

Abstract
Proline utilization A proteins (PutAs) are bifunctional enzymes that catalyze the oxidation of proline to glutamate using spatially separated proline dehydrogenase and pyrroline-5-carboxylate dehydrogenase active sites. Here we use the crystal structure of the minimalist PutA from Bradyrhizobium japonicum (BjPutA) along with sequence analysis to identify unique structural features of PutAs. This analysis shows that PutAs have secondary structural elements and domains not found in the related monofunctional enzymes. Some of these extra features are predicted to be important for substrate channeling in BjPutA. Multiple sequence alignment analysis shows that some PutAs have a 17-residue conserved motif in the C-terminal 20–30 residues of the polypeptide chain. The BjPutA structure shows that this motif helps seal the internal substrate-channeling cavity from the bulk medium. Finally, it is shown that some PutAs have a 100–200 residue domain of unknown function in the C-terminus that is not found in minimalist PutAs. Remote homology detection suggests that this domain is homologous to the oligomerization beta-hairpin and Rossmann fold domain of BjPutA.
Keywords: Proline Utilization A, PutA, Substrate Channeling, Proline Catabolism, Proline Metabolism, Proline Dehydrogenase, Pyrroline-5-Carboxylate Dehydrogenase, Pyrroline-5-Carboxylate, Glutamate Semialdehyde, Domain Repeat, Aldehyde Dehydrogenase, Flavoenzyme, Remote Homology Detection, Review
2. INTRODUCTION
The oxidation of proline to glutamate, i.e. proline catabolism, is catalyzed by two enzymes, proline dehydrogenase (PRODH) and pyrroline-5-carboxylate (P5C) dehydrogenase (P5CDH) (Figure 1). The former catalyzes the oxidation of L-proline to P5C with concomitant reduction of an enzyme-bound FAD cofactor. The latter enzyme catalyzes the oxidation of L-glutamate semi-aldehyde (GSA) to L-glutamate using NAD+ as the electron acceptor. Note that the product of the PRODH reaction is not the substrate for P5CDH. Instead, the two reactions are coupled by the hydrolysis of P5C, which has traditionally been thought of as a nonenzymatic process. As noted by Phang over two decades ago, researchers typically refer to P5C and GSA interchangeably, since the two species are not distinguished in most experiments (1). Phang's observation remains valid today.
Figure 1.
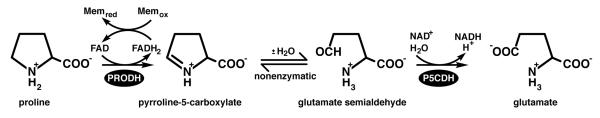
The reactions of proline catabolism.
One interesting feature of proline catabolism is that PRODH and P5CDH are combined into a single polypeptide in some organisms (Figure 2). The combined enzymes are known as proline utilization A (PutA) and were discovered by Roth's group in the late 1970s during their studies of proline utilization in Salmonella typhimurium (2). Analysis of genome sequence data suggests that PutAs are limited to Gram-negative bacteria (Figure 2, branches 1, 2), whereas PRODH and P5CDH are separate enzymes encoded by distinct genes in Gram-positive bacteria (branch 3B) (3). In eukaryotes, PRODH and P5CDH are also separate enzymes and are localized to mitochondria (branch 3A). Human PRODH is a p53-induced tumor suppressor protein localized to the inner mitochondrial membrane and is often referred to as POX to emphasize its role as a superoxide-generating oxidase (4–12). Human P5CDH (ALDH4 (13)) is also induced by p53 (14) and is located in the mitochondrial matrix. ALDH4 has been characterized biochemically, including elucidation of the oligomeric state in solution (dimer) and kinetic mechanism (15, 16).
Figure 2.

Phylogenetic tree representing the organization of proline catabolic enzymes in bacteria and eukaryotes. PutAs are found in branches 1 and 2. Monofunctional PRODH and P5CDH enzymes are found in branch 3. A cluster of trifunctional PutAs is indicated.
The PutA part of the PutA/PRODH/P5CDH family tree has two branches (3, 17). Branch 1 primarily consists of PutAs from alpha-, beta-, and gamma-proteobacteria. Branch 2 includes PutAs from delta- and epsilon-proteobacteria as well as cyanobacteria. The PutAs in branch 1 have chain lengths from 999 to almost 1400 residues, and the pairwise sequence identities are greater than 38 %. The polypeptide length for branch 2 PutAs ranges from around 980 to almost 1300 residues, and the pairwise sequence identity range can be as low as 23 %. Thus, branch 2 PutAs appear to be a more diverse group than branch 1 PutAs. Between branches 1 and 2, the pairwise sequence identities are typically less than 30 %. Nevertheless, the residues in the PRODH and P5CDH active sites are highly conserved, indicating that the three-dimensional structures of the catalytic domains are conserved by PutAs. Whether the three-dimensional arrangement of the catalytic and other domains is likewise conserved remains to be determined.
PutAs are further classified as bifunctional or trifunctional. Bifunctional PutAs exhibit only PRODH and P5CDH catalytic activities, have polypeptide chain lengths in the range of ~980 residues to over 1300 residues, and are found in both PutA branches. Bifunctional PutAs from Bradyrhizobium japonicum (BjPutA, (18–20)) and Helicobacter species (21–23) have been studied. Trifunctional PutAs constitute a subset of branch 1 PutAs and are distinguished by the presence of a DNA-binding domain (a ribbon-helix-helix domain) in the first ~50 residues of the polypeptide chain. The polypeptide chain length of trifunctional PutAs are in the range of ~1270–1361. In addition to functioning as dual PRODH/P5CDH enzymes, trifunctional PutAs have a third function of repressing transcription of the put regulon, which contains the genes encoding PutA and the proline transporter PutP, when proline levels are low (24–27). High levels of proline in the bacterium's environment cause PutA to disengage from the put control region thus activating transcription of putA and putP. Thus, trifunctional PutAs are remarkable proteins that link transcription and metabolism in response to an environmental cue (proline level). Trifunctional PutAs from S. typhimurium (25, 26, 28–31) and Escherichia coli (EcPutA) (24, 32–46) have been studied. PutA from E. coli is the most studied trifunctional PutA and is considered to be the archetypal trifunctional PutA.
The observation that enzymes catalyzing successive reactions in a metabolic pathway are combined into a single polypeptide chain as in PutA has intriguing implications. First, the covalent linking of the two active sites may allow the transfer of the reaction product of one enzyme to the next without equilibrating with the bulk medium. Substrate channeling is the term used for such kinetic mechanisms, and Arentson et al. provide a review of substrate channeling in proline metabolism in this issue (47). Two limiting channeling mechanisms are possible: direct transfer and proximity. In the former, the intermediate moves through an internal cavity or tunnel connecting the two active sites without leaving the confines of the protein. Proximity refers to a spectrum of cases in which the reaction product dissociates from the first enzyme but encounters a locally high concentration of the second enzyme and thus does not truly equilibrate with the bulk medium. There are potential advantages of substrate channeling, including decreased transit time between enzymes, protection of labile intermediates, and isolation of intermediates from competing enzymatic reactions (48–50). The latter point is relevant for proline metabolism, because P5C/GSA is common to proline catabolism, proline biosynthesis, and arginine biosynthesis. Another implication of fused enzymes was recognized by Eisenberg's group in 1999 and concerns the prediction of protein-protein interactions (51). The basic idea is that the observation that two proteins are separate in some organisms and fused in others implies that the two separate proteins form a functional association. Thus, protein fusion (or gene fusion) provides the `Rosetta stone' for identifying potential protein–protein interactions. In proline catabolism, for example, the Rosetta Stone hypothesis suggests the possibility that monofunctional PRODH and P5CDH interact and engage in intermolecular substrate channeling.
Three-dimensional structural studies have contributed to our understanding of proline catabolic proteins. Crystal structures have been solved for the monofunctional PRODH and P5CDH enzymes from Thermus thermophilus (TtPRODH (17, 52, 53), TtP5CDH (54, 55)), the DNA-binding domains of two trifunctional PutAs (56–58), a PRODH domain construct of EcPutA (EcPutA86-630, (46, 59–62)), and full-length BjPutA (63). Here, we use the BjPutA structure as a platform for identifying the unique features that distinguish PutAs from their monofunctional relatives and to gain insight into the structure and function of the C-terminal domains of PutAs.
3. COMPARISON OF A BIFUNCTIONAL PUTA WITH MONOFUNCTIONAL PRODH AND P5CDH
3.1. Structure of a minimalist PutA
PutA from Bradyrhizobium japonicum (BjPutA) is one of the simplest of bifunctional PutAs. At 999 residues in length, it is the shortest branch 1 PutA known and is thus considered to be a minimalist PutA. We recently reported the crystal structure of BjPutA (PDB code 3haz), which is the first structure of a full-length PutA. The structure shows that PutAs are more than simply a fusion of two catalytic domains. The protomer comprises seven domains: arm, alpha, PRODH barrel, linker, NAD+-binding, P5CDH catalytic, and oligomerization domain (Figure 3A). The PRODH active site is located in a distorted (beta-alpha)8 barrel. The barrel structure is very similar to that of EcPutA (59). The P5CDH active site is located in the interface between the NAD+-binding domain and the P5CDH catalytic domain. The two active sites are separated by 41 Å and connected by a large, irregularly shaped internal cavity (silver surface in Figure 3A). This cavity most likely functions in substrate channeling, as described in this issue (47).
Figure 3.

Structure of BjPutA. (A) Protomer structure with the domains colored according to the domain diagram. The silver surface represents the substrate-channeling cavity. FAD and NAD+ are represented as yellow and green sticks, respectively. Abbreviations used in the domain diagram are as follows: NBD, NAD+-binding domain; BH, beta-hairpin; CCM, conserved C-terminal motif. (B) The domain-swapped dimer of BjPutA. The domains are colored according to the domain diagram in panel A. The silver surfaces represent the substrate-channeling cavities of the two protomers. (C) Close-up view of the oligomerization domain covering the cavity of the other protomer. This figure and others were prepared with PyMol (72).
Oligomerization is essential for substrate channeling in BjPutA. The enzyme forms a U-shaped dimer (Figure 3B), and two of these dimers assemble into a ring-shaped dimer-of-dimers tetramer (see reference (63)). The dimer shown in Figure 3B is the relevant oligomeric state for understanding substrate channeling, so we will describe it in detail here. Dimerization is mediated by an oligomerization domain (orange in Figure 3) that protrudes from the NAD+-binding domain. The oligomerization domain consists of two elements that are far apart in sequence, a beta-hairpin formed by residues 629–647 and a beta-strand followed by a short helix at the C-terminus of polypeptide chain (residues 976–989). The C-terminal strand of the oligomerization domain of one protomer forms main chain hydrogen bonds with the beta-sheet of the P5CDH catalytic domain of the other protomer to form a large, twisted intermolecular beta-sheet (Figures 3B and 3C). This type of oligomerization is an example of domain swapping (64). Both the bipartite oligomerization domain and the domain-swapped dimer are conserved by aldehyde dehydrogenases. In BjPutA, the oligomerization domain not only mediates dimerization but also covers the cavity of the other protomer (Figure 3C). Without the lid provided by the oligomerization domain, the cavity would be open to the bulk medium. Thus dimerization appears to be essential for formation of the substrate channeling cavity. The sealed, internal cavity is consistent with direct transfer rather than proximity as the predominant mechanism of substrate channeling in BjPutA.
3.2. The PutA PRODH barrel has an extra helix involved in FAD binding and substrate channeling
Both the PutA PRODH domain and the monofunctional enzyme TtPRODH exhibit the distorted (beta-alpha)8 barrel that is characteristic of the PRODH/PutA family. As shown in Figure 4, the two folds are nearly identical; the RMSD between the two proteins is 2.0 Å for 256 aligned residues. In both structures, the FAD is bound at the C-terminal ends of the strands of the barrel. The fold is unusual in that the final helix, alpha8, sits atop the barrel rather than alongside beta8 as in the classical triosephosphate isomerase barrel. Helix 8 contains residues that are critical for substrate recognition, including a conserved Arg-Arg motif that has been shown to bind the substrate carboxylate group (60). Both PutA and the monofunctional PRODH exhibit this distortion of the classical (beta-alpha)8 barrel fold.
Figure 4.

Comparison of the monofunctional enzyme TtPRODH (white) and the PRODH barrel of BjPutA (cyan). Strands of the barrel are labeled 1–8 using the standard convention for (beta-alpha)8 barrels. The extra helix of the PutA barrel is denoted alpha5a. Trp346 of alpha5a is colored magenta. The three helices of TtPRODH that precede the barrel are labeled alphaA, alphaB, and alphaC. Two orthogonal views are shown.
Despite the similarities in overall fold and amino acid sequence (28 % identity), there is an important difference between monofunctional PRODH and PutA. The PutA PRODH barrel has an extra helix (alpha5a) inserted between beta5 and alpha5 (Figure 4). This additional secondary structural element is also present in EcPutA86-630, suggesting that it is conserved in branch 1 PutAs. Helix 5a contains a conserved tryptophan residue (Trp346 in BjPutA, Trp438 in EcPutA) that stacks against the FAD adenine (Figures 3C and 4), and this interaction presumably contributes to the different FAD conformations in PutA and TtPRODH, as described previously (17). The BjPutA structure reveals a new function for helix 5a. The helix forms a large section of the wall of the internal substrate-channeling cavity (Figure 3C), and its absence would leave a large hole to the bulk medium. Thus, helix 5a seems to be essential for channeling in PutA.
Finally, we note that TtPRODH has three additional helices that precede the start of the barrel (alphaA, alphaB, alphaC in Figure 4). These helices are located in the vicinity of the alpha domain of BjPutA, but they do not superimpose well with any of the helices of the alpha domain.
3.3. An extra helix at the C-terminus of PutA plugs the substrate-channeling cavity
The structure of the monofunctional enzyme TtP5CDH is very similar to that of the P5CDH half of BjPutA (Figure 5). The sequence identity of TtP5CDH to PutA is about 38 %, implying substantial structural similarity. Indeed, the RMSD between the two proteins is 1.4 Å for 463 residues. Both enzymes exhibit the characteristic aldehyde dehydrogenase fold (65), which consists of three domains, NAD+-binding, catalytic, and oligomerization. (The oligomerization is also referred to as the bridging domain (65) and beta-flap (63)). The NAD+-binding domain is structurally contiguous but actually consists of three separate sections of the polypeptide chain, as indicated in the domain diagram in Figure 3A. Sections 1 and 2 are separated in primary structure by the beta-hairpin of the oligomerization domain, while sections 2 and 3 are separated by the P5CDH catalytic domain. Section 2 exhibits a variation of the Rossmann dinucleotide binding fold. The classical Rossmann fold consists of two beta-alpha-beta-alpha-beta motifs that form a 6-stranded parallel beta sheet with relative strand order 321456 (66). The Rossmann domain of aldehyde dehydrogenase lacks the final strand and helix and is thus referred to as a non-classical Rossmann fold domain. The catalytic domains of TtP5CDH and BjPutA are also quite similar, as are the details of the P5CDH active sites (63).
Figure 5.

Comparison of the monofunctional enzyme TtP5CDH (white) and the P5CDH half of BjPutA. BjPutA is colored according to the legend in Figure 3A. NAD+ is drawn in green sticks.
Closer inspection of BjPutA and TtP5CDH, however, reveals an important difference at the C-terminus (Figure 5). Structure-based alignment of BjPutA and TtP5CDH shows that the BjPutA chain extends 14 residues past the final residue of TtP5CDH (Phe516). Some of these extra residues were resolved in the BjPutA structure, and they form a turn of helix that plugs a hole in the cavity wall (Figure 3C). Without this plug, there would be a significant hole in the cavity leading to the bulk medium. Interestingly, the extra residues of BjPutA are incompatible with the observed oligomeric state of TtP5CDH. TtP5CDH forms a hexamer, which can be thought of as a trimer of domain-swapped dimers. If the TtP5CDH chain were longer as in BjPutA, the C-terminus would clash with another dimer of the hexamer.
3.4. BjPutA has domains not found in the monofunctional enzymes
PutAs also have extra domains not found in the monofunctional enzymes, and the BjPutA structure exhibits three of them: arm, alpha domain, and linker (Figure 3A). The alpha-helical arm at the N-terminus (yellow in Figure 3A) wraps around the PRODH barrel and sits below the linker. The arm domain is observed in both BjPutA and EcPutA86-630, indicating that it is conserved by branch 1 PutAs. The arm connects to the alpha domain, which is a globular domain consisting of 6 helices (green in Figure 3A). The alpha domain contacts both the PRODH and P5CDH domains, as well as the oligomerization domain of the other protomer of the domain-swapped dimer (Figures 3B and 3C). Its strategic location at the confluence of three domains suggests that it is critical for properly orienting the two active sites for channeling and for formation of the internal cavity. We note that the alpha domain was disordered in the structure of EcPutA86-630, indicating that contacts with the P5CDH half of the enzyme are required for proper folding. As noted above, the alphaA, alphaB, and alphaC helices of TtPRODH are in the same general location as the alpha domain of BjPutA. Whether these helices play a similar role as the PutA alpha domain is unknown.
The polypeptide that links the PRODH barrel to the NAD+-binding domain is also unique to PutA. The linker is not simply a flexible tether that keeps the two catalytic domains in close proximity, but rather has a well-defined structure that appears to be essential for maintaining the tertiary and quaternary structure of the enzyme. The linker (residues 465–509, violet in Figure 3) joins the C-terminus of helix 8 of the PRODH barrel to the N-terminus of the NAD+-binding domain. The 45-residue linker consists of 5 short helical segments that form a meandering U-turn, effectively redirecting the chain toward the P5CDH domain. We note that a nearly identical meandering U-turn is also found in the structure of EcPutA86-630, suggesting that the linker structure is conserved by branch 1 PutAs. Because of its wide, curved path, the linker traverses 100 Å, although the two residues it connects are separated by only 30 Å. The linker forms extensive interactions with other domains, which likely are essential for maintaining its three-dimensional structure. The majority of these interactions are with the PRODH domain (2000 Å2 of inter-domain buried surface area) and the arm (1300 Å2). The large contact area with the arm reflects the fact that the linker sits atop the arm, essentially tracking its curved path around the barrel. In fact, it is possible that the primary role of the arm is to help stabilize the conformation of the linker. In summary, the linker is a structural element that interacts with disparate parts of the polypeptide chain via noncovalent interactions, and as a result is important for properly orienting the two active sites and creating the substrate channeling cavity.
4. THE CONSERVED C-TERMINAL MOTIF OF BRANCH 1 PUTAS
The second element of the oligomerization domain contains a conserved sequence motif, which to our knowledge has not been described previously. Multiple sequence alignment (MSA) analysis reveals a conserved stretch of 17-residues located in the C-terminal 20–30 residues of branch 1 PutAs (Figure 6). The motif is found in both bifunctional and trifunctional branch 1 PutAs, but it does not appear to be present in branch 2 PutAs and monofunctional P5CDHs. Based on the sequences analyzed, the consensus motif is Exxxxv[N or D]t[T or A]AaGGnaxL, where upper case denotes identity, lowercase denotes presence in over half of the sequences, and x denotes no significant conservation.
Figure 6.

Section of an MSA of branch 1 PutAs showing the conserved motif at the C-terminus. The trifunctional PutAs are EcPutA through Acidiphilium, plus Wigglesworthia. The other PutAs are bifunctional. The secondary structure elements are from the BjPutA structure (PDB code 3haz). This figure and others were prepared with ClustalW2 (73) and ESPript (74).
The BjPutA structure provides the three-dimensional context of the conserved motif. The first 12 residues of the motif were resolved in the structure. The motif begins at the N-terminus of the final strand of the enzyme and extends beyond the helical plug. The identically conserved Glu at position 1 forms an intersubunit hydrogen bond to the backbone of Lys965. The Asn at position 7, which is Asp in some sequences, forms an intersubunit hydrogen bond with Lys351 (Figure 3C). Lys351 is highly conserved in branch 1 PutAs (Arg in some sequences) and is part of helix 5a of the PRODH barrel. As described above, helix 5A is not found in monofunctional PRODHs. Positions 9–11 form the turn of helix that plugs a hole in the cavity wall. This analysis suggests that that the C-terminal motif is important for formation of the substrate-channeling cavity.
5. BEYOND THE MINIMALIST PUTA
Many PutAs have chain lengths that are much longer than that of the 999-residue BjPutA, so it is natural to ask how these extra residues beyond the minimalist PutA are incorporated into the polypeptide chain. MSAs provide information about which domains are shared among PutAs and the locations of the extra domains of long PutAs. We will focus here on branch 1 PutAs, because the sequence identity is high within this group and the resulting trends are obvious. Although we have analyzed many branch 1 PutAs using MSAs, we present an alignment of just three due to space limitations. The results presented here are valid for the larger group. Figure 7 shows an MSA of BjPutA with a representative long bifunctional branch 1 PutA (Azoarcus PutA) and the archetypal trifunctional PutA, EcPutA. The alignment exhibits a long region of high identity (48 – 59 % pairwise) corresponding to BjPutA residues 5–974. This region corresponds to the arm, alpha, PRODH, linker, NAD+-binding, and P5CDH catalytic domains of BjPutA. Note that there is a gap in the long PutAs corresponding to the beta-hairpin of the oligomerization domain (Figure 7, beta11). The second region of high identity corresponds to the conserved C-terminal motif. These results suggest that all the domains described here for BjPutA are also present in the long PutAs except for the beta-hairpin, which appears to be abbreviated or absent. The MSA also indicates an extra domain at the N-terminus of trifunctional PutAs and another domain immediately preceding the conserved C-terminal motif in long branch 1 PutAs. The N-terminal domain is the ribbon-helix-helix DNA-binding domain. The function of the extra C-terminal domain is unknown.
Figure 7.

MSA of three branch 1 PutAs: BjPutA (Bj, GenBank BAC52526.1), Azoarcus sp. BH72 PutA (Az, GenBank CAL96369.1), and EcPutA (Ec, GenBank AAB59985.1). BjPutA is a minimalist PutA. Az is a long bifunctional PutA. Ec is a trifunctional PutA. The secondary structure elements above the sequence blocks are from the BjPutA structure (PDB code 3haz). The secondary structure elements for the N-terminal ribbon-helix-helix domain are from a structure of the EcPutA DNA-binding domain (PDB code 2gpe). Symbols below the sequence blocks denote the following: green squares, substrate-channeling cavity; triangles, proline binding site; hexagons, FAD binding site; diamonds, NAD+ binding site; ovals, GSA binding site; stars, catalytic Cys of the P5CDH catalytic domain and the Glu that is predicted to assist in hydrolysis of the thioacylenzyme.
5.1. The DNA-binding domain of trifunctional PutA
The DNA-binding domains of EcPutA and Pseudomonas putida PutA have been extensively characterized using X-ray crystallography, NMR, and an array of biophysical and biochemical techniques (56–58, 67). The PutA DNA-binding domain has the ribbon-helix-helix (RHH) fold, which identifies PutA as a member of a large superfamily of transcription factors that includes Arc, MetJ, CopG, and NikR, among others. Schreiter and Drennan have written an excellent review on the RHH superfamily (68). The DNA-binding domain connects to the arm domain via a ~35-residue polypeptide (Figure 7). No structural information is available for these residues, thus it is not known whether this polypeptide is a flexible tether or has a well-defined three-dimensional structure. The sequence identity of the linker is relatively low (results not shown), which perhaps argues in favor of a flexible tether. Furthermore, analysis of the sequences of several trifunctional PutAs using the Disopred server suggests a high probability of disorder within residues 50–80 (69).
5.2. The C-terminal domain of unknown function
MSAs indicate that trifunctional PutAs have a ~200-residue domain near the C-terminus (Figure 7, EcPutA). Bifunctional PutAs of branch 1 that are longer than about 1100 residues also have this domain; the PutA from Azoarcus sp. BH72 is one example (Figure 7, Az). Interestingly, long bifunctional PutAs of branch 2 also have an extra domain in the C-terminus, but it is not clear whether it is related to the C-terminal domain of branch 1 PutAs. Thus, we will restrict our discussion here to the conserved C-terminal domain of branch 1 PutAs, which we denote by CTD. Using BjPutA as a reference, the CTD is inserted between the third section of the NAD+-binding domain and the conserved C-terminal motif of the oligomerization domain. The size of the CTD ranges from about 130 residues to 220 residues.
Although the function of the CTD is unknown, amino acid sequence analysis provides some intriguing ideas to test. Analysis of over 2 dozen branch 1 PutA CTDs using the remote homology detection algorithm HHSearch (70, 71) suggests that the CTD is homologous to the beta-hairpin and Rossmann fold regions of aldehyde dehydrogenases. For example, using the CTD of EcPutA as the query, HHSearch returned 33 aldehyde dehydrogenases with probability scores ranging from 99.1 % to 99.9 %. The top match was BjPutA with a probability score of 99.9 and E-value of 7.7E-26. The alignment shows that the CTD of EcPutA is 25 % identical to residues 551–761 of BjPutA. In the other cases tested, the BjPutA beta-hairpin/Rossmann domain was also identified as the closest homolog, and the probability score was in the range 99.7–100.0 %. These results are indicative of meaningful homology.
The homology is evident in an MSA of BjPutA with the CTDs of several branch 1 PutAs (Figure 8A). The core region of homology corresponds to the beta-hairpin of the oligomerization domain and the Rossmann fold domain of BjPutA. The structure of this region is highlighted in green, yellow, and red in Figure 8B. The highest similarity is found in the beta-hairpin, which contains the conserved motif lpGPtGExN. This result is significant, because multiple sequence alignments show that long branch 1 PutAs have a gap corresponding to the beta-hairpin of BjPutA (Figure 7, beta11). Thus, it appears that the beta-hairpin of the oligomerization domain has been shifted to the CTD in long PutAs.
Figure 8.

Homology of the CTD to the BjPutA beta-hairpin and Rossmann fold domain. (A) MSA of BjPutA with the CTDs of several branch 1 PutAs. (B) The structure of the NAD+-binding and oligomerization domains of BjPutA. The core region of homology with the CTD (residues 629–760) is colored according to secondary structure, with alpha helices in red, beta strands in yellow, and loops in green. Conserved residues of the CTD are indicated. Residues 510–628 are colored silver. Residues 955–989 are colored cyan.
In summary, remote homology detection suggests that the CTD includes a beta-hairpin that is homologous oligomerization domain of aldehyde dehydrogenases plus a Rossmann fold domain. Thus, long branch 1 PutAs are predicted to have two Rossmann fold domains, one that has high identity (~50 %) to the Rossmann fold domain of BjPutA and a second one in the CTD that has lower identity (14 – 33 %).
6. SUMMARY
Comparison of the PutA structures to those of the monofunctional enzymes is useful for thinking about the possibility of protein-protein interactions predicted by the Rosetta Stone hypothesis. Several unique features of PutAs are absent in the monofunctional enzymes, including helix alpha5a, the arm domain, the alpha domain, and the linker domain. These components appear to be important for orienting the catalytic domains of the PutA protomer so that the two active sites face each other and for sealing the substrate-channeling cavity from the bulk medium. The absence of these PutA-specific structural features in the monofunctional enzymes, at least those from branch 3B, perhaps argues against the formation of an efficient PRODH-P5CDH channeling complex. However, kinetic measurements of substrate channeling and biophysical measurements of protein-protein association are still needed to test the Rosetta Stone hypothesis for monofunctional proline catabolic enzymes.
The CTD is the only PutA domain that has not been structurally characterized. Remote homology detection suggests the tantalizing hypothesis that the CTD of branch 1 PutAs contains a beta-hairpin like the one in the oligomerization domain of aldehyde dehydrogenases and a Rossmann fold domain. Interestingly, remote homology detection did not find meaningful homology for the C-terminal domains of branch 2 enzymes. Thus, the C-terminal domains of branch 1 and 2 enzymes may differ in structure. These results raise several intriguing questions. Does the CTD bind NAD+ or is it a pseudo-NAD+-binding domain that primarily plays a structural role? We note that the oxidation of GSA to Glu requires only one equivalent of NAD+ and thus only one functional NAD+-binding domain is expected. Furthermore, the CTD is missing the NFP motif found in BjPutA residues Asn658-Phe659-Pro660 (Figure 8A). This motif is highly conserved by P5CDHs (including the first Rossmann domain of all PutAs), and the Asn residue is thought to help anchor the substrate in the oxyanion hole by forming a hydrogen bond with the epsilon O atom of GSA (54). The absence of this critical residue argues against the CTD participating in catalysis. If the CTD plays a structural role, does the predicted beta-hairpin interact with the C-terminal strand and participate in domain-swapped dimerization? And, does it help cover the substrate-channeling cavity as in BjPutA? New biochemical and structural studies on PutAs designed to answer these questions represent an exciting next phase of research in proline catabolism.
ACKNOWLEDGEMENTS
We thank Dr. Donald Becker for insightful discussions. This was supported in part by the National Institutes of Health Grants GM065546 and GM061068.
Abbreviations
- PutA
proline utilization A
- NAD+
nicotinamide adenine dinucleotide
- PRODH
proline dehydrogenase
- P5C
pyrroline-5-carboxylate
- P5CDH
pyrroline-5-carboxylate dehydrogenase
- FAD
flavin adenine dinucleotide
- GSA
glutamate semialdehyde
- BjPutA
PutA from Bradyrhizobium japonicum
- EcPutA
PutA from Escherichia coli
- TtPRODH
monofunctional PRODH from Thermus thermophilus
- TtP5CDH
monofunctional P5CDH from Thermus thermophilus
- MSA
multiple sequence alignment
- RHH
ribbon-helix-helix
- CTD
C-terminal domain of long branch 1 PutAs
8. REFERENCES
- 1.Phang JM. The regulatory functions of proline and pyrroline-5-carboxylic acid. Curr. Top. Cell. Reg. 1985;25:92–132. doi: 10.1016/b978-0-12-152825-6.50008-4. [DOI] [PubMed] [Google Scholar]
- 2.Ratzkin B, Roth J. Cluster of genes controlling proline degradation in Salmonella typhimurium. J. Bacteriol. 1978;133(2):744–54. doi: 10.1128/jb.133.2.744-754.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanner JJ. Structural biology of proline catabolism. Amino Acids. 2008;35(4):719–30. doi: 10.1007/s00726-008-0062-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Downing SJ, Phang JM, Kowaloff EM, Valle D, Smith RJ. Proline oxidase in cultured mammalian cells. J. Cell. Physiol. 1977;91(3):369–76. doi: 10.1002/jcp.1040910306. [DOI] [PubMed] [Google Scholar]
- 5.Kowaloff EM, Phang JM, Granger AS, Downing SJ. Regulation of proline oxidase activity by lactate. Proc. Natl. Acad. Sci. U S A. 1977;74(12):5368–71. doi: 10.1073/pnas.74.12.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donald SP, Sun XY, Hu CA, Yu J, Mei JM, Valle D, Phang JM. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res. 2001;61(5):1810–5. [PubMed] [Google Scholar]
- 7.Hu CA, Donald SP, Yu J, Lin WW, Liu Z, Steel G, Obie C, Valle D, Phang JM. Overexpression of proline oxidase induces proline-dependent and mitochondria-mediated apoptosis. Mol. Cell. Biochem. 2006 doi: 10.1007/s11010-006-9276-6. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates both intrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFAT and MEK/ERK signaling. Oncogene. 2006;25(41):5640–7. doi: 10.1038/sj.onc.1209564. [DOI] [PubMed] [Google Scholar]
- 9.Pandhare J, Cooper SK, Phang JM. Proline oxidase, a proapoptotic gene, is induced by troglitazone: evidence for both peroxisome proliferator-activated receptor gamma-dependent and -independent mechanisms. J. Biol. Chem. 2006;281(4):2044–52. doi: 10.1074/jbc.M507867200. [DOI] [PubMed] [Google Scholar]
- 10.Cooper SK, Pandhare J, Donald SP, Phang JM. A novel function for hydroxyproline oxidase in apoptosis through generation of reactive oxygen species. J. Biol. Chem. 2008;283(16):10485–92. doi: 10.1074/jbc.M702181200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Phang JM, Donald SP, Pandhare J, Liu Y. The metabolism of proline, a stress substrate, modulates carcinogenic pathways. Amino Acids. 2008;35(4):681–90. doi: 10.1007/s00726-008-0063-4. [DOI] [PubMed] [Google Scholar]
- 12.Phang JM, Pandhare J, Liu Y. The metabolism of proline as microenvironmental stress substrate. J Nutr. 2008;138(10):2008S–2015S. doi: 10.1093/jn/138.10.2008S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida A, Rzhetsky A, Hsu LC, Chang C. Human aldehyde dehydrogenase gene family. Eur. J. Biochem. 1998;251(3):549–57. doi: 10.1046/j.1432-1327.1998.2510549.x. [DOI] [PubMed] [Google Scholar]
- 14.Yoon KA, Nakamura Y, Arakawa H. Identification of ALDH4 as a p53-inducible gene and its protective role in cellular stresses. J. Hum. Genet. 2004;49(3):134–40. doi: 10.1007/s10038-003-0122-3. [DOI] [PubMed] [Google Scholar]
- 15.Forte-McRobbie CM, Pietruszko R. Purification and characterization of human liver “high Km” aldehyde dehydrogenase and its identification as glutamic gamma-semialdehyde dehydrogenase. J. Biol. Chem. 1986;261(5):2154–63. [PubMed] [Google Scholar]
- 16.Forte-McRobbie C, Pietruszko R. Human glutamic-gamma-semialdehyde dehydrogenase. Kinetic mechanism. Biochem. J. 1989;261(3):935–43. doi: 10.1042/bj2610935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.White TA, Krishnan N, Becker DF, Tanner JJ. Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus. J. Biol. Chem. 2007;282(19):14316–27. doi: 10.1074/jbc.M700912200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krishnan N, Becker DF. Characterization of a bifunctional PutA homologue from Bradyrhizobium japonicum and identification of an active site residue that modulates proline reduction of the flavin adenine dinucleotide cofactor. Biochemistry. 2005;44(25):9130–9. doi: 10.1021/bi050629k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuermann JP, White TA, Srivastava D, Karr DB, Tanner JJ. Three crystal forms of the bifunctional enzyme proline utilization A (PutA) from Bradyrhizobium japonicum. Acta Cryst. 2008;F64(Pt 10):949–53. doi: 10.1107/S174430910802842X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Straub PF, Reynolds PH, Althomsons S, Mett V, Zhu Y, Shearer G, Kohl DH. Isolation, DNA sequence analysis, and mutagenesis of a proline dehydrogenase gene (putA) from Bradyrhizobium japonicum. Appl. Environ. Microbiol. 1996;62(1):221–9. doi: 10.1128/aem.62.1.221-229.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krishnan N, Becker DF. Oxygen Reactivity of PutA from Helicobacter Species and Proline-Linked Oxidative Stress. J. Bacteriol. 2006;188(4):1227–35. doi: 10.1128/JB.188.4.1227-1235.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krishnan N, Doster AR, Duhamel GE, Becker DF. Characterization of a Helicobacter hepaticus putA mutant strain in host colonization and oxidative stress. Infect. Immun. 2008;76(7):3037–44. doi: 10.1128/IAI.01737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakajima K, Inatsu S, Mizote T, Nagata Y, Aoyama K, Fukuda Y, Nagata K. Possible involvement of put A gene in Helicobacter pylori colonization in the stomach and motility. Biomed. Res. 2008;29(1):9–18. doi: 10.2220/biomedres.29.9. [DOI] [PubMed] [Google Scholar]
- 24.Brown ED, Wood JM. Redesigned purification yields a fully functional PutA protein dimer from Escherichia coli. J. Biol. Chem. 1992;267(18):13086–92. [PubMed] [Google Scholar]
- 25.Menzel R, Roth J. Regulation of genes for Proline Utilization in Salmonella typhimurium: Autogenous Repression by the putA gene Product. J. Mol. Biol. 1981;148:21–44. doi: 10.1016/0022-2836(81)90233-3. [DOI] [PubMed] [Google Scholar]
- 26.Ostrovsky de Spicer P, O'Brien K, Maloy S. Regulation of proline utilization in Salmonella typhimurium: a membrane-associated dehydrogenase binds DNA in vitro. J Bacteriol. 1991;173(1):211–9. doi: 10.1128/jb.173.1.211-219.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vilchez S, Manzanera M, Ramos JL. Control of expression of divergent Pseudomonas putida put promoters for proline catabolism. Appl. Environ. Microbiol. 2000;66(12):5221–5. doi: 10.1128/aem.66.12.5221-5225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menzel R, Roth J. Enzymatic properties of the purified putA protein from Salmonella typhimurium. J. Biol. Chem. 1981;256(18):9762–6. [PubMed] [Google Scholar]
- 29.Ostrovsky de Spicer P, Maloy S. PutA protein, a membrane-associated flavin dehydrogenase, acts as a redox-dependent transcriptional regulator. Proc. Natl. Acad. Sci. USA. 1993;90(9):4295–8. doi: 10.1073/pnas.90.9.4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surber MW, Maloy S. The PutA protein of Salmonella typhimurium catalyzes the two steps of proline degradation via a leaky channel. Arch. Biochem. Biophys. 1998;354(2):281–287. doi: 10.1006/abbi.1998.0697. [DOI] [PubMed] [Google Scholar]
- 31.Surber MW, Maloy S. Regulation of Flavin Dehydrogenase Compartmentalization: Requirements for PutA-Membrane Association in Salmonella typhimurium. Biochim. Biophys. Acta. 1999;1421:5–18. doi: 10.1016/s0005-2736(99)00104-2. [DOI] [PubMed] [Google Scholar]
- 32.Wood JM, Zadworny D. Amplification of the put Genes and Identification of the put Gene Products in Escherichia coli K12. Can. J. Biochem. 1980;58:787–796. doi: 10.1139/o80-110. [DOI] [PubMed] [Google Scholar]
- 33.Wood JM. Genetics of L-proline Utilization in Escherichia coli. J. Bacteriol. 1981;146:895–901. doi: 10.1128/jb.146.3.895-901.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abrahamson JL, Baker LG, Stephenson JT, Wood JM. Proline dehydrogenase from Escherichia coli K12. Properties of the membrane-associated enzyme. Eur. J. Biochem. 1983;134(1):77–82. doi: 10.1111/j.1432-1033.1983.tb07533.x. [DOI] [PubMed] [Google Scholar]
- 35.Graham S, Stephenson JT, Wood JM. Proline Dehydrogenase from Escherichia coli K12, Reconstitution of Functional Membrane Association. J. Biol. Chem. 1984;259(4):2656–2661. [PubMed] [Google Scholar]
- 36.Wood JM. Membrane association of proline dehydrogenase in Escherichia coli is redox dependent. Proc. Natl. Acad. Sci. USA. 1987;84(2):373–377. doi: 10.1073/pnas.84.2.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown ED, Wood JM. Conformational change and membrane association of the PutA protein are coincident with reduction of its FAD cofactor by proline. J. Biol. Chem. 1993;268(12):8972–9. [PubMed] [Google Scholar]
- 38.Ling M, Allen SW, Wood JM. Sequence analysis identifies the proline dehydrogenase and delta 1-pyrroline-5-carboxylate dehydrogenase domains of the multifunctional Escherichia coli PutA protein. J. Mol. Biol. 1994;243(5):950–6. doi: 10.1006/jmbi.1994.1696. [DOI] [PubMed] [Google Scholar]
- 39.Becker DF, Thomas EA. Redox properties of the PutA protein from Escherichia coli and the influence of the flavin redox state on PutA-DNA interactions. Biochemistry. 2001;40(15):4714–21. doi: 10.1021/bi0019491. [DOI] [PubMed] [Google Scholar]
- 40.Vinod MP, Bellur P, Becker DF. Electrochemical and functional characterization of the proline dehydrogenase domain of the PutA flavoprotein from Escherichia coli. Biochemistry. 2002;41:6525–6532. doi: 10.1021/bi025706f. [DOI] [PubMed] [Google Scholar]
- 41.Zhu W, Gincherman Y, Docherty P, Spilling CD, Becker DF. Effects of proline analog binding on the spectroscopic and redox properties of PutA. Arch. Biochem. Biophys. 2002;408(1):131–6. doi: 10.1016/s0003-9861(02)00535-0. [DOI] [PubMed] [Google Scholar]
- 42.Zhu W, Becker DF. Flavin redox state triggers conformational changes in the PutA protein from Escherichia coli. Biochemistry. 2003;42(18):5469–77. doi: 10.1021/bi0272196. [DOI] [PubMed] [Google Scholar]
- 43.Baban BA, Vinod MP, Tanner JJ, Becker DF. Probing a hydrogen bond pair and the FAD redox properties in the proline dehydrogenase domain of Escherichia coli PutA. Biochim. Biophys. Acta. 2004;1701(1–2):49–59. doi: 10.1016/j.bbapap.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 44.Zhang W, Zhou Y, Becker DF. Regulation of PutA-membrane associations by flavin adenine dinucleotide reduction. Biochemistry. 2004;43(41):13165–74. doi: 10.1021/bi048596g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu W, Becker DF. Exploring the proline-dependent conformational change in the multifunctional PutA flavoprotein by tryptophan fluorescence spectroscopy. Biochemistry. 2005;44(37):12297–306. doi: 10.1021/bi051026b. [DOI] [PubMed] [Google Scholar]
- 46.Zhang W, Zhang M, Zhu W, Zhou Y, Wanduragala S, Rewinkel D, Tanner JJ, Becker DF. Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2'-OH group in regulating PutA--membrane binding. Biochemistry. 2007;46(2):483–91. doi: 10.1021/bi061935g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arentson BW, Sanyal N, Becker DF. Substrate channeling in proline metabolism. Front. Biosci. 2011 doi: 10.2741/3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anderson KS. Fundamental mechanisms of substrate channeling. Methods Enzymol. 1999;308:111–45. doi: 10.1016/s0076-6879(99)08008-8. [DOI] [PubMed] [Google Scholar]
- 49.Miles EW, Rhee S, Davies DR. The molecular basis of substrate channeling. J. Biol. Chem. 1999;274(18):12193–6. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 50.Huang X, Holden HM, Raushel FM. Channeling of substrates and intermediates in enzyme-catalyzed reactions. Annu. Rev. Biochem. 2001;70:149–80. doi: 10.1146/annurev.biochem.70.1.149. [DOI] [PubMed] [Google Scholar]
- 51.Marcotte EM, Pellegrini M, Ng HL, Rice DW, Yeates TO, Eisenberg D. Detecting protein function and protein-protein interactions from genome sequences. Science. 1999;285(5428):751–3. doi: 10.1126/science.285.5428.751. [DOI] [PubMed] [Google Scholar]
- 52.White TA, Tanner JJ. Cloning, purification and crystallization of Thermus thermophilus proline dehydrogenase. Acta Cryst. 2005;F61(Pt 8):737–739. doi: 10.1107/S1744309105019779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White TA, Johnson WH, Jr., Whitman CP, Tanner JJ. Structural basis for the inactivation of Thermus thermophilus proline dehydrogenase by N-propargylglycine. Biochemistry. 2008;47(20):5573–80. doi: 10.1021/bi800055w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Inagaki E, Ohshima N, Takahashi H, Kuroishi C, Yokoyama S, Tahirov TH. Crystal structure of Thermus thermophilus Delta1-pyrroline-5-carboxylate dehydrogenase. J. Mol. Biol. 2006;362(3):490–501. doi: 10.1016/j.jmb.2006.07.048. [DOI] [PubMed] [Google Scholar]
- 55.Inagaki E, Ohshima N, Sakamoto K, Babayeva ND, Kato H, Yokoyama S, Tahirov TH. New insights into the binding mode of coenzymes: structure of Thermus thermophilus [Delta]1-pyrroline-5-carboxylate dehydrogenase complexed with NADP+ Acta Cryst. 2007;F63:462–465. doi: 10.1107/S1744309107021422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Larson JD, Jenkins JL, Schuermann JP, Zhou Y, Becker DF, Tanner JJ. Crystal structures of the DNA-binding domain of Escherichia coli proline utilization A flavoprotein and analysis of the role of Lys9 in DNA recognition. Protein Sci. 2006;15:1–12. doi: 10.1110/ps.062425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y, Larson JD, Bottoms CA, Arturo EC, Henzl MT, Jenkins JL, Nix JC, Becker DF, Tanner JJ. Structural basis of the transcriptional regulation of the proline utilization regulon by multifunctional PutA. J. Mol. Biol. 2008;381(1):174–88. doi: 10.1016/j.jmb.2008.05.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Halouska S, Zhou Y, Becker DF, Powers R. Solution structure of the Pseudomonas putida protein PpPutA45 and its DNA complex. Proteins. 2009;75(1):12–27. doi: 10.1002/prot.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee YH, Nadaraia S, Gu D, Becker DF, Tanner JJ. Structure of the proline dehydrogenase domain of the multifunctional PutA flavoprotein. Nat. Struct. Biol. 2003;10(2):109–114. doi: 10.1038/nsb885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, White TA, Schuermann JP, Baban BA, Becker DF, Tanner JJ. Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors. Biochemistry. 2004;43(39):12539–48. doi: 10.1021/bi048737e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ostrander EL, Larson JD, Schuermann JP, Tanner JJ. A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate. Biochemistry. 2009;48(5):951–9. doi: 10.1021/bi802094k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Srivastava D, Zhu W, Johnson WH, Jr., Whitman CP, Becker DF, Tanner JJ. The structure of the proline utilization a proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction. Biochemistry. 2010;49(3):560–569. doi: 10.1021/bi901717s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Srivastava D, Schuermann JP, White TA, Krishnan N, Sanyal N, Hura GL, Tan A, Henzl MT, Becker DF, Tanner JJ. Crystal structure of the bifunctional proline utilization A flavoenzyme from Bradyrhizobium japonicum. Proc. Natl. Acad. Sci. U. S. A. 2010;107(7):2878–83. doi: 10.1073/pnas.0906101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bennett MJ, Schlunegger MP, Eisenberg D. 3D domain swapping: a mechanism for oligomer assembly. Protein Sci. 1995;4(12):2455–68. doi: 10.1002/pro.5560041202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu ZJ, Sun YJ, Rose J, Chung YJ, Hsiao CD, Chang WR, Kuo I, Perozich J, Lindahl R, Hempel J, Wang BC. The first structure of an aldehyde dehydrogenase reveals novel interactions between NAD and the Rossmann fold. Nat. Struct. Biol. 1997;4(4):317–26. doi: 10.1038/nsb0497-317. [DOI] [PubMed] [Google Scholar]
- 66.Bottoms CA, Smith PE, Tanner JJ. A structurally conserved water molecule in Rossmann dinucleotide-binding domains. Protein Sci. 2002;11(9):2125–37. doi: 10.1110/ps.0213502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gu D, Zhou Y, Kallhoff V, Baban B, Tanner JJ, Becker DF. Identification and characterization of the DNA-binding domain of the multifunctional PutA flavoenzyme. J. Biol. Chem. 2004;279(30):31171–6. doi: 10.1074/jbc.M403701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schreiter ER, Drennan CL. Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 2007;5(9):710–20. doi: 10.1038/nrmicro1717. [DOI] [PubMed] [Google Scholar]
- 69.Watanabe H, Hastings JW. Specificities and properties of three reduced pyridine nucleotide-flavin mononucleotide reductases coupling to bacterial luciferase. Mol. Cell. Biochem. 1982;44:181–187. doi: 10.1007/BF00238506. [DOI] [PubMed] [Google Scholar]
- 70.Soding J. Protein homology detection by HMM-HMM comparison. Bioinformatics. 2005;21(7):951–60. doi: 10.1093/bioinformatics/bti125. [DOI] [PubMed] [Google Scholar]
- 71.Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 72.DeLano WL. The PyMOL User's Manual. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- 73.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD. Thompson: Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31(13):3497–500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15(4):305–8. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]