021/ja077268b. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen S.G., DeVree B.T., Kobilka B.K. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Westfield G.H., Rasmussen S.G., Skiniotis G. Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc. Natl. Acad. Sci. USA. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Van Eps N., Preininger A.M., Hubbell W.L. Interaction of a G protein with an activated receptor opens the interdomain interface in the α-subunit. Proc. Natl. Acad. Sci. USA. 2011;108:9420–9424. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung K.Y., Rasmussen S.G., Sunahara R.K. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grant B.J., Gorfe A.A., McCammon J.A. Ras conformational switching: simulating nucleotide-dependent conformational transitions with accelerated molecular dynamics. PLOS Comput. Biol. 2009;5:e1000325. doi: 10.1371/journal.pcbi.1000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grant B.J., McCammon J.A., Gorfe A.A. Conformational selection in G-proteins: lessons from Ras and Rho. Biophys. J. 2010;99:L87–L89. doi: 10.1016/j.bpj.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortiz-Sanchez J.M., Nichols S.E., Grant B.J. Identification of potential small molecule binding pockets on Rho family GTPases. PLoS ONE. 2012;7:e40809. doi: 10.1371/journal.pone.0040809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sethi A., Eargle J., Luthey-Schulten Z. Dynamical networks in tRNA:protein complexes. Proc. Natl. Acad. Sci. USA. 2009;106:6620–6625. doi: 10.1073/pnas.0810961106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gorfe A.A., Grant B.J., McCammon J.A. Mapping the nucleotide and isoform-dependent structural and dynamical features of Ras proteins. Structure. 2008;16:885–896. doi: 10.1016/j.str.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zielinski T., Kimple A.J., Lowery R.G. Two Gα(i1) rate-modifying mutations act in concert to allow receptor-independent, steady-state measurements of RGS protein activity. J. Biomol. Screen. 2009;14:1195–1206. doi: 10.1177/1087057109347473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Remmers A.E., Engel C., Neubig R.R. Interdomain interactions regulate GDP release from heterotrimeric G proteins. Biochemistry. 1999;38:13795–13800. doi: 10.1021/bi990887f. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.