

Abstract
New series of pyrrolidine mercaptosulfide, 2-mercaptocyclopentane arylsulfonamide, and 3-mercapto-4-arylsulfonamido pyrrolidine matrix metalloproteinase inhibitors (MMPIs) were designed, synthesized, and evaluated. Exhibiting unique properties over other MMPIs (e.g., hydroxamates), these newly reported compounds are capable of modulating activities of several MMPs in the low nanomolar range, including MMP-2 (~2 to 50 nM), MMP-13 (~2 to 50 nM), and MMP-14 (~4 to 60 nM). Additionally these compounds are selective to intermediate- and deep-pocket MMPs but not shallow-pocketed MMPs (e.g., MMP-1, ~850 to >50,000 nM; MMP-7, ~4,000 to >25,000 nM). Our previous work with the mercaptosulfide functionality attached to both cyclopentane and pyrrolidine frameworks demonstrated that the cis-(3S,4R)-stereochemistry was optimal for all of the MMPs tested. However, in our newest compounds an interesting shift of preference to the trans-form of the mercaptosulfonamides was observed with increased oxidative stability and biological compatibility. We also report several kinetic and biological characteristics showing that these compounds may be used to probe the mechanistic activities of MMPs in disease.
INTRODUCTION
Matrix metalloproteinases (MMPs) are a family of metzincin endopeptidases chiefly responsible for remodeling the extracellular matrix (ECM). Further division of the family on the basis of domain arrangement and substrate specificity results in six subgroups: collagenases (MMPs -1, -8, -13, & -18); gelatinases (MMPs -2 & -9); stromelysins (MMPs -3, -10, & -11); matrilysins (MMPs -7 & -26); membrane type MMPs (MMPs -14, -15, -16, -17, -24, & -25); and others (MMPs -12, -19, -20, -21, -23, -27, & -28). Members of this family also process non matrix and cell surface signaling proteins to serve important roles in a number of physiological (e.g., epithelial morphogenesis, neurogenesis) and pathological processes (e.g., cancer, inflammation, cardiovascular disease).1-3 In light of these pathological roles, and in the pursuance of developing targeted treatments, biological and pharmacological regulation of MMPs continues to be extensively studied. Targeting particular MMPs however requires careful consideration and places a premium on selectivity so that side effects are minimized. For example in ischemic events (e.g., stroke), proteolytic activities of MMPs -2 and -9 result in initial blood-brain barrier disruption,4 while during later stages MMP-9 may exhibit neuroprotective effects.5 Whereas during atherosclerosis MMPs -1 and -8 are suggested to partake in plaque destabilization,6-7 while MMPs -2 and -9 facilitate smooth muscle cell migration and overall plaque stability.8 Hence, targeting specific MMPs is disease dependent.
The activity of MMPs is controlled through several mechanisms: gene expression; compartmentalization; zymogen activation; and important to this discussion, enzyme inhibition. Endogenously, the four tissue inhibitors of metalloproteinases (TIMPs -1, -2, -3, & -4) are primarily responsible for MMP inhibition. Since TIMPs exhibit picomolar to nanomolar affinities for each of the MMPs, they were initially proposed as the ideal inhibitors to block the pathological activities of MMPs.9 However, TIMPs are essentially broad-spectrum inhibitors, lacking selectivity for individual MMPs, and themselves are involved in various non-MMP related physiological processes (e.g., apoptosis).10-13 This led to a decade’s endeavor to develop small molecule MMP inhibitors (MMPIs). Generally MMPIs mimic substrate peptide structures, incorporating a non-cleavable zinc-binding group in place of the scissile bond, and thus presumably mimic substrate binding with the enzyme active site. Since the catalytic domains of all MMPs present a high degree of homology,14 the specificity and selectivity of inhibition by these early compounds was minimal, with numerous off target effects and binding promiscuity observed.15
Characterized by our group and others,16-17 the principal specificity pocket, designated the S1′ pocket, is a hydrophobic cavity positioned to the right (i.e., prime side) of the catalytic zinc. Critical to the shape of this pocket is the identity of one key residue in the catalytic domain’s second helix that determines the pocket’s relative depth.18 The S1′ pocket thus may be shallow (MMPs -1 & -7), intermediate (MMPs -2, -8, -9, & -26), or deep (MMPs -3, -12, & -14) and offers a degree of individuality to MMPs that can be exploited to ascertain specificity.16-17
The nature of the zinc-binding group (ZBG) is equally important to both potency and specificity of the MMPI. Numerous ZBGs have been utilized, including the widely incorporated hydroxamates and the lesser used carboxylates, phosphinyls, and mercaptans.19 Hydroxamate is the most potent ZBG, coordinating Zn2+ in a bidentate fashion to form a complex with distorted trigonal bipyramidal geometry. However, the hydroxamate affinity for Zn2+ seems to overwhelm specific protease binding contributions by other groups within the structure. Consequently, the selectivity of hydroxamate-based MMPIs was reduced and resulted in side effects such as tendonitis fibromyalgia and musculoskeletal abnormalities in oncology clinical trials.20-21 These results stimulated our own interest in developing MMPIs with alternative ZBGs that offer adequate levels of binding without sacrificing specificity.
The popularity of thiol-based angiotensin-converting enzyme inhibitors (e.g., (2S)-1-[(2S)-2-methyl-3-sulfanylpropanoyl]pyrrolidine-2-carboxylic acid)22 and the effectiveness of thiols (–SH) as metal binding ligands, despite some limitations,23 led us to investigate MMPIs containing the mercaptan ZBG. The earliest of our compounds were peptidomimetic mercaptosulfides; however, these MMPIs were readily deactivated by hydrolysis and by oxidation of the thiol.24-26 A second generation of mercaptosulfides attached to a cyclopentane or pyrrolidine core was produced.27-29 Incorporation of the 1,2-mercaptosulfide moiety into a ring system was designed from structure-based computer modeling using MacroModel (version 7) and chemical intuition to increase binding affinity by restricting rotation of the C-C bond connected to the two sulfurs; leading to development of the 1,2-cyclopentanemercaptosulfide moiety as a new pharmacophore. These novel inhibitors with a cyclopentane ring showed moderately improved inhibitory activities and significantly higher oxidation stability. Further modification of the ring from cyclopentane to pyrrolidine was carried out to enhance water solubility and allow for a greater degree of functionality (i.e., so that additional side chains that interact with the non-prime side of the enzyme active site may be appended) (Figure 1).28, 30 This structural change improved MMPI stability, leading to enhanced bioavailability and increased potency of inhibition.17, 31-32 A final modification of the peptidomimetic part to a mercaptosulfonamide, again attached to a cyclopentane or pyrrolidine core, led to further increases in ZBG stability and inhibitor potency and selectivity.33
Figure 1.
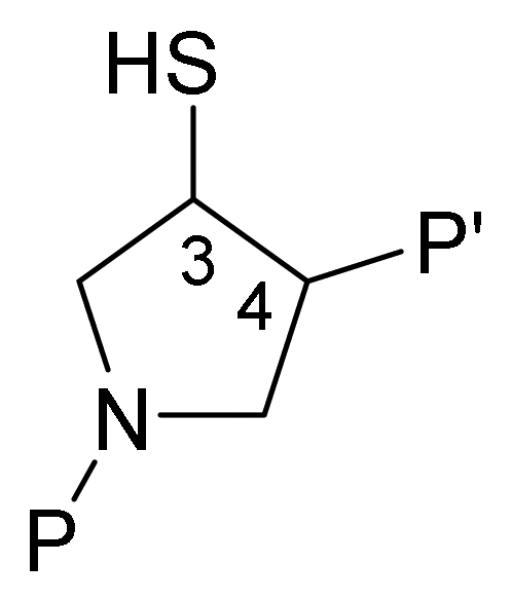
Representation of the pyrrolidine scaffold to functionalize the MMPIs for interaction with the non-prime side of the enzyme active site (P).
An obvious factor determining the effectiveness of these MMPIs, in addition to the nature of the P and P’ residues, is the relative and absolute stereochemistry at C-3 and C-4 needed to maximize MMP binding. Initial work with the mercaptosulfide functionality26 attached to both cyclopentane and pyrrolidine frameworks demonstrated that the cis-(3S,4R)-stereochemistry was optimal for all of the MMPs tested.17, 31-32 However, in the mercaptosulfonamides the trans-stereoisomer turned out to be the most potent, with little preference between absolute configurations.
We have produced an array of mercaptosulfonamide MMPIs capable of modulating the activity of numerous MMPs in the low nanomolar range and report several kinetic and biological characteristics to show how these compounds may be used for probing the mechanistic activities of MMPs in disease.
RESULTS
Structure-Activity Relationship Studies
Mercaptosulfide Inhibitors
The synthesis of mercaptosulfide inhibitors with a pyrrolidine pharmacophore is outlined in Scheme 1. Pyrroline (1) was Boc protected (2) and oxidized with m-CPBA to give the pyrrolidine epoxide (3) that was treated with thiolbenzoic acid over alumina to afford the benzoylthioalcohol intermediate (4). The benzoyl group of 4 was removed with EtONa in EtOH and t-Boc protecting group was introduced in one pot, affording compound 5, which was converted into the differentially protected cis-dithio compound (6) via the Mitsunobu reaction.34 The acetyl group of intermediate 6 was selectively removed with MeNH2 in MeOH and the resulting thiol was coupled with (S)-2-bromo-4-methylpentanoic acid to afford the S-alkylated acid intermediate (7). The acid (7) was coupled with phenylalanine N-methyl amide to give the key intermediate 8. N-Boc group of 8 was selectively removed in HCl ethyl acetate solution (9) and coupled with carboxylic acid to afford intermediate 10 that was treated with HCl in acetic acid to give the mercaptosulfide MMPIs (11). A series of pyrrolidine mercaptosulfides with varying N-substituents was initially synthesized to evaluate the ability of the P residue (Table 1, Figure 2) to confer potency and selectivity.33 These compounds were tested against MMPs -1, -2, -3, -7, -9, and -14. As the size of the P residue increased (11b vs. 11c-11e) potency against the enzymes was generally increased, but no substantial changes in selectivity were observed. Notably, potency against MMP-3 (stromelysin-1) was dramatically increased as the chain of the N-phthalimidoacyl group (11c-11e) was lengthened.
Scheme 1.

Synthesis of Pyrrolidine Mercaptosulfide Inhibitors.
Reagents and conditions: a)Boc2O, CH2Cl2, 0 °C; b) m-CPBA, CH2Cl2, 0 °C-rt; c) Al2O3, PhCOSH, Et2O, rt; d) EtONa/EtOH, Boc2O, 0 °C; e) Ph3P, DEAD, AcSH, 0 °C-rt; f) (2S)-2-bromo-4-methylpentanoic acid, K2CO3, DMF, rt; g) L-Phe, EDCI, HOBt, Et3N, 0 °C-rt; h) HCl-EtOAc; i) R1CO2H, DCC, HOBt, Et3N, CH2Cl2, 0 °C-rt; j) HCl/AcOH, rt.
Table 1.
Effect of N-Substituent on MMP Inhibition by Pyrrolidine Mercaptosulfides.
| Compounda | Kiapp (nM) |
|||||
|---|---|---|---|---|---|---|
| MMP-1 | MMP-2 | MMP-3 | MMP-7 | MMP-9 | MMP-14 | |
| 11a | 260 | 200 | 4100 | 230 | 5.3 | 310 |
| 11b | 99 | 14 | 990 | 91 | 5.7 | 6.2 |
| 11c | 110 | 17 | 300 | 50 | 4.9 | 70 |
| 11d | 75 | 8.5 | 31 | 12 | 3.9 | 6.0 |
| 11e | 52 | 1.7 | 1.9 | 11 | 0.98 | 7.0 |
Each inhibitor was a 1:1 mixture of cis-3R,4S and cis-3S,4R diastereomers.
Figure 2.

Structural representation of compounds 11a-11e presented in Table 1.
Cyclopentane Mercaptosulfonamide Inhibitors
To enhance the overall biological stability of these compounds, the P’ residue of the 3-Mercaptopyrrolidine inhibitors was changed from a peptidomimetic alkylsulfide group to an arylsulfonamide.35-36 This modification was intended to enhance enzyme inhibitor binding through hydrogen bonding to the enzyme backbone and guide the hydrophobic substituent into the S1′ pocket.37 On the non-prime side of the compounds, a cyclopentane scaffold was first employed to determine effective aryl substituents and the stereochemical requirements for inhibition in this new class of compounds.
The stereoisomeric 2-mercaptocyclopentane arylsulfonamides were synthesized from trans-2-azidocyclopentanol as outlined in Scheme 2. Kinetic resolution of racemic trans-2-azidocyclopentanol 12 using Lipase Amano AK-20 yielded (1S,2S) 13 and acetylated (1R, 2R) 14 that was treated with based to give (1R,2R) 15 as described similarly by Ami and Ohrui.38 Each of these chiral trans-2-azidocyclopentanols was reduced to aminoalcohol and then the amino group was protected with Boc to afford 16. Compound 16 was converted through Mitsunobu reaction34 into compound 17, which was first treated with HCl in MeOH to remove both protecting groups and then with arylsulfonyl chloride in DCM to give chiral cis-mercaptosulfonamide 18. For the synthesis of chiral trans-mercaptosulfonamide, chiral trans-2-azidocyclopentanol (12) was converted to chiral cis-2-azidocyclopentanol (20) through Mitsunobu reaction and hydrolysis. The azide group of compound 20 was reduced to amine and subsequently treated with arylsulfonyl chloride to give intermediate 21. The hydroxyl group of 21 was converted to acetylthio group through Mitsunobu reaction to give compound 22, which was treated with HCl in MeOH to give the desired chiral trans-mercaptosulfonamides (23). Compound 23f was obtained directly from trans-azidocyclopentanol through azide reduction and amine sulfonation.
Scheme 2.

Stereospecific Synthesis of 2-Mercaptocyclopentane Arylsulfonamides.
Reagents and conditions: a) Amano AK-20 lipase, isopropenyl acetate, tert-butyl methyl ether, rt; b) LiOH, THF-MeOH-H2O, rt; c) (1) PPh3, THF-H20, heat; (2) NaHCO3, Boc2O, 0 °C-rt; d) 4-Nitrobenzoic acid, PPh3, DEAD, THF, 0 °C-rt; e) AcSH, PPh3, DEAD, THF, 0 °C-rt; f) (1) 2N HCl in MeOH; (2) ArSO2Cl, Et3N, CH2Cl2, 0 °C-rt; g) LiOH, THF-MeOH-H2O, rt; h) (1) PPh3, THF-H20, heat; (2) NaHCO3, ArSO2Cl, 0°C-rt; i) AcSH, PPh3, DEAD, 0 °C-rt; j) MeNH2, MeOH, rt.
With this new set of inhibitors (Table 2, Figure 3), investigations into the effect of P’ aryl group substitution demonstrated that the 4-phenoxyphenyl (23c) maximized inhibition of intermediate (MMPs -2 & -9) and deep (MMPs -13 & -14) S1′ pocket MMPs when compared to those incorporating a phenyl (23a) or biphenyl (23b) group. In contrast to the preferred cis-stereochemistry observed with our mercaptosulfide compounds,17, 26, 31-32 the trans-stereochemistry was more potent (23c vs. 18a). Moreover, there was limited enantiomeric stereoselectivity by the trans-mercaptosulfonamides (23c vs. 23d and 23e), while there was a distinct preference in the less potent cis-mercaptocyclopentane sulfonamides for the (1R,2S)-enantiomer (18c). The sulfonamide functionality was also evaluated by modifying it to an amido functionality. This resulted in diminished potency as seen when the trans-2-(4-phenoxybenzamido)cyclopentanethiol (23g) is compared to the corresponding sulfonamide (23c). Expectedly, the structure lacking an effective ZBG (23f), showed no significant inhibitory activity when HO– was substituted for HS–.
Table 2.
MMP Inhibition by 2-Mercaptocyclopentane Arylsulfonamides.
| Compound | Kiapp (nM) |
||||||
|---|---|---|---|---|---|---|---|
| MMP-1 | MMP-2 | MMP-3 | MMP-7 | MMP-9 | MMP-13 | MMP-14 | |
| 18a | >1.0×105 | 810 | >2.0×105 | >2.0×104 | 970 | 200 | 410 |
| 18b | >2.0×105 | ~2.5×104 | >2.0×105 | >2.0×105 | >1600 | >1.0×105 | ~2.5×104 |
| 18c | 430 | >1.0×105 | 280 | 160 | 68 | ||
| 23a | >2.0×104 | 7200 | >2.0×105 | >5.0×104 | 9600 | 3400 | 3000 |
| 23b | ~1.0×104 | 240 | ~1.0×104 | 630 | 230 | 1200 | |
| 23c | 3400 | 18 | >2.0×105 | >2.0×104 | 25±7 | 15 | 18 |
| 23d | 2100 | 4.6 | >2.0×105 | >1.0×105 | 38±10 | 17 | 31 |
| 23e | 67 | >1.0×105 | 3.6×104 | 42±3 | 10 | 10 | |
| 23f | >2.0×105 | ~5.0×104 | >2.0×105 | >2.0×105 | ~2.0×105 | ~1.0×105 | >2.0×105 |
| 23g | >2.0×105 | 5700 | >2.0×105 | 5700 | 4300 | 2.8×104 | >2.5×104 |
Figure 3.

Structural representation of compounds 18a-18c and 23a-23g presented in Table 2.
Pyrrolidine Mercaptosulfonamide Inhibitors
Findings from the cyclopentane series of mercaptosulfonamides were applied to the target pyrrolidine scaffold. The synthetic route to the (±)-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine series of inhibitors is summarized in Scheme 3. Pyrrolidine epoxide (3) was treated with NaN3 to give azidoalcohol (24) that was first reduced to aminoalcohol then coupled with arylsulfonyl chloride to afford compound 25. N-Sulfonyl aziridine (26), which was obtained from 25 through Mitsunobu reaction,34 was treated with t-BuSH to give compound 27. N-Boc deprotection and subsequent N-acylation followed by removal of t-BuSH protecting group afforded the inhibitor 29. N-methylated inhibitor 29c was also made in the same way except the methylation step of 27. A different approach was required to prepare the individual enantiomers of these compounds, which is outlined in Scheme 4. Kinetic resolution of the 3,4-hydroxyazido pyrrolidine 24 gave (3S,4S)-enantiomer 30 and acetylated (3R,4R)-enantiomer 31 that was treated with base to afford (3R,4R)-enantiomer 32. The hydroxy group of each enantiomer was activated by mesylation then treated with AcOK to give the cis-acetoxy azide (33). The acetyl group of 33 was removed to give cis-3,4-hydroxyazido pyrrolidine 34 that was reduced and coupled with sulfonyl chloride to give compound 35. Compound 36, obtained from compound 35 through Mitsunobu reaction, was treated with MeNH2 to remove acetyl group to give thiol 37, which was re-protected with o-nitrobenzenethio group to afford disulfide 38. The final compound 39 was obtained by N-acylation after removing N-Boc group and reduction of the disulfide bond with TCEP.
Scheme 3.

Synthesis of (±)-trans-3-Mercapto-4-arylsulfonamidopyrrolidines.
Reagents and conditions: a) NaN3, NH4Cl, MeOH/H2O, 65 °C; b) PPh3, THF-H2O; NaHCO3, ArSO2Cl, 0 °C-rt; c) PPh3, DEAD, THF, rt; d) t-BuSH, t-BuOK, MeOH, 0 °C-rt; e) RX, t-BuOK, DMF, 0 °C-rt; f) (1) 1.5 M HCl/EtOAc, rt, (2) R1COCl, Et3N, CH2Cl2, rt; g) (1) 2-nitrobenzenesulfenyl chloride, HOAc, rt, (2) tri(carboxyethyl)phosphine, 1N NaOH, THF, rt.
Scheme 4.

Stereospecific Synthesis of trans-3-Mercapto-4-arylsulfonamidopyrrolidines.
Reagents and conditions: a) Amano AK-20 lipase, tert-butyl methyl ether, isopropenyl acetate, rt; b) LiOH, THF-MeOH-H2O, rt; c) (1) MeSO2Cl, Et3N, CHCl3, rt, (2) AcOK, DMF, 100 °C; d) LiOH, THF-MeOH-H2O, rt; e) PPh3, THF-H2O; NaHCO3, ArSO2Cl, 0 °C-rt; f) AcSH, PPh3, DEAD, 0 °C-rt; g) MeNH2, MeOH, rt; h) 2-nitrobenzenesulfenyl chloride, CH2Cl2, rt; i) (1) TFA, rt, (2) RCOCl, Et3N, CH2Cl2, 0 °C-rt; j) tri(carboxyethyl)phosphine, 1N NaOH, THF-H2O, rt.
Inhibition values for a selection of mercaptopyrrolidine compounds (Figure 4) are shown in Table 3. As with the trans-mercaptosulfonamide cyclopentanes, there was little enantiomeric stereoselectivity (29a vs. 39a and 39b). The N-carbamoylpyrrolidine derivative (29b) afforded good water solubility without the deleterious effect of the charged amino group of the unsubstituted pyrrolidine (29a). Similar to the earlier mercaptosulfides, attachment of a phthalimidoethylaminocarbonyl group to the pyrrolidine nitrogen (29d) as a non-primed side (P3) residue greatly improved selectivity for MMPs -3 and -9 (i.e., roughly ten fold) and moderately for MMPs -1 and -13.
Figure 4.

Structural representation of compounds 29a-29g, 39a, and 39b presented in Table 3.
Table 3.
MMP Inhibition by trans-3-Mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidines.
| Compound | Kiapp (nM) |
||||||
|---|---|---|---|---|---|---|---|
| MMP-1 | MMP-2 | MMP-3 | MMP-7 | MMP-9 | MMP-13 | MMP-14 | |
| 29a | ~2.5×104 | 27 | 2500 | >2.5×104 | 230 | ||
| 29b | 4100 | 3.9 | 460 | >2.5×104 | 15 | 50 | 11 |
| 29c | 850 | 2.3 | 350 | >2.5×104 | 1.1 | 28 | 62 |
| 29d | 2800 | 35 | 37 | >1.2×104 | 2.4 | 27 | 21 |
| 29e | >6000 | 3.8 | 810 | 3000 | 1.5 | 1.7 | 4 |
| 29f | >3000 | 3.1 | 2000 | 4000 | 3.6 | 2.0 | 6.6 |
| 29g | >5.0×104 | ~7000 | >1.0×105 | >1.0×105 | ~3.0×104 | >2.0×105 | 8.5×104 |
| 39a | 5.0×104 | 38 | 5900 | >2.0×105 | 210 | ||
| 39b | 6300 | 49 | 1600 | 4100 | 230 | ||
Biological Compatibility
Compound Stability
Although structurally distinct from the prototypical hydroxamate MMPIs, the mercaptan-based compounds exhibit comparable potencies, ranging from sub-micromolar down to single digit nanomolar Ki values.39-41 However, early generations of these compounds suffered from rapid oxidation of the thiol group, resulting in a reduced ability of these compounds to coordinate the active site Zn2+ due to disulfide formation.32 In order to assess the stability of these new compounds, studies were performed to evaluate the thiol’s zinc chelation over time.
Compared to the mercaptosulfide inhibitors, the mercaptosulfonamides showed increased oxidative stability in solution, moderately inhibiting MMP-9 for periods five-times longer than earlier compounds (Figure 5). In addition to increasing potency, incorporation of either the cyclopentyl or pyrrolidinyl ring to the non-primed side of the inhibitor enhanced resistance of the thiol to air oxidation. However, differences between ring types had no apparent effect on compound stability as 23c and 29b (Figure 5, Panels A & B) exhibited indistinguishable stability profiles. Evaluation of longer alkyl additions to the pyrrolidinyl nitrogen (29f and 29e) revealed direct correlation with reduced thiol stability (Figure 5, Panels C & D). Methylation of the sulfonamide nitrogen (29c) also resulted in reduced thiol stability despite increases in potency towards several MMPs (data not shown). An additional factor affecting the stability of these compounds is the presence of serum. Appearing to reduce oxidative degradation, higher percentages of serum in culture media prolong inhibitory activity of 23c and 29f (Figure 5, Panels A & C).
Figure 5.

Stability profiles of the mercaptosulfonamide MMPIs in cell culture media (αMEM). For panels A and C, serum concentration was varied to assess any sequestration of the compounds by albumin. Error bars represent the standard error in the mean (mean ± SEM).
Cytotoxicity
Before use in cellular assays, compound toxicity is assessed in human mesenchymal stem cells (hMSCs). This cell type was selected as current research surrounds the involvement of MMPs in the proliferation and differentiation of this cell line.42-44 Consistent with its use in other cell lines in our lab, 23c exhibited the greatest level of toxicity, reducing the number of cells by 40% at 100 LM (Figure 6, Panel A). The decrease in cell number was attributed to MMP inhibition since the non-inhibitory analogue (23f) revealed no difference in cell number. Similar trends were observed with 29b, 29d, and 29e (Figure 6, Panels B, C, & D).
Figure 6.

Cytotoxicity profiles of the mercaptosulfonamide MMPIs in hMSCs. Error bars represent standard error in the mean (mean ± SEM).
DISCUSSION AND CONCLUSIONS
Structure-Activity Relationship Studies
The principle goal of these studies was to develop potent and selective MMPIs that incorporate a thiol ZBG. This strategy was initially selected to circumvent the clinical side effects (e.g., poor plasma bioavailability, target promiscuity) that generally hampered early generation hydroxamate MMPIs, albeit recent hydroxamates are also avoiding such characteristics.39, 45-48 A recent report using a molecular dynamics approach to study the docking of various MMPIs with MMP-9 even indicates that thiol-based ZBGs are more energetically favorable than their hydroxamate counterparts for this enzyme.49
The inhibition of MMPs by mercaptosulfonamides is predicated upon coordination of the thiol with the enzyme’s catalytic zinc. For inhibitor 29c, where substitution of a methyl group directly upon the sulfonamide nitrogen did not reduce inhibitor potency, we speculate the possibility that hydrogen bonding between the sulfonamide –NH and enzyme is replaced by the methyl group’s hydrophobic interaction. In fact, potency was somewhat increased, perhaps by the methyl group interacting with the S1′* sub-site.50 Although our inhibitors showed somewhat lower binding affinities when compared with known bidentate ZBGs, the lower dissolution cost and amenable ionization are more favorable, resulting in only slightly lower potencies than hydroxamate-based MMPIs.51
Most efforts to establish MMPI selectivity have focused on modifying the prime side of compounds, with little effort evaluating non-prime substituents. The non-prime pockets of MMPs are primarily solvent exposed and display less segregation between subsites.52 For our purposes, substitution upon the pyrrolidinyl nitrogen was to enhance future functionalization of these compounds. However, exploration of varying N-substituents provided significant changes in potency. For example, addition of alkyl chains terminating with more polar head groups generally increased inhibitor potency (29e and 29f). With particular respect to MMP-3 (stromelysin-1), ring structures attached to long chains had significant effect on selectivity (11c-11e and 29d). Due to the S2 pocket’s location immediately adjacent to the catalytic zinc, we hypothesize that the linker and large pthalidimido group are likely interacting with the S3 pocket. As the S3 pocket is more hydrophobic, it may attract the phthalimido group, which may then position to participate in a previously noted π-π stacking interaction with the pocket’s Tyr155.53 Surprisingly, increased potency of these compounds against MMP-1 (collagenase-1) was considerably less dramatic (i.e., ~5-fold increase) than MMP-3 (i.e., ~2000-fold increase). This was unexpected as the S3 pocket in MMP-1, attributed to Ser155, is larger than those in MMPs -2, -3, -7, and -9, which all contain Tyr155 and are believed to energetically accommodate larger P residues.54
Looking at the prime side, a noticeable trend was observed with modulation of the diphenylether and sulfonyl group. Modifying the lone benzene (23a) to a biphenyl (23b) and finally a diphenylether (23c) dramatically increased inhibitory potency for MMP-13. Additionally, replacement of the sulfonyl group with a carbonyl (23g) not only reduced MMP-13 inhibition, but potency towards every MMP. These results are consistent with other reports of thiol-based MMP inhibitors, in that the diphenylether and sulfonyl groups are critical for maintaining potency.55
More surprising was the demonstration that, in contrast to the preferred cis-stereochemistry observed in our previous work with the mercaptosulfides,17, 26, 31,32 trans-mercaptosulfonamides were most effective (18a vs. 23c). It is possible that replacement of the prime side (i.e., S1′) peptidomimetic groups with 4-phenoxybenzene, as seen in 23c, necessitates different stereochemistry to maximize binding energy. Such modifications may have arranged the MMPIs in such a way that cis-enantiomers are no longer in the correct plain for coordination. We are currently further evaluating this finding.
Biological Studies
Early applications of thiol ZBGs were hampered by their metal-binding promiscuity and susceptibility to metabolic transformations. For example, stability assays with non-cyclic mercaptosulfides demonstrated significant sensitivity to air-oxidation. Yet, the incorporation of cyclopentyl and pyrrolidinyl rings at the P1 position of mercaptosulfonamides increased inhibitor half-life. When compared to another thiol-based chelator, N-(methyl)mercaptoacetamide, where 91% of the thiol was oxidized within the first 8 min,56 our mercaptosulfonamides appear dramatically more stable. It should be noted however, that in contrast to a previous report indicating that thiol stability varied depending on ring structure, i.e., position of endocyclic heteroatoms, and ring substituents,56 no preference was discovered during our investigation.
The culture media utilized with these assays contains approximately four times the amount of serum than other typical media. Consequently, it was not surprising to observe a “plateau” towards the later time points in each run. We attribute this to the MMPIs being sequestered by the serum albumin and subsequently released in a dynamic equilibrium of some form. Additional studies are being conducted to further investigate this effect.
Finally, cytotoxicity analyses revealed that these compounds exhibit only moderate toxicity (i.e., killing ≤ 20% of cells)--up to a 100 LM concentration. This differs from an earlier report, in part, which indicated that sulfur-containing compounds are generally more cytotoxic.56 In comparison to a recent study evaluating the effects of the aminobisphosphate alendronate, the mercaptosulfonamides display far less cytotoxicity, as bisphosphonates were toxic to all cells at concentrations ten-fold lower than those typically observed for the mercaptosulfonamides.57
Hydroxamate-based inhibitors are quite successful for some other zinc metalloproteinases, like SAHA (suberoylanilide hydroxamic acid) for histone deacetylases.47, 58-62 Even though the popularity of MMPIs as putative therapeutics was diminished due to the notable failures of hydroxamate-based MMPIs in oncology clinical trials, they are still valuable investigative tools for MMP functional studies. For example, preliminary evidence from our lab indicates that several of these mercaptosulfonamides are capable of reducing the adipogenic potential of human mesenchymal stem cells, suggesting unique roles of MMPs in modulating the fate of adult stem cells. Moreover, selectively targeting individual MMPs in a variety of disease models will inevitably provide vital information about the functions of these enzymes, information that may prove critical to identifying possible treatment options.
EXPERIMENTAL SECTION
General Information
All chemicals, reagents, and solvents for syntheses were analytical grade, purchased from commercial sources. Solvents were purified according to standard procedures, and all air and moisture-sensitive reactions were performed under nitrogen. 1H spectra were collected on a Bruker ARX 300 NMR spectrometer with chemical shifts in parts per million (ppm) downfield from tetramethylsilane as an internal standard. Mass spectrometry data was collected on a Jeol JMS-600H, applying ESI. Melting points were determined on a Buchi melting point apparatus and are uncorrected. Column chromatography was performed on Merck silica gel 60. The reactions were monitored by TLC using Merck 60 F254 silica gel glass-backed plates; zones were detected visually under UV irradiation (254 nm). Combustion analysis was performed by Atlantic Microlab, Inc., Norcross, GA, USA. Purity of key compounds was established by elemental analysis and/or Ellman’s reagent thiol titration, determined to be >95%.
Pyrrolidine Mercaptosulfides
The inhibitors listed in Table 1 (see Figure 2 for structures) were synthesized as diastereo meric mixtures as described,17 and as outlined in Scheme 1; their characterization data follows:
11a. 1H NMR (300 MHz, CD3OD) δ 7.25 (m, 5H), 4.66 (m, 1H), 4.05 (m, 0.5H), 3.83-3.00 (m, 7.5H), 2.87 (m, 1H), 2.72 (s, 1.5H), 2.71 (s, 1.5H), 1.57 (m, 1H), 1.26 (m, 1H), 1.16 (m, 1H), 0.76 (m, 6H).
11b. 1H NMR (300 MHz, CD3OD) δ 7.23 (m, 5H), 4.72 (m, 1H), 4.10-3.20 (m, 7H), 3.11 (m, 1H), 2.78 (m, 1H), 2.72 (m, 3H), 2.03 (m, 3H), 1.59 (m, 2H), 1.25 (m, 1H), 0.77 (m, 6H).
11c. 1H NMR (300 MHz, CDCl3) δ 7.87 (m, 2H), 7.74 (m, 2H), 7.40-7.15 (m, 5H), 7.01 (m, 0.5H), 6.93 (m, 0.5H), 6.00 5.78 (m, 1H), 4.67 (m, 1H), 4.40 (s, 1H), 4.36 (s, 1H), 4.00-3.00 (m, 8H), 2.76 (m, 3H), 2.06 (m, 0.5H, SH), 1.95 (m, 0.5H, SH), 1.67 (m, 2H), 1.50 (m, 1H), 0.88 (m, 6H). Anal. Calc’d for C30H36N4O5S2•¼H2O: C, 59.93; H, 6.12; N, 9.32; S, 10.66. Found: C, 59.94; H, 5.92; N, 9.10; S, 10.41.
11d. 1H NMR (300 MHz, CD3OD) δ 7.82 (m, 4H), 7.22 (m, 5H), 4.67 (m, 1H), 3.84-3.0 (m, 9H), 3.10 (m, 1H), 2.85 (m, 1H), 2.70 (s, 1.5H), 2.69 (s, 1.5H), 2.36 (m, 2H), 1.98 (m, 2H), 1.59 (m, 1H), 1.24 (m, 2H), 0.77 (m, 6H).
11e. 1H NMR (300 MHz, CD3OD) δ 7.80 (m, 4H), 7.24 (m, 5H), 4.69 (m, 1H), 3.85-2.80 (m, 11H), 2.70 (s, 1.5H), 2.69 (s, 1.5H), 2.28 (m, 2H), 1.65 (m, 5H), 1.32 (m, 4H), 0.78 (m, 6H).
2-Mercaptocyclopentane Arylsulfonamides (Scheme 2 and Figure 3)
(1S,2S)-(+)- and (1R,2R)-(−)-trans-2-Azidocyclopentanol
A modified version of the procedure of Ami and Ohrui was used.38 To a stirred solution of (±)--trans-2-azidocyclopentanol (1.90 g, 14.9 mmol) in tert-butyl methyl ether (75 mL) was added isopropenyl acetate (3.3 g, 33.0 mmol) and Lipase Amano AK-20 (4.5 g) (Amano Enzyme Inc, Japan), and the mixture was stirred at room temperature for three days. The reaction mixture was filtered, the filtrate was evaporated under reduced pressure, and the residue was subjected to flash chromatography on silica gel. Elution with 50% ethyl acetate in hexane gave (1R,2R)-trans-2-azidocyclopentyl acetate (14) as an oil (1.26 g, 7.45 mmol, 50%): 1H NMR (300 MHz, CDCl3) δ 4.96 (dt, J=7, 4, 1H), 3.87 (m, 1H), 2.05 (s, 3H), 1.92 2.18 (m, 2H), 1.57-1.83 (m, 4H); [α]20D = 56.7° (c 1.2, CH2Cl2). Continued elution with 50% ethyl acetate in hexane afforded (1S,2S)-trans-2-azidocyclopentanol (13) as an oil (0.93 g, 49%): 1H NMR δ 4.2 (m, 1H), 3.7 (m, 1H) 1.9-2.2 (m, 2H), 1.5 1.9 (m, 5H); [α]20D = +76.0° (c 1.1, CH2Cl2), (lit.63 [α]20D = +84.0°, (c 1.1, CH2Cl2)).
To a stirred solution of (1R,2R)-(−)-trans-2-azidocyclopentyl acetate (14) (610 mg, 3.61 mmol) in THF-MeOH (3:1, 15 mL) at 0°C was added 1N LiOH (6 mL) dropwise. The reaction mixture was stirred at room temperature until no starting material was detectable by TLC. The reaction mixture was evaporated under reduced pressure and ethyl ether (50 mL) was added to the residue. The solution was washed with water, dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (1R,2R)-trans-2-azidocyclopentanol (15) as an oil (450 mg, 98%): 1H NMR identical to that of the (1S,2S) enantiomer; [α]20D = −84.3° (c 1.1, CH2Cl2).
(1R,2S)-(+)- and (1S,2R)-(−)-cis-2-Azidocyclopentanol
To a stirred solution of Ph3P (1.55 g, 5.91 mmol) in THF (25 mL) at 0°C was added DEAD (1.03 mL, 6.54 mmol) dropwise. After 10 min, a solution of (1S,2S)-(+)-trans-2-azidocyclopentanol (13) (500 mg, 3.93 mmol) and p-nitrobenzoic acid (985 mg, 5.89 mmol) in THF (5 mL) was added dropwise and the reaction mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to give (1R,2S)-cis-2-azidocyclopentyl p-nitrobenzoate (19) as a solid (1.00 g, 92%): mp 51.5-52.0°C; 1H NMR (300 MHz, CDCl3) δ 8.33 (d, J = 9, 2H), 8.27 (d, J = 9, 2H), 5.42 (dt, J = 6, 5, 1H), 4.00 (dt, J = 5, 6, 1H), 1.88-2.25 (m, 5H), 1.65 1.83 (m, 1H); [α]20D = −97.3° (c 0.59, CH2Cl2).
To a stirred solution of (1R,2S)-(−)-cis-2-azidocyclopentyl p-nitrobenzoate (19) (500 mg, 1.81 mmol) in THF-MeOH (3:1, 10 mL) at 0°C was added 1N LiOH (3 mL) dropwise. The reaction mixture was stirred at room temperature until no starting material was detectable by TLC. The reaction mixture was evaporated under reduced pressure and ethyl ether (25 mL) was added to the residue. The solution was washed with water, dried over Na2SO4, and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (1R,2S)-cis-2-azidocyclopentanol (20) as an oil (225 mg, 98%): 1H NMR (300 MHz, CDCl3) δ 4.13 (br p, J = 5, 1H), 3.78 (dt, J = 5, 6, 1H), 1.78-2.00 (m, 4H), 1.53-1.75 (m, 3H); [α]20D = +66.7° (c 1.44, CH2Cl2).
Using the same procedures, (1R,2R)-(−)-trans-2-azidocyclopentanol (15) was converted to (1S,2R)-cis-2-azidocyclopentyl p-nitrobenzoate (19 enantiomer) (mp 51.5°C, [α]20D = +97.9° (c 1.22, CH2Cl2)) and then to (1S,2R)-cis-2-azidocyclopentanol (20 enantiomer): 1H NMR identical to that of the (1R,2S) enantiomer; [α]20D = −64.4° (c 1.12, CH2Cl2).
(±)-, (1S,2S)-(−)-, and (1R,2R)-(+)-trans-2-(tert-Butoxycarbonylamino)cyclopentanol
To a stirred solution of (±)-trans-2-azidocyclopentanol (12) (1.27 g, 10.0 mmol) in THF-H2O (4:1, 10 mL) was added Ph3P (2.88 g, 12.0 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C for 2 h. The solution was cooled with stirring to 0°C and Et3N (1.4 mL, 10.0 mmol) was added, then a solution of di-tert-butyl dicarbonate (2.18 g, 10.0 mmol) in THF (5 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue and the organic layer was washed with 10% citric acid and saturated aq NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (±)-trans-2-(tert-butoxycarbonylamino)cyclopentanol ((±)-16) (1.8 g, 90%): 1H NMR (300 MHz, CDCl3) δ 4.67 (br s, 1H), 3.98 (m, 2H), 3.62 (m, 1H), 1.93-2.18 (m, 2H), 1.59-1.85 (m, 3H), 1.45 (s, 9H), 1.27-1.41 (m, 1H).
By the same procedure, (1S,2S)-(+)-trans-2-azidocyclopentanol (13) gave (1S,2S)-trans-2-(tert-butoxycarbonylamino)cyclopentanol (16) as a solid (370 mg, 73%): mp 81°C; 1H NMR (300 MHz, CDCl3) identical to that of (±)-16; [α]20D = −23.6° (CH2Cl2) (lit.63 mp 87°C, [α]20D = −21.0° (c 1.0, CH2Cl2)).
By the same procedure, (1R,2R)-(−)-trans-2-azidocyclopentanol (15) (160 mg, 1.26 mmol) afforded (1R,2R)-trans-2-(tert-butoxycarbonylamino)cyclopentanol (16 enantiomer) (180 mg, 71%): mp 81°C; 1H NMR (300 MHz, CDCl3) identical to that of (±)-16; [α]20D = +23.4° (CH2Cl2) (lit.63 mp 87°C, [α]20D = +21.0° (c 1.0, CH2Cl2)).
(1R,2S)-(+)-cis-2-(4-Phenoxybenzenesulfonamido)cyclopentanethiol (18c)
To a stirred solution of Ph3P (393 mg, 1.50 mmol) in THF (5 mL) at 0°C was added DEAD (236 μL, 1.50 mmol). After 10 min, solutions of (1S,2S)-(−)-trans-2-(tert-butoxycarbonylamino)cyclopentanol (16) (166 mg, 0.826 mmol) and thiolacetic acid (107 μL, 1.50 mmol) in THF (2 mL) were added. The reaction mixture was stirred at room temperature overnight, then it was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (10% ethyl acetate in hexane) to give (1R,2S)-(+)-cis-2-(tert-butoxycarbonylamino)cyclopentanethioacetate (17) (170 mg, 79%) as a solid: mp 72°C; 1H NMR (300 MHz, CDCl3) δ 4.57 (br s, 1H), 4.18 (br s, 1H), 3.95 (q, J = 6, 1H), 2.33 (s, 3H), 2.09-2.21 (m, 1H), 1.93-2.09 (m, 1H), 1.59-1.81 (m, 3H), 1.44 (s, 9H), 1.35 1.53 (m, 1H); [α]20D = +34.7° (CH2Cl2).
To a stirred solution of 17 (35 mg, 0.135 mmol) in THF (0.5 mL) was added 40% aqueous MeNH2 (120 μL, 3.47 mmol). After 20 min the reaction mixture was concentrated under reduced pressure and the residue was dissolved in trifluoroacetic acid (0.5 mL), and the mixture was stirred for 2 h. The solvent was removed under reduced pressure and the residue was dissolved in CH2Cl2 (2 mL). The solution was cooled to 0°C and Et3N (38 μL, 0.273 mmol) and 4-phenoxy benzenesulfonyl chloride (31.5 mg, 0.135 mmol) were added successively. The reaction mixture was stirred at 0°C for 3 h, then it was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to give 18c as a solid (41 mg, 87%): mp 112-113°C; 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J= 9, 2H), 7.42 (t, J = 8, 2H), 7.23 (t, J = 8, 1H), 7.06 (m, 4H), 5.02 (d, J = 8, 1H), 3.62 (m, 1H), 3.23 (m, 1H), 2.06 (m, 1H), 1.5-1.8 (m, 5H), 1.33 (d, J = 7, 1H); [α]20D = +1.30° (CH2Cl2); HRMS (ESI) calculated for C17H19NO3S2Na+ 372.0699, found 372.0703.
(1S,2R)-(−)-cis-2-(4-Phenoxybenzenesulfonamido)cyclopentanethiol (18b)
Following the Ph3P/DEAD procedure described above, (1R,2R)-(+)-trans-2-(tert-butoxycarbonylamino)cyclopentanol (151 mg, 0.75 mmol) was converted to (1S,2R)-cis-2-(tert-butoxycarbonylamino)cyclopentanethioacetate as a solid (177 mg, 91%): mp 72°C; 1H NMR (300 MHz, CDCl3) identical to that of the (1R,2S) enantiomer 17; [α]20D = −34.8° (CH2Cl2).
To a stirred solution of 1S,2R-(−)-cis-2-(tert-butoxycarbonylamino)cyclopentanethioacetate (50 mg, 0.193 mmol) in MeOH (0.5 mL) was added 2N HCl in MeOH (2 mL), and the mixture was stirred at room temperature for 2 h. The solvent was removed under reduced pressure and the residue was dissolved in CH2Cl2 (2 mL). The solution was cooled to 0°C and Et3N (65 μL, 0.466 mmol) and 4-phenoxybenzenesulfonyl chloride (45.0 mg, 0.193 mmol) were added successively. The mixture was stirred at 0°C for 3 h, then it was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to give 18b as a solid (52 mg, 77%): mp 112-113°C; 1H NMR identical to that of 18c; [α]20D = −1.30° (CH2Cl2); HRMS (ESI) calculated for C17H19NO3S2Na+ 372.0699, found 372.0703.
(±)-cis-2-(4-Phenoxybenzenesulfonamido)cyclopentanethiol (18a)
By the same procedures, (±)-trans-2-azidocyclopentanol (12) afforded the racemic thiol 18a as a solid: mp 113-114°C; 1H NMR (300 MHz, CDCl3) identical to that of each of the enantiomers.
(±)-trans-2-(Benzenesulfonamido)cyclopentanethiol (23a)
To a stirred solution of Ph3P (1.4 g, 5.3 mmol) in THF (25 mL) at 0°C was added DEAD (0.87 g, 5.0 mmol) dropwise. After 10 min, (±)-trans-2-(tert-butoxycarbonylamino)cyclopentanol ((±)-16) (1.0 g, 5.0 mmol) was added dropwise and the reaction mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel (10% ethyl acetate in hexane) to give the aziridine N tert-butoxycarbonyl-6-azabicyclo[3.1.0]pentane as an oil (0.80 g, 87%): 1H NMR (300 MHz, CDCl3) δ 2.04-2.16 (m, 2H), 1.53-1.64 (m, 4H), 1.46 (s, 9H), 1.15-1.32 (m, 2H).
To a stirred solution of the aziridine (550 mg, 3.0 mmol) in MeOH (20 mL) were added tert-butylthiol (0.5 mL, 4.4 mmol) and a solution of sodium methoxide (162 mg, 3.0 mmol) in MeOH (5 mL), and the reaction mixture was stirred at room temperature overnight. Water was added to the solution and the mixture was evaporated under vacuum. The residue was dissolved in ethyl acetate (50 mL) and the organic layer was washed with water, dried over Na2SO4, and evaporated to give the crude (±)-trans-S-tert-butyl-2-(tert-butoxycarbonylamino)cyclopentane thiol as an oil (820 mg, 100%): 1H NMR (300 MHz, CDCl3) δ 4.60 (broad, 1H), 3.64 (m, 1H). 2.80 (m, 1H), 2.10-2.25 (m, 2H), 1.60 1.75 (m, 4H), 1.45 (s, 9H), 1.36 (s, 9H).
A sample of the oil (82 mg, 0.30 mmol) was dissolved in trifluoroacetic acid (1 mL) and the solution was stirred at room temperature until no starting material remained by TLC analysis. The trifluoroacetic acid was evaporated under vacuum and the residue was dissolved in CH2Cl2 (5 mL). The solution was cooled to 0°C and Et3N (124 μL, 0.88 mmol) and benzenesulphonyl chloride (42 μL, 0.33 mmol) were added. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue and the organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (±)-trans-S-tert-butyl-2-(benzenesulphonamido)cyclopentanethiol (90 mg, 100%): 1H NMR (300 MHz, CDCl3) δ 7.85-7.89 (m, 2H), 7.48-7.61 (m, 3H), 4.75 (br d, J = 3, 1H, NH), 3.01 (dq, J = 4, 8, 1H), 2.81 (q, J = 8, 1H), 2.00-2.22 (m, 2H), 1.44 1.70 (m, 4H), 1.28 (s, 9H).
To a solution of the (±)-trans-S-tert-butyl-2-(benzenesulphonamido)cyclopentanethiol (90 mg, 0.30 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (62 mg, 0.33 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum and to the residue (82 mg, ~0.2 mmol) was added THF (2 mL), 1N NaOH (0.4 mL), and tris(2-carboxyethyl)phosphine (TCEP, 63.1 mg, 0.22 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was condensed under vacuum. The residue was dissolved in ethyl acetate, dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (25% ethyl acetate in hexane) to give the thiol 23a (45 mg, 58%): 1H NMR (300 MHz, CDCl3) δ 7.90-7.94 (m, 2H), 7.50-7.63 (m, 3H), 4.67 (br d, J = 6, 1H, NH), 3.25 (p, J = 7.5, 1H), 2.90 (p, J = 7.5, 1H), 1.99-2.19 (m, 2H), 1.63-1.74 (m, 2H), 1.59 (d, J = 7, 1H, SH), 1.33-1.52 (m, 2H); HRMS (ESI) calculated for C11H15NO2S2Na+ 280.0436, found 280.0442.
(±)-trans-2-(4-Phenylbenzenesulfonamido)cyclopentanethiol (23b)
A sample of the crude (±)-trans-S-tert-butyl-2-(tert-butoxycarbonylamino)cyclopentanethiol (59 mg, 0.214 mmol) from the previous experiment was dissolved in trifluoroacetic acid (1 mL) and was stirred at room temperature until no starting material remained by TLC analysis. The trifluoroacetic acid was evaporated under vacuum and the residue was dissolved in CH2Cl2 (5 mL). The solution was cooled to 0°C and Et3N (65.6 μl, 0.47 mmol) and 4-phenylbenzenesulphonyl chloride (46.6 mg, 0.214 mmol) were added. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue and the organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was filtered through a short column of silica gel (50% ethyl acetate in hexane). The resulting crude product was dissolved in AcOH (1 mL), 2-nitrobenzenesulfenyl chloride (41 mg, 0.24 mmol) was added, and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum to give the crude disulfide that was used without further purification. To the crude disulfide was added THF (1 mL), 1 N NaOH (0.23 mL), and TCEP (63.1 mg, 0.22 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was condensed under vacuum. To the residue was added ethyl acetate, and the mixture was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (25% ethyl acetate in hexane) to give 23b (66 mg, 0.198 mmol, 93%): 1H NMR (300 MHz, CDCl3) δ 7.96 (d, J = 8, 2H), 7.74 (d, J = 8, 2H), 7.62 (d, J = 8, 2H), 7.39-7.52 (m, 3H), 4.69 (br d, J = 5, 1H, NH), 3.29 (p, J = 8, 1H), 2.93 (p, J = 8, 1H), 2.03 2.22 (m, 2H), 1.64-1.75 (m, 2H), 1.62 (d, J = 7, 1H, SH), 1.36-1.54 (m, 2H); HRMS (ESI) calculated for C17H19NO2S2Na+ 356.0749, found 356.0755.
(1R,2S)-(−)- and (1S,2R)-(+)-cis-2-(4-Phenoxybenzenesulfonamido)cyclopentanol
To a stirred solution of (1R,2S)-(+)-cis-2-azidocyclopentanol (20) (414 mg, 3.26 mmol) in THF-H2O (4:1, 5 mL) was added Ph3P (897 mg, 3.42 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C for 2 h. The solution was cooled with stirring to 0°C and NaHCO3 (310 mg, 3.69 mmol) was added, then a solution of 4-phenoxybenzenesulfonyl chloride (760 mg, 3.26 mmol) in THF (5 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue and the organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (1R,2S)-cis-2-(4-phenoxybenzenesulfonamido)cyclopentanol as a solid (420 mg, 75%): mp 110°C; 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 9, 2H), 7.42 (t, J = 8, 2H), 7.23 (t, J = 8, 1H), 7.06 (m, 4H), 4.87 (d, J = 8, 1H), 4.03 (m, 1H), 3.42 (m, 1H), 1.7-1.9 (m, 4H), 1.4 1.7 (m, 3H); [α]20D = −22.0° (c 1.00, CH2Cl2).
Application of the same procedure to (1S,2R)-(−)-cis-2-azidocyclopentanol (20 enantiomer) afforded (1S,2R)-cis-2-(4-phenoxybenzenesulfonamido)cyclopentanol: mp 110°C; 1H NMR identical to that of the (1R,2S) enantiomer; [α]20D = +22.3° (c 1.05, CH2Cl2).
(±)-, (1S,2S)-(+)-, and (1R,2R)-(−)-trans-2-(4-Phenoxybenzenesulfonamido)cyclopentanethiol (23c, 23d, and 23e)
To a stirred solution of Ph3P (262 mg, 1.0 mmol) in THF (5 mL) at 0°C was added DEAD (157 μL, 1.0 mmol). After 10 min, a solution of (1R,2S)-(−)-cis-2-(4-phenoxybenzenesulfonamido)cyclopentanol (166 mg, 0.50 mmol) and thiobenzoic acid (118 μL, 1.0 mmol) in THF (2 mL) was added. The reaction mixture was stirred at room temperature overnight, then it was concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to give (1S,2S)-trans-2-(4-phenoxy benzenesulfonamido)cyclopentanethiobenzoate as a solid (180 mg, 79%): mp 132-133°C; 1H NMR (300 MHz, CDCl3) δ 7.82 (d, J = 8, 2H), 7.72 (d, J = 9, 2H), 7.59 (t, J = 8, 1H), 7.43 (t, J = 8, 2H), 7.36 (t, J = 8, 2H), 7.19 (t, J = 8, 1H), 6.93 (d, J = 9, 2H), 6.77 (d, J = 9, 2H), 5.54 (br d, 1H), 3.73 (q, J = 9, 1H), 3.42 (m, 1H), 2.1-2.3 (m, 2H), 1.6-1.9 (m, 4H); [α]20D = −140.6° (c 0.65, CH2Cl2).
To a stirred solution of (1S,2S)-(−)-trans-2-(4-phenoxybenzenesulfonamido)cyclopentanethio benzoate (80 mg, 0.176 mmol) in THF (1 mL) under N2 was added 40% aqueous MeNH2 (900 μL, 10.5 mmol) and the reaction mixture was stirred for 20 min. The solution was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to give (1S,2S)-(+)-trans-2-(4-phenoxybenzenesulfonamido)cyclo pentanethiol (23d) as a solid (60 mg, 98%): mp 133-134°C; 1H NMR (300 MHz, CDCl3) δ 7.84 (d, J = 9, 2H), 7.42 (t, J = 8, 2H), 7.23 (t, J = 8, 1H), 7.06 (m, 4H), 4.53 (br d, 1H), 3.23 (pentet, J = 7.5, 1H), 2.91 (p, J = 7.5, 1H), 2.03 2.21 (m, 2H), 1.64-1.76 (m, 2H), 1.60 (d, J = 7, 1H), 1.34-1.53 (m, 2H); [α]20D = +16.9° (c 1.00, CH2Cl2).
By the same procedure, (1S,2R)-(+)-cis-2-(4-phenoxybenzenesulfonamido)cyclopentanol (180 mg, 0.54 mmol) was converted to (1R,2R)-trans-2-(4-phenoxybenzenesulfonamido)cyclopentanethiobenzoate (170 mg, 69%): mp 132-133°C; [α]20D = +142.4° (1.05, CH2Cl2). NMR analysis of the product in presence of the chiral shift reagents (europium[3] tris-trifluoromethylhydroxymethylene-(+)-camphorate and europium[3] tris-heptafluoropropylhydroxymethylene-(+)-camphorate) at both 300 MHz and 400 MHz indicated it to be in >90% ee.
The (1R,2R)-(+)-trans-2-(4-phenoxybenzenesulfonamido)cyclopentanethiobenzoate (85 mg, 0.187 mmol) was hydrolyzed as above to give (1R,2R)-(−)-trans-2-(4-phenoxybenzenesulfonamido)cyclopentanethiol 23e as a solid (61 mg, 93%): mp 133-134°C; 1H NMR (300 MHz, CDCl3) identical to that of 23d; [α]20D = −17° (c 1.00, CH2Cl2).
By the same procedures, (±)-trans-2-azidocyclopentanol afforded the racemic thiol 23c as a solid: mp 156-157°C; 1H NMR (300 MHz, CDCl3) identical to that of each of the enantiomers; HRMS (ESI) calculated for C17H19NO3S2Na+ 372.0699, found 372.0704.
(±)-trans-2-(4-Phenoxybenzamido)cyclopentanethiol (23g)
To a stirred solution of cyclopentane oxide (1.57 g, 18.7 mmol) in MeOH (20 mL) was added t-Bu thiol (2.1 mL, 18.7 mmol) and a solution of sodium methoxide (1.01 g, 18.7 mmol) in MeOH (10 mL), and the reaction mixture was stirred at room temperature overnight. Water was added to the solution and the mixture was evaporated under vacuum. The residue was dissolved in ethyl acetate (50 mL) and the organic layer was washed with water, dried over Na2SO4, and evaporated to give (±)-trans-2-tert-butylmercaptocyclopentanol (3.00 g, 92%): 1H NMR (300 MHz, CDCl3) δ 3.90 (q, J = 7, 1H), 2.84 (dt, J = 7, 8, 1H), 2.24 (m, 1H), 1.99 (m, 1H), 1.94 (br s, 1H, OH), 1.73 (m, 2H), 1.61 (m, 2H), 1.36 (s, 9H).
To a stirred solution of Ph3P (2.7 g, 10.3 mmol) in THF (25 mL) at 0°C was added DEAD (1.8 g, 10.3 mmol) dropwise. After 10 min, (±)-trans-2-tert-butylmercaptocyclopentanol (1.64 g, 9.36 mmol) in THF (5 mL) and hydrazoic acid (1.0 g in 10 mL of benzene, 25 mmol) were added dropwise, and the reaction mixture was stirred at room temperature overnight. The mixture was concentrated under reduced pressure and the residue was purified by flash chromatography on silica gel (20% ethyl acetate in hexane) to (±)-trans-2-tert-butylmercaptocyclopentyl azide as an oil (1.3 g, 70%): 1H NMR (300 MHz, CDCl3) δ 3.75 (m, 1H), 2.98 (m, 1H), 2.25 (m, 1H), 2.02 (m, 1H), 1.70 (m, 4H), 1.37 (s, 9H).
To a stirred solution of the azide (470 mg, 2.35 mmol) in THF-H2O (4:1, 10 mL) was added Ph3P (696 mg, 2.65 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C for 2 h. The solution was cooled with stirring to 0°C and Et3N (3.93 μL, 4.79 mmol) was added, then a solution of 4-phenoxybenzoyl chloride (546 mg, 2.35 mmol) in THF (5 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue and the organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give (±)-trans-S-tert-butyl-2-(4-phenoxybenzamido)cyclopentanethiol (0.67 g, 77%): 1H NMR (300 MHz, CDCl3) δ 7.73 (d, J = 9, 2H), 7.38 (t, J = 8, 2H), 7.17 (t, J = 7, 1H), 7.04 (d, J = 8, 2H), 7.00 (d, J = 9, 2H), 6.10 (br d, J = 4, 1H, NH), 4.06 (p, J = 7, 1H), 3.08 (q, J = 7, 1H), 2.20-2.40 (m, 2H), 1.66 1.87 (m, 3H), 1.52-1.66 (m, 1H), 1.38 (s, 9H).
To a solution of (±)-trans-S-tert-butyl-2-(4-phenoxybenzamido)cyclopentanethiol (211 mg, 0.57 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (108 mg, 0.57 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum, and to the residue (212 mg, 0.455 mmol) were added THF (4 mL), 1N NaOH (0.91 mL), and TCEP (144 mg, 0.50 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was condensed under vacuum. To the residue was added ethyl acetate and it was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (25% ethyl acetate in hexane) to give 23g (135 mg, 76%): 1H NMR (300 MHz, CDCl3) δ 7.75 (d, J = 9, 2H), 7.38 (t, J = 8, 2H), 7.17 (t, J = 7.5, 1H), 7.02 (m, 4H), 6.01 (br d, J = 7, 1H, NH), 4.19 (p, J = 7.5, 1H), 3.10 (dt, J = 7, 8, 1H), 2.30-2.42 (m, 1H), 2.15-2.29 (m, 1H), 1.88 (d, J = 6, 1H, SH), 1.45-1.85 (m, 4H); HRMS (ESI) calculated for C18H19NO2SNa+ 336.1029, found 336.1034.
(±)-trans-2-(4-Phenoxybenzenesulfonamido)cyclopentanol (23f)
To a stirred solution of (±)-trans-2-azidocyclopentanol (553 mg, 4.35 mmol) in THF-H2O (4:1, 10 mL) was added Ph3P (1.26 g, 4.79 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C for 2 h. The solution was cooled with stirring to 0°C and Et3N (0.67 mL, 4.79 mmol) was added, then a solution of 4-phenoxybenzenesulfonyl chloride (1.01 g, 4.35 mmol) in THF (5 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure and ethyl acetate was added. The organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography on silica gel (50% ethyl acetate in hexane) to give 23f as a solid (0.93g, 71%): mp 98°C; 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 9, 2H), 7.42 (t, J = 8, 2H), 7.24 (t, J = 8, 1H), 7.06 (m, 4H), 4.8 (br, 1H, NH), 4.06 (q, J = 6, 1H), 3.24 (m, 1H), 1.88-2.03 (m, 2H), 1.49-1.72 (m, 4H), 1.30-1.43 (m, 1H); HRMS (ESI) calculated for C17H19NO4SNa+ 356.0927, found 356.0933.
trans-3-Mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidines (Scheme 3 and Figure 4, Ar = -C6H4-O-C6H5)
(±)-N-Boc-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (25)
A mixture of N-Boc-3-pyrroline oxide (3) (0.925 g, 5.0 mmol), NaN3 (0.65 g, 10 mmol), and NH4Cl (5.0 mmol) in MeOH-H2O (6:1, 16 mL) was stirred at 65°C overnight. The reaction mixture was concentrated under reduced pressure and the residue was extracted thoroughly with ethyl acetate. The combined ethyl acetate extract was washed with water and saturated brine, dried over anhydrous Na2SO4, and evaporated. The crude (±)-N-Boc-trans-3-hydroxy-4-azidopyrrolidine (24) was purified by recrystallization from ether-hexane: mp 34-36°C; 1H NMR (300 MHz, CD3OD) δ 4.16 (m, 1H), 3.95 (m, 1H), 3.62 (m, 1H), 3.51 (m, 1H), 3.35 (m, 2H), 1.47 (s, 9H).
To a stirred solution of the azidoalcohol 24 (1.02 g, 4.45 mmol) in THF-H2O (10:1, 10 mL) was added Ph3P (1.28 g, 4.88 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C for 2 h. The solution was cooled with stirring to 0°C, Et3N (0.74 mL, 5.34 mmol) was added, then a solution of 4-phenoxybenzenesulfonyl chloride (1.04 g, 4.45 mmol) in THF (5 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue, and the organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (±)-N-Boc-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (25) as a solid (1.52 g, 79%): mp 98 99°C; 1H NMR (300 MHz, CD3OD) δ 7.85 (d, J = 8, 2H), 7.45 (m, 2H), 7.24 (t, J = 7, 1H), 7.10 (m, 4H), 4.09 (m, 1H), 3.95 (m, 1H), 3.70 (m, 2H), 3.40 (m, 2H), 1.42 (s, 9H); MS, ESI m/e 434.1 (M+); HRMS (ESI) calculated for C21H26N2O6SNa+ 457.1391, found 457.1391.
(±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27)
To a stirred solution of Ph3P (0.88 g, 3.35 mmol) and (±)-N-Boc-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (25) (1.316 g, 3.04 mmol) in THF (20 mL) at 0°C was added DEAD (0.58 mL, 3.33 mmol) dropwise. The reaction mixture was stirred at room temperature overnight, then it was evaporated under reduced pressure. The residue was purified by flash chromatography (30% ethyl acetate in hexane) to give the N-(4-phenoxybenzenesulfonyl)aziridine 26 as a solid (1.23 g, 98%): mp 136-138°C; 1H NMR (300 MHz, CDCl3) δ 7.88 (m, 2H), 7.43 (m, 2H), 7.22 (m, 1H), 7.10 (m, 4H), 3.72 (m, 2H), 3.59 (m, 1H), 3.47 (m, 1H), 3.38 (m, 2H), 1.42 (s, 9H); MS, ESI m/e 416.1 (M+); HRMS (ESI) calculated for C21H24N2O5SNa+ 439.1290, found 439.1290.
To a stirred solution of the aziridine 26 (416 mg, 1.0 mmol) and tert-butyl mercaptan (0.15 mL, 1.2 mmol) in anhydrous MeOH (5 mL) at 0°C was added t-BuOK (0.2 mmol). The mixture was stirred at room temperature for 4 h, then it was evaporated under reduced pressure. Ethyl acetate was added to the residue and the solution was extracted with water and saturated brine, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography (20% ethyl acetate in hexane) to give (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) as a solid (506 mg, 98%): mp 85-86°C; 1H NMR (300 MHz, CD3OD) δ 7.85 (d, J = 7, 2H), 7.45 (t, J = 7, 2H), 7.25 (t, J = 7, 1H), 7.09 (d, J = 7, 4H), 3.81 (m, 2H), 3.58 (m, 1H), 3.47 (m, 1H), 3.22 (m,1H), 3.09 (m, 1H), 1.45 (s, 9H), 1.29 (s, 9H); MS, ESI m/e 506.2 (M+); HRMS (ESI) calculated for C25H34N2O5S2Na+ 529.1796, found 529.1795.
(±)-trans-3-Mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine hydrochloride (29a)
To a solution of (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) (117 mg, 0.226 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (46.0 mg, 0.243 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum, and to the residue were added THF (4 mL), 1N NaOH (0.25 mL), and TCEP (67 mg, 0.234 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was condensed under vacuum. To the residue was added ethyl acetate and it was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (ethyl acetate) to give (±)-N-Boc-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine as a white solid (82 mg, 81%): mp 78-79°C; 1H NMR (300 MHz, CD3OD) δ 7.88 (m, 2H), 7.45 (m, 2H), 7.25 (t, J = 7, 1H), 7.10 (m, 4H), 3.74 (m, 1H), 3.55 (m, 2H), 3.17 (m, 2H), 3.00 (m, 1H), 1.44 (s, 9H); MS (ESI) m/e 450.2 (M+); HRMS (ESI) calculated for C21H26N2O5S2Na+ 473.1172, found 473.1170.
To a solution of 2.5M HCl in AcOH (1 mL) was added (±)-N-Boc-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (45 mg, 0.10 mmol) and the mixture was stirred at room temperature for 3 h. Liophylization of the solution gave the product 29a as a white powder (38.7 mg, 100%): 1H NMR (300 MHz, CD3OD) δ 7.87 (d, J = 12, 2H), 7.44 (m, 2H), 7.24 (t, J = 7, 1H), 7.12 (m, 4H), 3.72 (m, 1H), 3.62 (m, 2H), 3.37 (m, 1H), 3.19 (m, 2H); MS (ESI) m/e 351.1 (MH+); HRMS (ESI) calculated for C16H19N2O3S2+ 351.0837, found 351.0837.
(±)-N-Carbamoyl-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (29b)
To a stirred solution of 1.5 M HCl in ethyl acetate (5 mL) was added (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) (155 mg, 0.3 mmol) and stirring was continued at room temperature until no starting material was detected by TLC (50% ethyl acetate in hexane). The solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2. The solution was cooled to 0°C and Et3N (46 μl, 33 mmol) and N-trimethylsilyl isocyanate (TMSNCO) (100 μL, 0.74 mmol) were added successively. The solution was stirred at room temperature overnight. The reaction mixture was extracted with water and saturated brine, and was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (5% MeOH in ethyl acetate) to give (±)-N-carbamoyl-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (130 mg, 96%): mp 132-133°C; 1H NMR (300 MHz, CD3OD) δ 7.85 (d, J = 9, 2H), 7.44 (t, J = 8, 2H), 7.24 (t, J = 8, 1H), 7.10 (m, 2H), 7.09 (d, J = 9, 2H), 3.86 (m, 1H), 3.59 (m, 2H), 3.27 (m, 2H), 3.15 (m,1H), 1.29 (s, 9H); MS (ESI) m/e 449.2 (M+); HRMS (ESI) calculated for C21H27N3O4S2Na+ 472.1322, found 472.1326.
To a solution of (±)-N-carbamoyl-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (110 mg, 0.245 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (51 mg, 0.269 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum, and to the residue were added THF (4 mL), 1N NaOH (0.3 mL), and TCEP (77 mg, 0.269 mmol) with stirring under argon. Stirring under argon was continued for 3 h at room temperature, then the mixture was concentrated under vacuum. The residue was dissolved in ethyl acetate and the solution was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (5% MeOH in ethyl acetate) to give the mercaptan 29b as a white solid (89 mg, 96%): mp 170 171°C; 1H NMR (300 MHz, CD3OD) δ 7.87 (m, 2H), 7.44 (m, 2H), 7.24 (m, 1H), 7.10 (m, 4H), 3.78 (m, 1H), 3.68 (m, 1H), 3.59 (m, 1H), 3.20 (m, 2H), 3.08 (m, 1H); MS (ESI) m/e 393.1 (M+); HRMS (ESI) calculated for C17H19N3O4S2Na+ 416.0701, found 416.0699.
(±)-N-Carbamoyl-trans-3-mercapto-4-(N-methyl-4-phenoxybenzenesulfonamido)pyrrolidine (29c)
To a solution of (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) (236 mg, 0.466 mmol) in DMF (2 mL) at 0°C was added t-BuOK (0.69 mmol). The mixture was stirred at 0°C for 10 min, then MeI (0.69 mmol) was added. The solution was stirred at 0°C until TLC analysis showed no starting material remained. Ethyl acetate (20 mL) was added and the solution was extracted with water, then was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (ethyl acetate) to give (±)-N-Boc-trans-3-tert-butylmercapto-4-(N-methyl-4-phenoxybenzenesulfonamido)pyrrolidine (28, R = CH3) as a white powder (240 mg, 99%): 1H NMR (300 MHz, CD3OD) δ 7.84 (d, J = 8, 2H), 7.44 (t, J = 8, 2H), 7.24 (t, J = 8, 1H), 7.10 (m, 2H), 7.09 (d, J = 9, 2H), 4.22 (m, 1H), 3.90 (m, 1H), 3.37 (m, 2H), 3.08 (m, 2H), 2.86 (s, 3H), 1.44 (s, 9H), 1.28 (s, 9H); MS (ESI) m/e 520.2 (M+); HRMS (ESI) calculated for C26H36N2O5S2Na+ 543.1950, found 543.1950.
To a stirred solution of 1.5 M HCl in ethyl acetate (5 mL) was added (±)-N-Boc-trans-3-tert-butylmercapto-4-(N-methyl-4-phenoxybenzenesulfonamido)pyrrolidine (28, R = CH3) (130 mg, 0.25 mmol), and stirring was continued at room temperature until no starting material was detected by TLC. The solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2. The solution was cooled to 0°C and Et3N (46 μL, 0.33 mmol) and TMSNCO (100 μL, 0.74 mmol) were added successively. The solution was stirred at room temperature overnight. The reaction mixture was extracted with water and saturated brine, and was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (5% MeOH in ethyl acetate) to give (±)-N-carbamoyl-trans-3-tert-butylmercapto-4-(N-methyl-4-phenoxybenzenesulfonamido)pyrrolidine as a white powder (110 mg, 95%): 1H NMR (300 MHz, CD3OD) δ 7.85 (m, 2H), 7.42 (m, 2H), 7.22 (m, 1H), 7.10 (m, 4H), 4.24 (m, 1H), 3.96 (m, 1H), 3.42 (m, 2H), 3.08 (m, 2H), 2.87 (s, 3H), 1.28 (s, 9H); MS (ESI) m/e 463.2 (M+); HRMS (ESI) calculated for C22H29N3O4S2Na+ 486.1479, found 486.1481.
To a solution of (±)-N-carbamoyl-trans-3-tert-butylmercapto-4-(N-methyl-4-phenoxybenzenesulfonamido)pyrrolidine (90 mg, 0.194 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (38.6 mg, 0.204 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum, and to the residue were added THF (4 mL), 1N NaOH (0.25 mL), and TCEP (67 mg, 0.234 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was condensed under vacuum. To the residue was added ethyl acetate and it was dried over Na2SO4. The solvent was evaporated and the residue was purified by flash chromatography (5% MeOH in ethyl acetate) to give the mercaptan 29c as a white powder (75.1 mg, 95%): 1H NMR (300 MHz, CD3OD) δ 7.87 (m, 2H), 7.44 (m, 2H), 7.24 (m, 1H), 7.10 (m, 4H), 4.32 (q, J = 9, 1H), 3.83 (dd, J = 10, 8 Hz, 1H), 3.42 (m, 2H), 3.13 (q, J = 10, 2H), 2.83 (s, 3H); MS (ESI) m/e 407.2 (M+); HRMS (ESI) calculated for C22H29N3O4S2Na+ 430.0864, found 430.0867.
(±)-N-(2-Phthalimidoethylaminocarbonyl)-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (29d)
To a stirred solution of 1.5 M HCl in ethyl acetate (3 mL) was added (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) (77.5 mg, 0.15 mmol) and stirring was continued at room temperature until no starting material was detected by TLC (50% ethyl acetate in hexane). The solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2 (3 mL). The solution was cooled to −10°C and diisopropylethylamine (63 μL, 0.36 mmol) and triphosgene (14.9 mg, 0.05 mmol) in CH2Cl2 (3 mL) were added. The mixture was stirred at 10°C for 20 min, then diisopropylethylamine (63 μL, 0.36 mmol) and 2-phthalimidoethylamine hydrochloride (34 mg, 0.15 mmol) were added successively. The solution was warmed to room temperature and was stirred for 4 h. The reaction mixture was extracted with water, dried over Na2SO4, and evaporated under reduced pressure. The residue was purified by flash chromatography (5% MeOH in ethyl acetate) to give (±)-N-(2-phthalimidoethylaminocarbonyl)-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine as a white solid (79 mg, 84%): mp 105 106°C; 1H NMR (300 MHz, CDCl3) δ 7.92 (m, 2H), 7.85 (m, 2H), 7.72 (m, 2H), 7.42 (m, 2H), 7.24 (m, 1H), 7.10 (m, 4H), 5.62 (d, J = 9, 1H), 4.62 (m, 1H), 3.98-3.70 (m, 3H), 3.62 (m, 1H), 3.48 (m, 3H), 3.24 (m, 2H), 3.14 (m, 1H), 1.32 (s, 9H); MS (ESI) m/e 622.2 (M+); HRMS (ESI) calculated for C31H34N4O6S2Na+ 645.1812, found 645.1814.
To a solution of (±)-N-(2-phthalimidoethylaminocarbonyl)-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (62.3 mg, 0.10 mmol) in AcOH (1 mL) was added 2-nitrobenzenesulfenyl chloride (21 mg, 0.11 mmol) and the mixture was stirred at room temperature for 2 h. The AcOH was evaporated under vacuum, and to the residue were added THF (2 mL), 1 N NaOH (0.2 mL), and TCEP (34 mg, 0.12 mmol). The mixture was stirred at room temperature under argon for 3 h, then it was concentrated under vacuum. To the residue was added ethyl acetate and the solution was dried over Na2SO4 and filtered. The solvent was evaporated and the residue was purified by flash chromatography (ethyl acetate) to give the mercaptan 29d as a white solid (48 mg, 87%): mp 140-141.5°C; 1H NMR (300 MHz, CDCl3) δ 7.93-7.80 (m, 4H), 7.73 (m, 2H), 7.41 (m, 2H), 7.23 (t, J = 8, 1H), 7.15-7.00 (m, 4H), 5.73 (br, 1H), 4.73 (m, 1H), 3.87 (m, 2H), 3.71 (m, 2H), 3.55 (m, 1H), 3.47 (m, 2H), 3.37 (m, 1H), 3.24 (m, 1H), 3.13 (m, 1H), 1.74 (d, J = 7, 1H); MS (ESI) m/e 566.2 (M+); HRMS (ESI) calculated for C27H26N4O6S2Na+ 589.1191, found 589.1190.
(±)-N-(6-Acetamidohexanoyl)-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)-pyrrolidine (29e) and (±)-N-(6-Aminohexanoyl)-trans-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine Hydrochloride (29f)
To a stirred solution of 1.5 M HCl in ethyl acetate (1 mL) was added (±)-N-Boc-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (27) (200 mg, 0.39 mmol) and stirring was continued at room temperature until no starting material was detected by TLC (50% ethyl acetate in hexane). The solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2. The solution was cooled to 0°C and Et3N (60 μL, 0.43 mmol), HOBt•H2O (54 mg, 0.40 mmol), 6-(tert-butoxycarbonylamino)hexanoic acid (90 mg, 0.39 mmol), and DCC (80 mg, 0.39 mmol) were added. The mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure and the residue was dissolved in ethyl acetate (10 mL). The organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (201 mg, 84%): 1H NMR (300 MHz, CDCl3) δ 7.82 (dd, J = 9, 3, 2H), 7.43 (t, J = 8, 2H), 7.24 (t, J = 7, 1H), 7.08 (m, 4H), 5.13 (br m, 1H, NH), 4.57 (br s, 1H, NH), 3.72-4.10 (m, 2H), 3.00-3.45 (m, 6H), 2.21 (t, J = 7, 2H), 1.64 (m, 2H), 1.48 (m, 2H), 1.44 (s, 9H), 1.36 and 1.30 (s, 9H), 1.26 (m, 2H); HRMS (ESI) calculated for C31H45N3O6S2Na+ 642.2642, found 642.2654.
To a solution of (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-tert-butylmercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (130 mg, 0.21 mmol) in AcOH CH2Cl2 (1:1, 1 mL) was added 2-nitrobenzenesulfenyl chloride (42 mg, 0.22 mmol) and the mixture was stirred at room temperature for 2 h. The solvents were evaporated under vacuum and the residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-(2-nitrobenzenedisulfido) 4 (4 phenoxybenzenesulfon amido)pyrrolidine: 1H NMR (300 MHz, CDCl3) δ 8.28 (m, 1H), 8.22 and 8.11 (d, J = 8, 1H), 7.75 (m, 3H), 7.42 (m, 3H), 7.24 (m, 1H), 7.10 (m, 2H), 7.02 (m, 2H), 5.40 and 5.12 (br s, 1H, NH), 4.57 (br s, 1H, NH), 3.75-4.00 (m, 3H), 3.21-3.53 (m, 3H), 2.95-3.14 (m, 2H), 2.19 (m, 2H), 1.61 (m, 2H), 1.47 (m, 2H), 1.43 (s, 9H), 1.33 (m, 2H).
A solution of (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-(2-nitrobenzenedisulfido)-4-(4-phenoxybenzenesulfonamido)pyrrolidine (78 mg, 0.11 mmol) in trifluoroacetic acid (1 mL) was stirred at room temperature for 1 h. The trifluoroacetic acid was evaporated under vacuum and the residue was dissolved in CH2Cl2 (1 mL). The solution was cooled to 0°C and Et3N (34 μL, 0.244 mmol) and acetyl chloride (8.6 μL, 0.12 mmol) were added, and the mixture was stirred for 2 h. The solution was evaporated under vacuum; the residue was dissolved in ethyl acetate (5 mL), washed with water, dried over Na2SO4, and the solvent was evaporated. The residue was purified by flash chromatography (100% ethyl acetate) to give (±)-N-(6-acetamidohexanoyl)-trans-3-(2-nitrobenzenedisulfido)-4-(4-phenoxybenzenesulfonamido)pyrrolidine (68 mg, 94%): 1H NMR (300 MHz, CD3OD) δ 8.20-8.33 (m, 2H), 7.76 (m, 3H), 7.47 (m, 3H), 7.25 (t, J = 8, 1H), 7.10 (d, J = 9, 2H), 6.99 (m, 2H), 3.92 (m, 2H), 3.68-3.86 (m, 2H), 3.52 (m, 1H), 3.39 (m, 1H), 3.14 (m, 2H), 2.24 (m, 2H), 1.93 and 1.92 (s, 3H), 1.54 (m, 4H), 1.34 (m, 2H). To a solution of the 2-nitrobenzenedisulfide (10 mg, 0.015 mmol) in THF-H2O (4:1, 1 mL) were added TCEP (5 mg, 0.017 mmol) and 1 N NaOH (20 μL). The mixture was stirred until no starting material was detected by TLC (100% ethyl acetate). The solvent was evaporated under reduced pressure and the residue was purified by short flash column chromatography (100% ethyl acetate) to give the mercaptan 29e (7 mg, 91%): 1H NMR (300 MHz, CD3OD) δ 7.88 (m, 2H), 7.45 (t, J = 8, 2H), 7.25 (t, J = 8, 1H), 7.10 (m, 4H), 3.81-3.98 (m, 2H), 3.53-3.72 (m, 2H), 3.05-3.40 (m, 4H), 2.26 (m, 2H), 1.92 (s, 3H), 1.46-1.65 (m, 4H), 1.30-1.41 (m, 2H); HRMS (ESI) calculated for C24H32N3O5S2+ 506.1778, found 506.1786.
To a solution of (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-(2-nitrobenzenedisulfido)-4-(4-phenoxybenzenesulfonamido)pyrrolidine (50 mg, 0.7 mmol) in THF-H2O (4:1, 1 mL) were added TCEP (23 mg, 0.8 mmol) and 1 N NaOH (100 μL). The mixture was stirred until no starting material was detected by TLC (50% ethyl acetate in hexane). The solution was evaporated under reduced pressure and the residue was purified by a short flash column chromatography (50% ethyl acetate in hexane) to give the thiol. The latter was dissolved in 2 M HCl in acetic acid (1 mL) and the mixture was stirred at room temperature for 1 h. Liophylization of the solution gave the mercaptan 29f as a white hygroscopic solid (33 mg, 94%): 1H NMR (300 MHz, CD3OD) δ 7.88 (m, 2H), 7.45 (t, J = 8, 2H), 7.25 (t, J = 8, 1H), 7.10 (m, 4H), 3.84-3.97 (m, 2H), 3.71 (dd, J = 12, 7, 1H), 3.52-3.64 (m, 2H), 3.06-3.18 (m, 1H), 2.93 (br m, 2H), 2.31 (q, J = 7, 2H), 1.66 (br m, 4H), 1.42 (br m, 2H); HRMS (ESI) calculated for C22H30N3O4S2+ 464.1672, found 464.1672.
(±)-N-(6-Aminohexanoyl)-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine Hydrochloride (29g)
To a stirred solution of 1.5 M HCl in ethyl acetate (3 mL) was added (±)-N-Boc-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (25) (430 mg, 0.99 mmol) and stirring was continued until no starting material was detected by TLC (50% ethyl acetate in hexane). The solvent was evaporated under reduced pressure and the residue was dissolved in CH2Cl2. The solution was cooled to 0°C and Et3N (153 μL, 1.1 mmol), HOBt*H2O (135 mg, 1.0 mmol), 6-(tert-butoxycarbonylamino)hexanoic acid (231 mg, 1.0 mmol), and DCC (206 mg, 1.0 mmol) were added. The mixture was stirred at room temperature overnight. The solvent was evaporated under reduced pressure and the residue was dissolved in ethyl acetate (10 mL). The organic layer was washed with 10% citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (±)-N-(6-(tert-butoxycarbonylamino)hexanoyl)-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (400 mg, 74%). The latter was dissolved in 2M HCl in acetic acid (3 mL) and the mixture was stirred at room temperature for 1 h. Liophylization of the solution gave (±)-N-(6-aminohexanoyl)-trans-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine hydrochloride (29g) as a white hygroscopic solid (353 mg, 100%): 1H NMR (300 MHz, CD3OD) δ 7.86 (dd, J = 9, 3, 2H), 7.46 (t, J = 8, 2H), 7.25 (t, J = 8, 1H), 7.10 (m, 4H), 4.24 and 4.12 (m, 1H), 3.70-3.76 (m, 1H), 3.47-3.63 (m, 3H), 3.34-3.42 (m, 1H), 2.93 (br t, J = 7, 2H), 2.35 and 2.31 (t, J = 7, 2H), 1.67 (m, 4H), 1.44 (m, 2H); HRMS (ESI) calculated for C22H30N3O5S+ 448.1906, found 448.1904.
Enzymatic Resolution of (±)-N-Boc-trans-3-hydroxy-4-azidopyrrolidine (Scheme 4)
To a solution of (±)-N-Boc-trans-3-hydroxy-4-azidopyrrolidine-(24) (2.28 g, 10 mmol) in tert-butyl methyl ether (50 mL) were added isopropenyl acetate (3 g, 3 mmol) and Lipase Amano AK-20 (3 g), and the mixture was stirred at room temperature for 72 h. The mixture was filtered, the filtrate was evaporated under reduced pressure, and the product was separated by flash chromatography (25% ethyl acetate in hexane). First eluted was (3R,4R)-trans-N-Boc-3-acetoxy-4-azidopyrrolidine (31) (1.30 g): 1H NMR (300 MHz, CDCl3) δ 5.09 (br s, 1H), 4.04 (m, 1H), 3.57-3.74 (m, 2H), 3.34-3.55 (m, 2H), 2.09 (s, 3H), 1.47 (s, 9H); [α]20D = −30.4° (c = 1.0, CHCl3).
Second to be eluted was (3S,4S)-trans N-Boc-3-hydroxy-4-azidopyrrolidine (30) (1.07 g): 1H NMR (300 MHz, CDCl3) δ 4.25 (br s, 1H), 3.93 (br s, 1H), 3.55-3.75 (m, 2H), 3.30-3.50 (m, 2H), 2.05 (br s, 1H), 1.46 (s, 9H); [α]20D = +26.8° (c = 0.5, CHCl3).
To a stirred solution of (3R,4R)-trans-N-Boc-3-acetoxy-4-azidopyrrolidine (31) (1.0 g, 3.4 mmol) in THF-MeOH (3:1, 8 mL) at 0°C was added 1N LiOH (4 mL) dropwise. The reaction mixture was stirred at room temperature overnight, then it was evaporated under reduced pressure. Ethyl acetate (20 mL) was added to the residue and the solution was washed with brine, dried over Na2SO4, and evaporated. The residue was purified by flash column chromatography (50% ethyl acetate in hexane) to give (3R,4R)-trans-N-Boc-3-hydroxy-4-azidopyrrolidine (32): 1H NMR (300 MHz, CDCl3) identical to that of the (3S,4S) isomer; [α]20D = −26.8° (c = 1.0, CHCl3).
A sample of the (3S,4S)-trans-N-Boc-3-hydroxy-4-azidopyrrolidine (30) (80 mg, 0.35 mmol) was dissolved in trifluoroacetic acid (1 mL) and was stirred until no starting material was detected on TLC (25% ethyl acetate in hexane). The solution was evaporated under reduced pressure and the residue was dissolved in CH2Cl2 (2 mL). The solution was cooled to 0°C, Et3N (117 μL, 0.84 mmol) and Cbz-Cl (60 mg, 0.35 mmol) were added, and the resulting solution was stirred for 2 h at room temperature. The reaction mixture was washed with water, dried over Na2SO4, and evaporated. The residue was purified by flash chromatography (25% ethyl acetate in hexane) to give (3S,4S)-trans-N-Cbz-3-hydroxy-4-azidopyrrolidine (89 mg, 97%): [α]20D = +16.1° (c = 1.00, CHCl3) (lit.64: [α]25D = +14.3° (c = 1.06, CHCl3)).
(3S,4S)-trans-3-Mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine Hydrochloride (39a)
A solution of (3S,4S)-trans-N-Boc-3-hydroxy-4-azidopyrrolidine (30) (850 mg, 3.72 mmol) and Et3N (622 μL, 4.46 mmol) in CH2Cl2 (10 mL) was cooled with stirring to 0°C, and methanesulfonyl chloride (331 μl, 4.28 mmol) was added dropwise. The solution was stirred at room temperature until no starting material was detected by TLC (25% ethyl acetate in hexane). The reaction mixture was washed with 10% aqueous citric acid and water, then was dried over Na2SO4, and evaporated. The residual crude methanesulfonate was dissolved in anhydrous DMF (20 mL), potassium acetate (1.10 g, 11.2 mmol) was added, and the mixture was stirred at 105°C under N2 for 6 h. The reaction mixture was poured into ice water and was extracted with ethyl acetate. The organic layer was washed with brine, dried over Na2SO4, and evaporated. The residue was purified by flash chromatography (25% ethyl acetate in hexane) to give (3R,4S)-cis-N-Boc-3-acetoxy-4-azidopyrrolidine (33) (860 mg, 86%): 1H NMR (300 MHz, CDCl3) δ 5.31 (br m, 1H), 4.07 (m, 1H), 3.60-3.71 (m, 2H), 3.36 3.50 (m, 2H), 2.16 (s, 3H), 1.46 (s, 9H); [α]20D = −34.5° (c = 1.00, CHCl3).
To a stirred solution of the (3R,4S)-azidoacetate 33 (560 mg, 2.07 mmol) in THF-MeOH (3:1, 8 mL) at 0°C was added 1N LiOH (3 mL) dropwise. The reaction mixture was stirred at room temperature overnight, then it was evaporated under reduced pressure. Ethyl acetate (20 mL) was added to the residue and the solution was washed with brine, dried over Na2SO4, and evaporated. The residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (3R,4S)-cis-N-Boc-3-hydroxy-4-azidopyrrolidine (34) (460 mg, 97%): 1H NMR (300 MHz, CDCl3) δ 4.35 (br m, 1H), 4.02 (br m, 1H), 3.25-3.72 (m, 4H), 2.16 (br s, 1H), 1.46 (s, 9H); [α]20D = +32.3° (c = 0.69, CHCl3).
To a solution of the (3R,4S)-azidoalcohol 34 (470 mg, 2.06 mmol) in THF-H2O (10:1, 10 mL) was added Ph3P (540 mg, 2.06 mmol), and the mixture was stirred at room temperature for 2 h and at 65°C until no starting material was detected on TLC (25% ethyl acetate in hexane). The solution was cooled with stirring to 0°C and Et3N (345 μL, 2.48 mmol) was added, then a solution of 4-phenoxybenzenesulfonyl chloride (529 mg, 2.26 mmol) in THF (2 mL) was added dropwise. The reaction mixture was warmed to room temperature and was stirred for 3 h. The mixture was concentrated under reduced pressure, ethyl acetate was added to the residue, and the organic layer was washed with 10% aqueous citric acid and saturated aqueous NaHCO3, then it was dried over Na2SO4 and evaporated. The residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (3R,4S)-cis-N-Boc-3-hydroxy-4-(4-phenoxybenzenesulfonamido)pyrrolidine (35) (670 mg, 75%): 1H NMR (300 MHz, CDCl3) δ 7.83 (d, J = 7, 2H), 7.42 (t, J = 8, 2H), 7.24 (t, J = 8, 1H), 7.06 (m, 4H), 5.14 (br s, 1H), 4.25 & 4.11 (br s, 1H), 3.76 (m, 1H), 3.55 (m, 1H), 3.43 (m, 2H), 3.07 (m, 1H), 2.36 and 2.23 (br s, 1H), 1.43 (s, 9H); [α]20D = +15.6° (c = 0.80, CHCl3).
To a stirred solution of Ph3P (525 mg, 2.0 mmol) in THF (3 mL) at 0°C was added DEAD (315 μL, 2.0 mmol) dropwise. After 10 min, a solution of alcohol 35 (435 mg, 1.0 mmol) and thiolacetic acid (228 μL, 2.29 mmol) in THF (3 mL) was added. The resulting reaction mixture was stirred at room temperature for 6 h, then it was concentrated under reduced pressure. The residue was purified by flash chromatography (15% ethyl acetate in hexane) to give (3S,4S)-trans-N-Boc-3-acetylthio-4-(4-phenoxybenzenesulfonamido)pyrrolidine (36) (400 mg, 81%): 1H NMR (300 MHz, CDCl3) δ 7.80 (d, J = 9, 2H), 7.42 (m, 2H), 7.23 (t, J = 8, 1H), 7.06 (m, 4H), 5.21 and 5.00 (br s, 1H, NH), 3.75 (m, 4H), 3.16 (m, 2H), 2.31 and 2.25 (br s, 3H), 1.44 (s, 9H); [α]20D = +22.8° (c = 0.62, CHCl3).
To a stirred solution of the acetylthiopyrrolidine 36 (60 mg, 0.122 mmol) in MeOH (1 mL) under nitrogen was added 40% aqueous MeNH2 (100 μL, 1.16 mmol) and the mixture was stirred for 10 min. The solution was concentrated under vacuum and the residue was purified by flash chromatography (50% ethyl acetate in hexane) to give (3S,4S)-trans-N-Boc-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (37) (54 mg, 98%) as an oil: 1H NMR (300 MHz, CD3OD) identical to that of the (±)-modification described earlier; [α]20D = +39.2° (c = 0.54, MeOH).
To a solution of 2.0 M HCl in AcOH (1 mL) was added (3S,4S)-trans-N-Boc-3-mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine (37) (45 mg, 0.10 mmol) and the mixture was stirred at room temperature for 1 h. Liophylization of the reaction mixture gave the amine hydrochloride 39a as a white hygroscopic solid (37 mg, 100%): 1H NMR (300 MHz, CD3OD) identical to that of the racemic inhibitor 29a; [α]20D = +21.9° (c = 0.7, MeOH); HRMS (ESI) calculated for C16H19N2O3S +2 351.0832, found 351.0824.
(3R,4R)-3-Mercapto-4-(4-phenoxybenzenesulfonamido)pyrrolidine Hydrochloride (39b).
By the procedures used for the 3S,4S enantiomer, (3R,4R)-trans-N-Boc-3-hydroxy-4-azidopyrrolidine was converted to the title inhibitor 39b as a white hygroscopic solid: 1H NMR (300 MHz, CD3OD) identical to those of 29a and 39a; [α]20D = −21.0° (c = 1.0, MeOH); HRMS (ESI) calculated for C16H19N2O3S +2 351.0832, found 351.0820.
Enzyme Kinetics
Enzymatic assays to characterize inhibitor potency were performed as previously described.17, 65 Assays were consistently carried out at 25°C in 50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) buffer, pH 7.5, with 10 mM CaCl2, 200 mM NaCl, and 0.01% (w/v) Brij-35. The reducing agent TCEP was also included at a concentration of 5 μM to maintain the fully reduced form of the MMPIs. Briefly, 10 μL of varying concentrations of MMPIs dissolved in DMSO, 176 μL of HEPES buffer, and 10 μL of enzyme stock solution were mixed and incubated for 30 min. The assay was started by addition of 4 μL of a 1 μM solution of the synthetic substrate Mca-PLGLDpa-AR-NH2 (M-1895, Bachem)66 in 50% DMSO and the release of product was monitored by fluorescence (λex = 328 nm and λem = 393 nm) with a PerkinElmer luminescence spectrophotometer LS50B (FL WinLab v3.0) connected to a temperature controlled water bath. Enzyme concentrations ranged from 0.2 to 7 nM during the assays.
Stability Assays
To assess the relative stability of the thiol inhibitors over time, the inhibitory potencies of these compounds were assessed at varying time points. The compounds were initially dissolved in DMSO and diluted in cell culture medium (containing either 20%, 10%, or 0% fetal bovine serum) to a concentration approximately 10,000 times their IC50 value; they were then incubated in a humidified environment at 37°C and 5% CO2 for varying periods up to 72 hours. After the initial incubation period, 10 μL of the inhibitor solution was further incubated with 176 μL of HEPES buffer and 10 μL of MMP-9 enzyme (final concentration ~1 nM) for 30 min prior to initiation of the assay. The relative rates (νi/νo) for each compound at its various time points were obtained in triplicate as previously described and plotted versus time to indicate their corresponding stability.
Cell Culture and Compound Toxicity
Low passage hMSCs were routintely cultured in α-modified minimum essential medium (αMEM) supplemented with L-glutamine, penicillin, streptomycin, and 20% fetal bovine serum. Prior to the initial seeding of the cells at low density (~60 cells/cm2), cells were plated for 24 h of recovery and maintained at 37°C in a humidified atmosphere containing 5% CO2. The addition of fresh culture media was provided every three days until a confluency of 75% was reached. At this point varying dosages of inhibitor (maintaining a constant final DMSO concentration) were used to supplement fresh media and added to the cells for a period of 24 h. The logarithmic panel of inhibitor concentrations ranged from 100 μM to 1 μM. At the end of the treatment period the inhibitor conditioned media was removed and the cells were trypsinized (0.25% Trypsin, Hyclone) after being washed with phosphate buffered saline (pH 7.4).
To quantify the levels of toxicity these compounds elicited on hMSC growth, cells were pelleted by centrifugation at 450 × g and resuspended in 1 mL of media. An aliquot of the suspension was then diluted 1:1 with a 0.4% (w/v) Trypan Blue solution and counted with a hemocytometer. Determined by normalizing the number of cells counted for each treatment against its respective control, the relative groups were plotted with error bars representing the standard error in the mean (mean ± SEM). Each of the treatments and hemocytometer counts were performed in triplicate, and compounds at a particular concentration killing more than 20% of the cells were deemed cytotoxic.
Acknowledgment
This work was supported in part by grants from the Department of Defense (DoD) Prostate Cancer Research Program (DAMD17-02-1-0238 and W81XWH-07-1-0225), Florida Department of Health James and Esther King Biomedical Research Program (1KF04) and Florida State University (to Q.-X.S.), by grants 1 R21 NS066418 and 2 R01 NS045847-05A2 from the National Institutes of Health (NIH) (to Gary A. Rosenberg and Q.-X.S.), and the Molecular Design and Synthesis (MDS) Foundation (to M.A.S.). The authors would also like to gratefully acknowledge Dr. Umesh Goli, Director of the MASS/BASS Facilities at Florida State University, for his assistance in mass spectrometry characterization of the compounds.
Abbreviations Used
- DEAD
diethyl azodicarboxylate
- ECM
extracellular matrix
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- hMSC
human mesenchymal stem cell
- MMPI
matrix metalloproteinase inhibitor
- TCEP
tris(2-carboxyethyl)phosphine
- TIMP
tissue inhibitor of metalloproteinase
- TMSNCO
trimethylsilyl isocyanate
- ZBG
zinc-binding group
Footnotes
Author Contributions:
Y.J. performed compound synthesis and characterization; M.D.R. was responsible for cell culture, cytotoxicity, and stability results; D.B.B., Q.C., & M.H.C. determined inhibition constants and IC50 values; M.A.S. and Q.-X.S. conceived the study and experimental designs; and Y.J., M.D.R., M.A.S., and Q. X.S. wrote the manuscript.
REFERENCES
- 1.Decock J, Thirkettle S, Wagstaff L, Edwards DR. Matrix metalloproteinases: protective roles in cancer. J. Cell. Mol. Med. 2011;15:1254–1265. doi: 10.1111/j.1582-4934.2011.01302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Iyer RP, Patterson NL, Fields GB, Lindsey ML. The history of matrix metalloproteinases: milestones, myths, and misperceptions. Am. J. Physiol. Heart Circ. Physiol. 2012;303:H919, 930. doi: 10.1152/ajpheart.00577.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martins VL, Caley M, O’Toole EA. Matrix metalloproteinases and epidermal wound repair. Cell Tissue Res. 2013;351:255–268. doi: 10.1007/s00441-012-1410-z. [DOI] [PubMed] [Google Scholar]
- 4.Candelario-Jalil E, Yang Y, Rosenberg GA. Diverse roles of matrix metalloproteinases and tissue inhibitors of metalloproteinases in neuroinflammation and cerebral ischemia. Neuroscience. 2009;158:983–994. doi: 10.1016/j.neuroscience.2008.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosell A, Lo EH. Multiphasic roles for matrix metalloproteinases after stroke. Curr. Opin. Pharmacol. 2008;8:82–89. doi: 10.1016/j.coph.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Lehrke M, Greif M, Broedl UC, Lebherz C, Laubender RP, Becker A, von Ziegler F, Tittus J, Reiser M, Becker C, Goke B, Steinbeck G, Leber AW, Parhofer KG. MMP-1 serum levels predict coronary atherosclerosis in humans. Cardiovasc. Diabetol. 2009;8:50. doi: 10.1186/1475-2840-8-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tuomainen AM, Nyyssonen K, Laukkanen JA, Tervahartiala T, Tuomainen TP, Salonen JT, Sorsa T, Pussinen PJ. Serum matrix metalloproteinase-8 concentrations are associated with cardiovascular outcome in men. Arterioscler. Thromb. Vasc. Biol. 2007;27:2722–2728. doi: 10.1161/ATVBAHA.107.154831. [DOI] [PubMed] [Google Scholar]
- 8.Johnson JL, George SJ, Newby AC, Jackson CL. Divergent effects of matrix metalloproteinases 3, 7, 9, and 12 on atherosclerotic plaque stability in mouse brachiocephalic arteries. Proc. Natl. Acad. Sci. U. S. A. 2005;102:15575–15580. doi: 10.1073/pnas.0506201102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker AH, Edwards DR, Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J. Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 10.Ahonen M, Poukkula M, Baker AH, Kashiwagi M, Nagase H, Eriksson JE, Kahari VM. Tissue inhibitor of metalloproteinases 3 induces apoptosis in melanoma cells by stabilization of death receptors. Oncogene. 2003;22:2121–2134. doi: 10.1038/sj.onc.1206292. [DOI] [PubMed] [Google Scholar]
- 11.Bond M, Murphy G, Bennett MR, Newby AC, Baker AH. Tissue inhibitor of metalloproteinase 3 induces a Fas-associated death domain-dependent type II apoptotic pathway. J. Biol. Chem. 2002;277:13787–13795. doi: 10.1074/jbc.M111507200. [DOI] [PubMed] [Google Scholar]
- 12.Guedez L, Courtemanch L, Stetler-Stevenson M. Tissue inhibitor of metalloproteinase (TIMP)-1 induces differentiation and an antiapoptotic phenotype in germinal center B cells. Blood. 1998;92:1342–1349. [PubMed] [Google Scholar]
- 13.Valente P, Fassina G, Melchiori A, Masiello L, Cilli M, Vacca A, Onisto M, Santi L, Stetler-Stevenson WG, Albini A. TIMP-2 over-expression reduces invasion and angiogenesis and protects B16F10 melanoma cells from apoptosis. Int. J. Cancer. 1998;75:246–253. doi: 10.1002/(sici)1097-0215(19980119)75:2<246::aid-ijc13>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 14.Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ. Res. 2003;92:827–839. doi: 10.1161/01.RES.0000070112.80711.3D. [DOI] [PubMed] [Google Scholar]
- 15.Whittaker M, Floyd CD, Brown P, Gearing AJ. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999;99:2735–2776. doi: 10.1021/cr9804543. [DOI] [PubMed] [Google Scholar]
- 16.Bode W, Fernandez-Catalan C, Tschesche H, Grams F, Nagase H, Maskos K. Structural properties of matrix metalloproteinases. Cell. Mol. Life Sci. 1999;55:639–652. doi: 10.1007/s000180050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park HI, Jin Y, Hurst DR, Monroe CA, Lee S, Schwartz MA, Sang QX. The intermediate S1′ pocket of the endometase/matrilysin-2 active site revealed by enzyme inhibition kinetic studies, protein sequence analyses, and homology modeling. J. Biol. Chem. 2003;278:51646–51653. doi: 10.1074/jbc.M310109200. [DOI] [PubMed] [Google Scholar]
- 18.Babine RE, Bender SL. Molecular Recognition of Protein Ligand Complexes: Applications to Drug Design. Chem. Rev. 1997;97:1359–1472. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 19.Sang QX, Jin Y, Newcomer RG, Monroe SC, Fang X, Hurst DR, Lee S, Cao Q, Schwartz MA. Matrix metalloproteinase inhibitors as prospective agents for the prevention and treatment of cardiovascular and neoplastic diseases. Curr. Top. Med. Chem. 2006;6:289–316. doi: 10.2174/156802606776287045. [DOI] [PubMed] [Google Scholar]
- 20.Kontogiorgis CA, Papaioannou P, Hadjipavlou-Litina DJ. Matrix metalloproteinase inhibitors: a review on pharmacophore mapping and (Q)SARs results. Curr. Med. Chem. 2005;12:339–355. doi: 10.2174/0929867053363243. [DOI] [PubMed] [Google Scholar]
- 21.Peterson JT. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 2004;9:63–79. doi: 10.1023/B:HREV.0000011395.11179.af. [DOI] [PubMed] [Google Scholar]
- 22.White CM. Pharmacologic, pharmacokinetic, and therapeutic differences among ACE inhibitors. Pharmacotherapy. 1998;18:588–599. [PubMed] [Google Scholar]
- 23.Duchin KL, McKinstry DN, Cohen AI, Migdalof BH. Pharmacokinetics of captopril in healthy subjects and in patients with cardiovascular diseases. Clin. Pharmacokinet. 1988;14:241–259. doi: 10.2165/00003088-198814040-00002. [DOI] [PubMed] [Google Scholar]
- 24.Sang QA, Schwartz MA, Li H, Chung LW, Zhau HE. Targeting matrix metalloproteinases in human prostate cancer. Ann. N. Y. Acad. Sci. 1999;878:538–540. doi: 10.1111/j.1749-6632.1999.tb07720.x. [DOI] [PubMed] [Google Scholar]
- 25.Sang QX, Jia MC, Schwartz MA, Jaye MC, Kleinman HK, Ghaffari MA, Luo YL. New thiol and sulfodiimine metalloproteinase inhibitors and their effect on human microvascular endothelial cell growth. Biochem. Biophys. Res. Commun. 2000;274:780–786. doi: 10.1006/bbrc.2000.3212. [DOI] [PubMed] [Google Scholar]
- 26.Schwartz MA, Van Wart H. Mercaptosulfide metalloproteinase inhibitors. 1995 Oct. 5,455,262. [Google Scholar]
- 27.Cheng M, De B, Almstead NG, Pikul S, Dowty ME, Dietsch CR, Dunaway CM, Gu F, Hsieh LC, Janusz MJ, Taiwo YO, Natchus MG, Hudlicky T, Mandel M. Design, synthesis, and biological evaluation of matrix metalloproteinase inhibitors derived from a modified proline scaffold. J. Med. Chem. 1999;42:5426–5436. doi: 10.1021/jm9904699. [DOI] [PubMed] [Google Scholar]
- 28.Cheng XC, Wang Q, Fang H, Xu WF. Advances in matrix metalloproteinase inhibitors based on pyrrolidine scaffold. Curr. Med. Chem. 2008;15:374–385. [PubMed] [Google Scholar]
- 29.Natchus MG, Bookland RG, De B, Almstead NG, Pikul S, Janusz MJ, Heitmeyer SA, Hookfin EB, Hsieh LC, Dowty ME, Dietsch CR, Patel VS, Garver SM, Gu F, Pokross ME, Mieling GE, Baker TR, Foltz DJ, Peng SX, Bornes DM, Strojnowski MJ, Taiwo YO. Development of new hydroxamate matrix metalloproteinase inhibitors derived from functionalized 4-aminoprolines. J. Med. Chem. 2000;43:4948–4963. doi: 10.1021/jm000246e. [DOI] [PubMed] [Google Scholar]
- 30.Li X, Li J. Recent advances in the development of MMPIs and APNIs based on the pyrrolidine platforms. Mini Rev. Med. Chem. 2010;10:794–805. doi: 10.2174/138955710791608334. [DOI] [PubMed] [Google Scholar]
- 31.Hurst DR, Schwartz MA, Ghaffari MA, Jin Y, Tschesche H, Fields GB, Sang QX. Catalytic- and ecto-domains of membrane type 1 matrix metalloproteinase have similar inhibition profiles but distinct endopeptidase activities. Biochem. J. 2004;377:775–779. doi: 10.1042/BJ20031067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurst DR, Schwartz MA, Jin Y, Ghaffari MA, Kozarekar P, Cao J, Sang QX. Inhibition of enzyme activity of and cell mediated substrate cleavage by membrane type 1 matrix metalloproteinase by newly developed mercaptosulphide inhibitors. Biochem. J. 2005;392:527–536. doi: 10.1042/BJ20050545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin YH, Ghaffari MA, Schwartz MA. A practical synthesis of differentially-protected cis-1,2-cyclopentanedithiols and cis-3,4-pyrrolidinedithiols. Tetrahedron Lett. 2002;43:7319–7321. [Google Scholar]
- 34.Mitsunobu O, Yamada M. Preparation of esters of carboxylic and phosphoric acid via quaternary phosphonium salts. Bull. Chem. Soc. Jpn. 1967;40:2380–2382. [Google Scholar]
- 35.DeCrescenzo G, Abbas ZS, Freskos JN, Getman DP, Heintz RM, Mischke BV, McDonald JJ. Thiol sulfonamide metalloprotease inhibitors. 2004 Jun 8; 6,747,027. [Google Scholar]
- 36.Rao BG, Bandarage UK, Wang T, Come JH, Perola E, Wei Y, Tian SK, Saunders JO. Novel thiol-based TACE inhibitors: rational design, synthesis, and SAR of thiol-containing aryl sulfonamides. Bioorg. Med. Chem. Lett. 2007;17:2250–2253. doi: 10.1016/j.bmcl.2007.01.064. [DOI] [PubMed] [Google Scholar]
- 37.Cheng XC, Wang Q, Fang H, Xu WF. Role of sulfonamide group in matrix metalloproteinase inhibitors. Curr. Med. Chem. 2008;15:368–373. doi: 10.2174/092986708783497300. [DOI] [PubMed] [Google Scholar]
- 38.Ami E, Ohrui H. Lipase catalyzed Kinetic Resolution of ( 2-Azidocycloalkanols. Biosci. Biotechnol. Biochem. 1999;63:2150–2156. doi: 10.1271/bbb.63.2150. [DOI] [PubMed] [Google Scholar]
- 39.Hugenberg V, Breyholz HJ, Riemann B, Hermann S, Schober O, Schafers M, Gangadharmath U, Mocharla V, Kolb H, Walsh J, Zhang W, Kopka K, Wagner S. A new class of highly potent matrix metalloproteinase inhibitors based on triazole-substituted hydroxamates: (radio)synthesis and in vitro and first in vivo evaluation. J. Med. Chem. 2012;55:4714–4727. doi: 10.1021/jm300199g. [DOI] [PubMed] [Google Scholar]
- 40.Kolodziej SA, Hockerman SL, DeCrescenzo GA, McDonald JJ, Mischke DA, Munie GE, Fletcher TR, Stehle N, Swearingen C, Becker DP. MMP-13 selective isonipecotamide alpha-sulfone hydroxamates. Bioorg. Med. Chem. Lett. 2010;20:3561–3564. doi: 10.1016/j.bmcl.2010.04.111. [DOI] [PubMed] [Google Scholar]
- 41.Tommasi RA, Weiler S, McQuire LW, Rogel O, Chambers M, Clark K, Doughty J, Fang J, Ganu V, Grob J, Goldberg R, Goldstein R, Lavoie S, Kulathila R, Macchia W, Melton R, Springer C, Walker M, Zhang J, Zhu L, Shultz M. Potent and selective 2-naphthylsulfonamide substituted hydroxamic acid inhibitors of matrix metalloproteinase-13. Bioorg. Med. Chem. Lett. 2011;21:6440–6445. doi: 10.1016/j.bmcl.2011.08.087. [DOI] [PubMed] [Google Scholar]
- 42.Lu C, Li XY, Hu Y, Rowe RG, Weiss SJ. MT1-MMP controls human mesenchymal stem cell trafficking and differentiation. Blood. 2010;115:221–229. doi: 10.1182/blood-2009-06-228494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mannello F. Multipotent mesenchymal stromal cell recruitment, migration, and differentiation: what have matrix metalloproteinases got to do with it? Stem Cells. 2006;24:1904–1907. doi: 10.1634/stemcells.2005-0608. [DOI] [PubMed] [Google Scholar]
- 44.Ries C, Egea V, Karow M, Kolb H, Jochum M, Neth P. MMP-2, MT1-MMP, and TIMP-2 are essential for the invasive capacity of human mesenchymal stem cells: differential regulation by inflammatory cytokines. Blood. 2007;109:4055–4063. doi: 10.1182/blood-2006-10-051060. [DOI] [PubMed] [Google Scholar]
- 45.Janusz MJ, Hookfin EB, Brown KK, Hsieh LC, Heitmeyer SA, Taiwo YO, Natchus MG, Pikul S, Almstead NG, De B, Peng SX, Baker TR, Patel V. Comparison of the pharmacology of hydroxamate- and carboxylate-based matrix metalloproteinase inhibitors (MMPIs) for the treatment of osteoarthritis. Inflamm. Res. 2006;55:60–65. doi: 10.1007/s00011-005-0014-4. [DOI] [PubMed] [Google Scholar]
- 46.Abdul-Hay SO, Lane AL, Caulfield TR, Claussin C, Bertrand J, Masson A, Choudhry S, Fauq AH, Maharvi GM, Leissring MA. Optimization of peptide hydroxamate inhibitors of insulin-degrading enzyme reveals marked substrate-selectivity. J. Med. Chem. 2013;56:2246–2255. doi: 10.1021/jm301280p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marek L, Hamacher A, Hansen FK, Kuna K, Gohlke H, Kassack MU, Kurz T. Histone deacetylase (HDAC) inhibitors with a novel connecting unit linker region reveal a selectivity profile for HDAC4 and HDAC5 with improved activity against chemoresistant cancer cells. J. Med. Chem. 2013;56:427–436. doi: 10.1021/jm301254q. [DOI] [PubMed] [Google Scholar]
- 48.Choi E, Lee C, Cho M, Seo JJ, Yang JS, Oh SJ, Lee K, Park SK, Kim HM, Kwon HJ, Han G. Property-based optimization of hydroxamate-based gamma-lactam HDAC inhibitors to improve their metabolic stability and pharmacokinetic profiles. J. Med. Chem. 2012;55:10766–10770. doi: 10.1021/jm3009376. [DOI] [PubMed] [Google Scholar]
- 49.Tandon A, Sinha S. Structural insights into the binding of MMP9 inhibitors. Bioinformation. 2011;5:310–314. doi: 10.6026/97320630005310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heim-Riether A, Taylor SJ, Liang S, Gao DA, Xiong Z, Michael August E, Collins BK, Farmer BT, 2nd, Haverty K, Hill-Drzewi M, Junker HD, Mariana Margarit S, Moss N, Neumann T, Proudfoot JR, Keenan LS, Sekul R, Zhang Q, Li J, Farrow NA. Improving potency and selectivity of a new class of non Zn chelating MMP 13 inhibitors. Bioorg. Med. Chem. Lett. 2009;19:5321–5324. doi: 10.1016/j.bmcl.2009.07.151. [DOI] [PubMed] [Google Scholar]
- 51.Tu G, Xu W, Huang H, Li S. Progress in the development of matrix metalloproteinase inhibitors. Curr. Med. Chem. 2008;15:1388–1395. doi: 10.2174/092986708784567680. [DOI] [PubMed] [Google Scholar]
- 52.Gupta SP, Patil VM. Specificity of binding with matrix metalloproteinases. EXS. 2012;103:35–56. doi: 10.1007/978-3-0348-0364-9_2. [DOI] [PubMed] [Google Scholar]
- 53.Jacobsen EJ, Mitchell MA, Hendges SK, Belonga KL, Skaletzky LL, Stelzer LS, Lindberg TJ, Fritzen EL, Schostarez HJ, O’Sullivan TJ, Maggiora LL, Stuchly CW, Laborde AL, Kubicek MF, Poorman RA, Beck JM, Miller HR, Petzold GL, Scott PS, Truesdell SE, Wallace TL, Wilks JW, Fisher C, Goodman LV, Kaytes PS, et al. Synthesis of a series of stromelysin-selective thiadiazole urea matrix metalloproteinase inhibitors. J. Med. Chem. 1999;42:1525–1536. doi: 10.1021/jm9803222. [DOI] [PubMed] [Google Scholar]
- 54.Terp GE, Cruciani G, Christensen IT, Jorgensen FS. Structural differences of matrix metalloproteinases with potential implications for inhibitor selectivity examined by the GRID/CPCA approach. J. Med. Chem. 2002;45:2675–2684. doi: 10.1021/jm0109053. [DOI] [PubMed] [Google Scholar]
- 55.Bruttomesso AC, Doller D, Gros EG. Mechanistic studies of the rearrangements of steroidal 16,17-ketols and syntheses of 20-->16-cis-gamma-carbolactones. Bioorg. Med. Chem. 1999;7:943–947. doi: 10.1016/s0968-0896(99)00042-5. [DOI] [PubMed] [Google Scholar]
- 56.Puerta DT, Griffin MO, Lewis JA, Romero-Perez D, Garcia R, Villarreal FJ, Cohen SM. Heterocyclic zinc-binding groups for use in next-generation matrix metalloproteinase inhibitors: potency, toxicity, and reactivity. J. Biol. Inorg. Chem. 2006;11:131–138. doi: 10.1007/s00775-005-0053-x. [DOI] [PubMed] [Google Scholar]
- 57.Sun J, Song F, Zhang W, Sexton BE, Windsor LJ. Effects of alendronate on human osteoblast like MG63 cells and matrix metalloproteinases. Arch. Oral Biol. 2012;57:728–736. doi: 10.1016/j.archoralbio.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 58.Flipo M, Charton J, Hocine A, Dassonneville S, Deprez B, Deprez-Poulain R. Hydroxamates: relationships between structure and plasma stability. J. Med. Chem. 2009;52:6790–6802. doi: 10.1021/jm900648x. [DOI] [PubMed] [Google Scholar]
- 59.Oger F, Lecorgne A, Sala E, Nardese V, Demay F, Chevance S, Desravines DC, Aleksandrova N, Le Guevel R, Lorenzi S, Beccari AR, Barath P, Hart DJ, Bondon A, Carettoni D, Simonneaux G, Salbert G. Biological and biophysical properties of the histone deacetylase inhibitor suberoylanilide hydroxamic acid are affected by the presence of short alkyl groups on the phenyl ring. J. Med. Chem. 2010;53:1937–1950. doi: 10.1021/jm901561u. [DOI] [PubMed] [Google Scholar]
- 60.Salmi-Smail C, Fabre A, Dequiedt F, Restouin A, Castellano R, Garbit S, Roche P, Morelli X, Brunel JM, Collette Y. Modified cap group suberoylanilide hydroxamic acid histone deacetylase inhibitor derivatives reveal improved selective antileukemic activity. J. Med. Chem. 2010;53:3038–3047. doi: 10.1021/jm901358y. [DOI] [PubMed] [Google Scholar]
- 61.Hendricks JA, Keliher EJ, Marinelli B, Reiner T, Weissleder R, Mazitschek R. In vivo PET imaging of histone deacetylases by 18F-suberoylanilide hydroxamic acid (18F-SAHA) J. Med. Chem. 2011;54:5576–5582. doi: 10.1021/jm200620f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Guerrant W, Patil V, Canzoneri JC, Oyelere AK. Dual targeting of histone deacetylase and topoisomerase II with novel bifunctional inhibitors. J. Med. Chem. 2012;55:1465–1477. doi: 10.1021/jm200799p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Govindaraju T, Kumar VA, Ganesh KN. (1S,2R/1R,2S)-cis-cyclopentyl PNAs (cpPNAs) as constrained PNA analogues: synthesis and evaluation of aeg-cpPNA chimera and stereopreferences in hybridization with DNA/RNA. J. Org. Chem. 2004;69:5725–5734. doi: 10.1021/jo049442+. [DOI] [PubMed] [Google Scholar]
- 64.Kamal A, Sandbhor M, Ali Shaik, A. Chemoenzymatic synthesis of (S) and (R)-propranolol and sotalol employing one-pot lipase resolution protocol. Bioorg. Med. Chem. Lett. 2004;14:4581–4583. doi: 10.1016/j.bmcl.2004.05.084. [DOI] [PubMed] [Google Scholar]
- 65.Park HI, Turk BE, Gerkema FE, Cantley LC, Sang QX. Peptide substrate specificities and protein cleavage sites of human endometase/matrilysin-2/matrix metalloproteinase-26. J. Biol. Chem. 2002;277:35168–35175. doi: 10.1074/jbc.M205071200. [DOI] [PubMed] [Google Scholar]
- 66.Knight CG, Willenbrock F, Murphy G. A novel coumarin-labelled peptide for sensitive continuous assays of the matrix metalloproteinases. FEBS Lett. 1992;296:263–266. doi: 10.1016/0014-5793(92)80300-6. [DOI] [PubMed] [Google Scholar]