

Abstract
While analyzing plasmids of Acinetobacter sp. DS002 we have detected a circular DNA molecule pTS236, which upon further investigation is identified as the genome of a phage. The phage genome has shown sequence similarity to the recently discovered Sphinx 2.36 DNA sequence co-purified with the Transmissible Spongiform Encephalopathy (TSE) particles isolated from infected brain samples collected from diverse geographical regions. As in Sphinx 2.36, the phage genome also codes for three proteins. One of them codes for RepA and is shown to be involved in replication of pTS236 through rolling circle (RC) mode. The other two translationally coupled ORFs, orf106 and orf96, code for coat proteins of the phage. Although an orf96 homologue was not previously reported in Sphinx 2.36, a closer examination of DNA sequence of Sphinx 2.36 revealed its presence downstream of orf106 homologue. TEM images and infection assays revealed existence of phage AbDs1 in Acinetobacter sp. DS002.
Acinetobacter sp. are of great interest owing to the diverse habitats they colonize, the involvement of certain strains in epidemic outbreaks in hospitals and their various metabolic capabilities like aromatic catabolism, degradation of hydrocarbons in oil spills etc1,2,3. Acinetobacter baumannii is known to be an opportunistic pathogen and is implicated in nosocomial infections, bacteremia, secondary meningitis, urinary tract infections and ventilator-associated pneumonia etc4. Since 1970's A. baumannii has been known to have a clear role in multi-drug resistance. The ever-increasing resistance of A. baumannii against many clinically important antibiotics has been attributed to its proclivity in acquiring the drug resistance genes from among the members of the microbial community5,6. Dissemination of the resistance genes occurs mainly through horizontal gene transfer (HGT)7,8,9,10. Plasmids play a predominant role in HGT and the existence of plasmids carrying genes of high ecological and physiological relevance has been reported for A. baumannii5,11,12,13. In view of its clinical significance, the complete genome sequence has been determined for a number of A. baumannii strains isolated from different geographical regions and in most of them the existence of multiple plasmids has been reported14,15,16,17.
Transmissible spongiform encephalopathies (TSEs) such as Creutzfeldt–Jakob Disease (CJD) and kuru in humans, scrapie in sheep, and BSE in cows are caused by a group of related, but incompletely characterized infectious agents. Prions are responsible for the TSE in a variety of mammals18,19. In all healthy animals, prions (PrPC) are found in the membranes of all the cells and distributed throughout the body. However, Prions (PrPSc) found in TSE-affected animals have an amyloid fold and are resistant to proteases18. Although the precise reasons for conversion of PrPC to PrPSc are unknown, the PrPSc has been shown to induce conversion of PrPC into PrPSc. The prion-only theory suggests that no external agent is involved in conversion of PrPC to PrPSc18. In contrast to this notion, however, a number of studies have implicated an environmental origin of TSE agents due to geographic prevalence and occurrence20,21,22. Studies have also indicated their transmission through the gastrointestinal tract and blood23,24,25. A recent study has identified two circular DNA molecules in the TSE particles purified from infected samples collected from diverse geographical regions26. These two circular DNA molecules were designated as SPHINX sequences using an acronym given for Slow Progressive Hidden INfections of variable (X) latency as they were enriched in infectious preparations. Of these two Sphinx sequences, Sphinx 1.76 has shown 70% sequence similarity to a plasmid, p1ABTCDC0715 isolated from A. baumannii TCDC-AB071527 and a comparable similarity to a plasmid, p2ABAYE found in A. baumannii AYE13. However, no good homologues were found for Sphinx 2.36, the second Sphinx sequence isolated from BSE-infected samples. Sequence similarity was only seen in the DNA region that codes for a replication protein RepA26. In the present study we report the existence of a circular double-stranded DNA in Acinetobacter sp. DS002, with high sequence similarity to Sphinx 2.36. Sequence similarity of 67% between these two DNA molecules was found both in coding and non-coding sequences. There was absolute sequence identity in the region containing replicative elements, such as the double-stranded origin of replication (DSO) and the RepA-coding sequence. The experimental evidence presented in this study suggests that the circular DNA is the genome of a phage capable of replicating via the rolling-circle mode.
Results
A number of Acinetobacter sp. have been shown to have multiple plasmids with different sizes17. Some of them, especially strains of A. baylyi, have the ability to take up linear and circular DNA molecules under natural conditions28. Such an unique ability is attributed to the acquisition of multiple plasmids and resistance genes for multiple drugs from microbial communities6. We have been studying regulation and horizontal mobility of organophosphate-degrading (opd) genes among soil bacteria29,30. Acinetobacter sp. DS002 was isolated from OP pesticide-polluted agricultural soils and the strain name was given based on 16 S rRNA gene sequence identity (100%) with the type strain Acinetobacter baumannii MDR-TJ. The culture deposited in Microbial Type Culture Collection Center (MTCC), IMTECH, Chandigarh, India is available as Acinetobacter sp. DS002 MTCC11451. Acinetobacter sp. DS002 was originally analyzed to detect extra chromosomal genetic material, to establish a link, if any, between the plasmids and OP pesticide degradation. Four indigenous plasmid molecules were detected (Fig. 1a) in this soil isolate and were designated as pTS13 (13 kb), pTS11 (11 kb), pTS5 (5 kb) and pTS236 (2.2 kb). Initial hybridization studies using a well-conserved organophosphate-degrading (opd) gene, gave no signal with any of the detected plasmids or with the chromosomal DNA suggesting that the OP degradation mechanism of Acinetobacter sp. DS002 might be novel. In the process of identifying opd plasmids through complementation, we rescue-cloned the plasmids of Acinetobacter sp. DS002 into E. coli pir116 cells after tagging with a mini-transposon (EZ-Tn5<R6Kγori/KAN-2>), which has a R6Kγ replicative origin. The unique restriction profiles and size indicated that rescue cloning of all identified circular DNA molecules from Acinetobacter sp. DS002 had been successful (Fig. 1, panel b). When sequenced, one of the rescue-cloned plasmids, pTS236, showed significant sequence similarity (67%) to the recently reported Sphinx sequence, Sphinx 2. 36.
Figure 1.

In panel (a) an agarose gel shows the existence of multiple plasmids in Acinetobacter sp. DS002. Lane 1 and 2 represent a kilobase DNA ladder as standard and plasmid preparations from the plasmid-free E. coli strain used as negative control, respectively. The plasmids found in Acinetobacter sp. DS002 are shown in lane 3. Panel (b) shows the restriction profile of rescue-cloned plasmids of Acinetobacter sp. DS002 isolated from E. coli pir116 cells. The plasmids were independently digested with XhoI and BamHI and analyzed on 0.8% (w/v) agarose gels. The total size of the rescue-cloned plasmids was calculated based on the electrophoretic mobility of the DNA fragments generated upon restriction digestion. A portion of the minitransposon released as a XhoI and BamHI fragment is shown with an arrow. While calculating the total size of rescue-cloned plasmids a size of 2 kb based on the size of the minitransposon was deduced. Panel (c) shows the restriction map of the minitransposon-EZ-Tn5<R6Kγori/KAN-2> inserted into rescue-cloned plasmids. The open box represents the minitransposon and the lines flanking the open box indicate the rescue-cloned plasmid. The size of the minitransposon and the 1.0 kb portion released upon digesting with BamHI and XhoI is indicated with inverted arrows.
Sequence analysis of pTS236
The 2252 bp sequence of pTS236 is deposited in the EMBL GenBank and can be accessed with the number JN872565. The open reading frames (ORFs), putative promoter and inverted and imperfect repeats predicted in pTS236 are shown in Supplementary Fig. S1. Only three ORFs were identified in the sequence of pTS236. One of them showed sequence similarity to repA, which is generally found in plasmids that exhibit rolling-circle replication. The other two ORFs that code for proteins 106 and 96 amino acids in length had no sequence similarities to any known protein sequences available in the databases and hence were designated as orf106 and orf96. These two ORFs appear to be translationally linked, as the predicted translational stop codon of orf106 overlaps with the start codon of orf96. Moreover, a consensus σ70-dependent promoter was identified only in the upstream region of orf106 and no predicted promoter sequences were found upstream of orf96 (Supplementary Fig. S1). The absence of promoter elements upstream of orf96 and the existence of translational coupling strongly suggested these two ORFs formed an operon (Supplementary Fig. S1). While gaining experimental evidence, the total RNA extracted from Acinetobacter sp. DS002 cells was used as template and RT-PCR was performed using orf106-specific forward and orf96 reverse primers. An amplicon equivalent to the size of these two ORFs was amplified indicating that these two adjacent ORFs are organized as an operon (Supplementary Fig. S2).
A 12 bp long sequence that matched perfectly the double-stranded origin of replication (DSO) typically found in rolling-circle (RC) replicating plasmids was found in the intergenic region between orf96 and repA (Supplementary Fig. S1). Indeed, the −10 hexameric sequence of a putative σ70-dependent promoter of the repA gene was located in the middle of the DSO sequence motif (Supplementary Fig. S1).
RepA translation starts from the UUG codon
The bioinformatic tool GLIMMER, which is used to predict ORFs, has indicated that initiation of RepA translation occurs at an UUG start codon, which is located 21 bp downstream of the putative promoter element of repA. The first AUG codon was found 513 bp downstream of the putative promoter element. The predicted amino acid sequence coded by the DNA region present between these two codons shows strong sequence similarity to the N-terminal portion of the RepA sequence coded by a well-characterized RC plasmid pPP8131, strengthening the supposition of a possible initiation of RepA synthesis from the UUG codon. As this is an unusual observation, we next determined whether RepA is indeed translated from the UUG codon. We initially collected all rescue-cloned pTS236 plasmids with a single mini-transposon insertion and established their restriction profile to locate precisely the point of mini-transposon insertion. In one of the rescue-cloned pTS236 derivatives, the mini-transposon was inserted between predicted start codon UUG of RepA and the conventional start codon AUG (Fig. 2a). This pTS236 variant, pTS236-K failed to replicate in pir negative E. coli cells (Fig. 2e). This derivative was therefore used to develop a complementation assay to assess the functional status of RepA expressed from these two putative translational start codons. Two expression plasmids were constructed by complementing with an expression plasmid encoding RepA either from the predicted start codon UUG (pTRW1) or from the conventional start codon AUG (pTRW2). Initially these expression plasmids were transformed into E. coli BL21(DE3) cells and were used as host to co-transform with plasmid pTS236-K. When selected on kanamycin plates, which indicates the ability of pTS236-K to replicate, growth occurred only with E. coli BL21 (pTRW1) cells encoding RepA initiating from codon UUG (Fig. 2d, e). No growth was found in E. coli cells with RepA encoded from AUG, suggesting that the N-terminal portion of RepA specified by the sequence found upstream of the AUG is essential for the RepA function. Supporting this result, a signal similar to the size of RepA coded from codon UUG was obtained when total proteins of Acinetobacter sp. DS002 were probed with RepA specific antibodies (Fig. 2f).
Figure 2.

Panel (a) shows the pTS236-K map with a minitransposon insertion between the predicted start codon UUG and a single AUG codon of repA specifying 165-methionine residue. Arrows indicate the transcriptional orientation of repA, orf106 and orf96. The DSO found upstream of repA start codon UUG is shown as a filled square. Panel (b) shows the physical map of the DSO and repA region of pTS236. DSO is shown as a hatched box. The extent of repA cloned to generate expression plasmids coding RepA (pTRW1) from predicted start codon UUG and from the AUG specifying 165-methionine (pTRW2) are indicated with solid lines. Panel (c) shows an SDS-PAGE and the corresponding western blot probed using anti-His antibody. Lane 1 represents the protein molecular mass marker. Lanes 2 and 3 show protein extracts prepared from uninduced E. coli BL21(DE3) cells having either pTRW1(lane 2) or pTRW2 (lane 3). Lanes 4 and 5 represent similar extracts prepared from induced cultures. RepA-specific signals seen in induced cultures are shown with arrows. Panel (d) and (e) represent replication of pTS236-K in permissive (E. coli pir116) and non-permissive (E. coli BL21) hosts. Panel (d) shows growth on LB with ampicillin and kanamycin plates of E. coli pir116 (pTS236-K) containing expression vector pET23b (sector 1), pTRW1 (sector 2), pTRW2 (sector 3), pTRM (sector 4). Sectors 5 and 6 represent growth of E. coli pir116 (pTRW1) having plasmids pT106M and pT96M, respectively. Growth of E. coli BL21 carrying the same plasmids is shown in panel (e). Panel (f) shows an SDS-PAGE and the corresponding western blots generated using RepA specific antibodies. Lane 1 represents total proteins of Acinetobacter sp. DS002. Lanes 2 and 3 are protein extracts prepared from E. coli BL21 cells expressing RepA from predicted start codon with or without C-terminal histidine tag, respectively.
Tyrosine 265 is essential for RepA function
Plasmids that replicate using the rolling-circle mechanism have a functionally conserved tyrosyl residue near the C-terminus (Supplementary Fig. S3). This invariant tyrosyl residue has been implicated in initiation of the replication process by generating a nick at DSO32. The RepA of pTS236, when aligned with the RepA of pPP8131, a well characterized RC plasmid, as well as with RepA proteins from other RC plasmids isolated from Staphylococcus aureus33,34 and Lactobacillus hilgardii35, have a high similarity throughout the protein, particularly in the segment with the conserved tyrosyl residue (Supplementary Fig. S3). In order to assess if the conserved tyrosine of RepA plays a role in replication of pTS236, we performed a complementation assay by transforming plasmid pTS236-K into E. coli BL21 cells having expression plasmids coding for either wild type (pTRW1) or mutant (pTRM) RepA proteins (Fig 2 d, e). Only BL21 with the plasmid pTRW1 exhibited kanamycin resistance suggesting that replication of plasmid pTS236-K was dependent on the invariant tyrosine residue. A similar complementation assay was performed to determine whether the ORFs orf106 and orf96 have any role in the replication process. Variants of pTS236-K with a translational termination codon incorporated immediately downstream of the predicted start codon of either orf106 or of orf96 successfully replicated in E. coli BL21cells (pTRW1) (Fig. 2e). This indicates that neither ORF is required for replication of pTS236-K.
RepA DSO interactions
In order to assess any interaction of RepA with the predicted DSO of pTS236, recombinant RepA6His was purified and used to perform two independent in vitro studies. Initially recombinant RepA6His was incubated with the supercoiled form of pTS236-K and the ability of it to form the OC species of the plasmid was analyzed. Generation of the OC form of pTS236-K increased both in a time- and concentration-dependent manner upon incubation with RepA and no conversion was seen in control samples prepared if RepA was omitted (Fig. 3a). Though the data clearly suggests a possible RepA-dependent generation of the OC species from the supercoiled plasmid, it does not indicate whether the generated nick was in the DSO region. Initially, about 100 bp of sequence flanking the predicted DSO was amplified using primers DS00101and DS00102 and the 32P end-labeled amplicon was used to perform electrophoretic mobility shift assay (EMSA) by incubating with pure recombinant RepA6His. An apparent shift was observed in the mobility of the amplicon due to mobility retardation of the DSO-RepA complex (Fig. 3b). The complex was not dissociated in the presence of BSA or herring sperm DNA. Suggesting that the interactions between DSO and RepA were specific, the labeled DSO dissociated from DSO-RepA complex when challenged with cold DSO. Thus, it is likely that generation of OC of pTS236-K was due to introduction of a nick in the predicted DSO (Fig. 4b). In plasmids replicating through RC mode, a considerable amount of single-stranded (SS) intermediate is always seen in actively grown cultures36. Before designating pTS236 as RC plasmid with reasonable confidence, experiments were conducted to detect the SS form of pTS236 in cultures of Acinetobacter sp. DS002 following established procedures37. As seen in the RC plasmids, a strong signal was obtained only in the plasmid samples that were not treated with S1 nuclease. Since S1 nuclease treatment preferentially eliminated SS form of pTS236 no such signal was seen, giving a clear indication that pTS236 is a RC plasmid (Fig. 5).
Figure 3. RepA-mediated generation of open-circular (OC) form of plasmid pTS236-K from a super-coiled (SC) form.

Panel (a) indicates SC to OC conversion of pTS236-K against time (I) or with increasing concentration of RepA (II). The pTS236-K incubated with increasing concentrations of RepA(Y265F) is shown in panel (a) III. Electrophoretic Mobility Shift Assay (EMSA) for DSO and RepA is shown in panel (b). A shift in DSO mobility was observed due to the formation of DSO-RepA complex when labelled DSO was incubated with increased concentrations of RepA. Control represents 32P-labelled DSO without RepA. The DSO mobility shift is shown with an arrow (C) while F represents the free probe.
Figure 4. Alignment of Double-Stranded Origin of replication (DSO) of plasmid pTS236 with the known rolling circle replicating plasmids (Panel a).
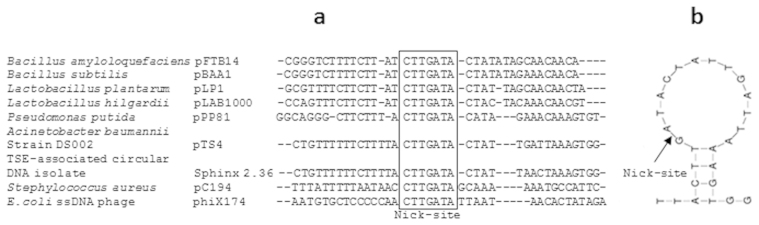
Panel (b) shows the secondary structure of DSO. The proposed site of the nick is shown with an arrow.
Figure 5. Detection of the single-stranded intermediate of pTS236: Lane 1 represents 1 kb DNA ladder, lanes 2 and 3 represent total plasmid preparation treated with (2) and without (3) S1 nuclease.
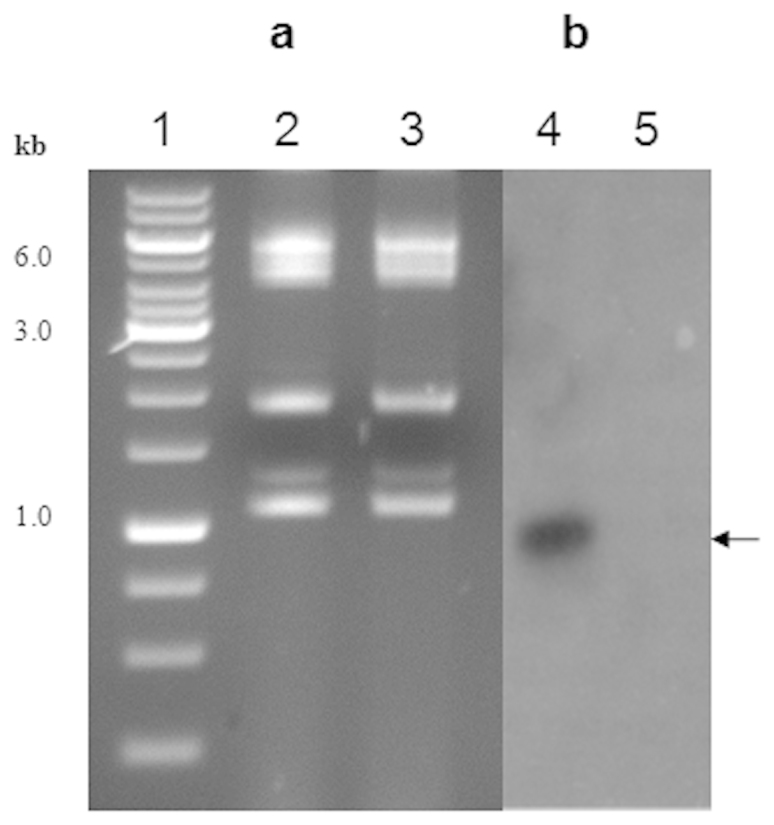
Lanes 4 and 5 represent the corresponding autoradiogram developed using labelled pTS236 as probe. Single-stranded pTS236 is shown with an arrow mark.
The pTS236 is a homologue of Sphinx 2.36
Although database searches to find homologues of pTS236 identified repA genes from similar RC plasmids such as pPP8131, a clear overall sequence similarity was only seen with the recently reported Sphinx 2.36 DNA sequences. About 67% identity was found between these two circular DNA molecules. The DSO sequences of both plasmids were identical and they were found to be highly similar to the DSOs of other RC plasmids (Fig. 4a). Interestingly, the Sphinx 2.36 plasmid also codes for Orf106 and Orf96 homologues (Supplementary Fig. S4). Though existence of orf96 was not predicted in the reported sequence, a thorough analysis of the Sphinx 2.36 sequence revealed its presence immediately downstream of the orf106 gene. As seen in pTS236, the two ORFs in Sphinx 2.36 appear to be translationally coupled, suggesting functional conservation of the gene products.
Despite sharing identical DSO sequences, the RepA sequences were not so highly conserved between pTS236 and Sphinx 2.36 (Supplementary Fig. S1, S4). Only 61% similarity was seen between these two RepA amino acid sequences encoded by these two circular DNA molecules. This observation prompted us to reanalyze the repA sequence of Sphinx 2.36 to identify possible frame-shifts in the sequence. Initially, the proteins coded in all six frames of the repA sequence were aligned with the RepA sequence of pTS236. The protein encoded by frame + 2 was 47% similar to the N-terminal part of pTS236 RepA. Similarly, the protein encoded by frame + 1 was again similar (43%) but with the C-terminal portion of pTS236 RepA. The RepA sequence reported by Manuelidis was encoded by frame + 3 and it matched the RepA of pTS236 from amino acids 48 to 28126. Reconstruction of the RepA-coding sequences into a single frame by arbitrarily changing the sequence revealed a gene product with 57% identity to the RepA of pTS236. Interestingly, the Sphinx 2.36 repA also has a predicted UUG translation initiation codon, along with a putative promoter overlapping DSO (Supplementary Fig. S4). Taken together, these data suggest possible frame-shifts in the coding region, which could result either from sequencing errors or mis-incorporation of bases while amplifying the Sphinx 2.36 sequence by using ø29-polymerase from infected brain samples26. Although no data is available to prove that the predicted RepA of Sphinx 2.36 is functional, nevertheless, if the experimental evidence shown in this study is taken into consideration, it suggests that RepA initiated from the alternative UUG codon might be needed for replication of Sphinx 2.36 in neuronal cells. A better understanding will only be possible through a new analysis of the sequence of Sphinx 2.36 repA.
pTS236 is the genome of a phage
Due to existence of a pTS236 homologue in brain samples of TSE-infected animals we have done further experiments to determine whether pTS236 is a genome of a phage infecting Acinetobacter sp. DS002 cells. The PEG precipitate obtained from the culture supernatant was taken and immuno-purified (IP) by passing through a protein-A column conjugated with polyclonal antibodies raised against Orf96. The pure affinity-purified particles were then examined by transmission electron microscopy (TEM). Figure 6 shows the electron micrograph of icosahedral virions (Panel - Ia). From among these virions, a total of about 50 isolated particles (some selected virions are shown in Fig. 6, panel Ic) were used for generating a class average using the EMAN software tool38 and this is shown in Fig. 6, panel - Ib. The icosahedral features that are seen in the class average (Fig. 6, panel Ib) confirms the formation of virions. In support of this data the immuno-purified phage particles also gave a signal when western blots were performed using Orf96 and Orf106 specific antibodies (Fig. 6, panels II, III). The obtained signals were found to be bigger than the recombinant Orf96 and Orf106, indicating formation of SDS-resistant Orf96 and Orf106 multimers (Fig. 6, panels II, III). Multimerization of Orf96 and Orf106 was also evident when western blots were performed for cellular proteins extracted from Acinetobacter sp. DS002 cells. A Orf96-specific ladder-like signals were seen in westerns blots (Supplementary Fig. S5). Even the PCR reaction performed using immuno-purified phage particles as template gave pTS236-specific amplicons suggesting the existence of pTS236 in the icosahedral-shaped phage particles (Fig. 6, panel IV). Identical results were obtained when a PIP (phage immuno-purification) assay was repeated using Orf106 antibodies. However, neither phage particles nor pTS236-specific amplifications were seen when similar assays were performed using an unspecific anti-organophosphate hydrolase (OPH) antibody. All these results confirm the specificity of the PIP (phage immuno-purification) assays and clearly indicate that the Orf96 and Orf106 are the coat proteins of isolated phage.
Figure 6.

Panel I (a): TEM image of phage AbDs1. Out of the 50 isolated particles used for generating a class average, a few selected virions are shown in panel (c). The icosahedral features that are seen in the class average generated using the EMAN software tool, confirms the formation of virions (panel b). Panel (II) shows a western blot analysis of immune-purified phage AbDs1 using Orf96-specific antibodies. Lane 1 represents the molecular mass marker, lanes 2 and 3 are loaded with phage proteins and recombinant Orf96, respectively. Lanes 4 and 5 show the corresponding western blots. The western blot performed using Orf106-specific antibodies is shown in panel (III). Lanes 2 and 3 represent recombinant Orf106 and proteins of phage AbDs1, respectively. Lanes 4 and 5 show the corresponding western blots. Amplification of pTS236-specific ORFs from immune-purified AbDs1 phage particles is shown in panel (IV). Lane 1 represents kilo base ladder. Lanes 2, 3 and 4 show amplification of orf96, orf106 and repA, respectively.
While gaining further information on the infectivity of identified phage, the Acinetobacter sp. DS002 was transformed with pTS236 variant pTS236-K1. The immuno-purified phage particles from the culture supernatant of Acinetobacter sp. DS002 (pTS236-K1) cells were re-infected into wild type cells of Acinetobacter sp. DS002. The wild type Acinetobacter sp. DS002 gave kanamycin-resistant colonies after 11 hours of infection (Fig. 7b). No such colonies were observed in Acinetobacter sp. DS002 cells incubated with purified pTS236-K1 DNA even at a concentration of 1 μg/ml in LB medium.
Figure 7.

Panel (a) indicates physical map of pTS236-K1. Panel (b) indicates the generation of kanamycin resistant Acinetobacter sp. DS002 colonies after infection with AbDs1 (pTS236-K1). Amplification of the kanamycin gene from immune-purified phage particles, AbDs1 (pTS236-K1), is shown in panel (c). Lane 1 represents the kilobase ladder, lanes 2 and 3 are loaded with the PCR-amplified kanamycin gene from AbDs1 and AbDs1 (pTS236-K1).
Discussion
We initiated the current study to gain clearer insights into the plasmid profile of Acinetobacter sp. DS002 isolated from agricultural soils polluted with OP insecticides. The circular DNA, pTS236 was the smallest of the rescued plasmids of Acinetobacter sp. DS002. Analysis of its sequence led to the discovery of a link between pTS236 and Sphinx 2.36. The Sphinx 2.36 sequence matches throughout its length the sequence of pTS236 and shows an overall 67% match at the nucleotide sequence level. In a recent network analysis, only the pTS236 homologue, p4ABAYE, showed no apparent link with existing Acinetobacter plasmids13. This lack of evolutionary linkage prompted the question as to whether pTS236 might be a replicative form of a phage genome propagating using Acinetobacter sp. DS002 as a host. We have therefore examined a polyethylene glycol precipitate of the spent medium by TEM. Our initial observations under TEM gave no indication of the presence of phage particles and hence it was assumed that pTS236 is a unique plasmid found in Acinetobacter sp. DS002 cells. However, further purification steps of the PEG precipitate of the spent medium enriched phage particles and the existence of icosahedral phage particles was confirmed (Fig. 6, panel I). These pure phage particles have also cross-reacted with Orf106 and Orf96 antibodies and the viral immune-precipitation (VIP) assay performed to amplify pTS236 from immune-purified viruses amplified pTS236-specific sequences (Fig. 6 II, III, IV). If all these results are taken into consideration, encapsulation of pTS236 DNA is evident in an icosahedral protein coat generated by Orf106 and Orf96.
Acinetobacter sp. are robust microbes that can adapt to a variety of habitats, including soil. They can easily gain entry into animals while grazing or drinking water. In fact, they were implicated in TSE due to the existence of high levels of Acinetobacter-specific antibodies in infected animals that tested BSE-positive39. An amino acid sequence similarity has also been identified between a bovine prion sequence (RPVDQ) and uridine diphosphate-N-acetyl glucosamine-1-carboxy-vinyl transferase of A. calcoaceticus. In further support of such an unusual observation, even the class specific antibodies were significantly elevated against structurally related synthetic peptides40. Following these important observations, serious efforts were made to find a possible epidemiological link between BSE and Acinetobacters. However, no solid evidence was found to support this decade-old proposition.
Prion involvement in BSE has been proved beyond doubt18,19, and in all TSE-infected animals, the prion protein has an amyloid fold resistant to proteases. A major unsolved question in BSE is the identification of the factor or factors responsible for its misfolding. Though a ‘protein only’ theory has gained wider acceptance, detection of nucleic acids in TSE preparations supported the hypothesis that contradicts this otherwise established notion25,41. One school of thought which argues against the protein-only theory suggests involvement of a nucleic acid-based ‘cofactor’ during conversion of the cellular form of the prion, PrPC into the disease-causing, protease-resistant PrPSc 42,43. In support of this notion, a vertebrate single-stranded RNA was shown to act as a ‘cofactor’ for the in vitro amplification of PrPSc 44. Though no viral involvement is shown in BSE, virus-like particles were found associating with BSE-infected samples43.
The present study was neither designed to gather evidence in support of a particular school of thought nor do the findings support any of the existing hypotheses. It rather provides strong evidence to link the Sphinx 2.36 sequence found in BSE-infected samples to the genome of Acinetobacter sp. DS002 phage. These results should be taken into account when considering the findings of Manuelidis who identified the existence of viral particles in BSE-infected samples with Sphinx 2.36 as its genome26. Moreover, BSE-infected samples should be thoroughly examined to determine whether samples have Acinetobacter contamination. Even if the possibility of Acinetobacter contamination can be ruled out, nevertheless the presence of Acinetobacter phage-specific circular DNAs in brain samples of infected animals is too convincing to be ignored. Though no direct involvement of Sphinx has been demonstrated in the generation of PrPSc, their amplification, particularly in infected brain samples is potentially significant. Even in the absence of a causal link between Sphinx and TSE, the existence of a circular DNA in mammalian brain, which has a resemblance to a genome of an Acinetobacter phage is an important observation.
The existence of plasmid sequences homologous to the second Sphinx sequence, Sphinx 1.76, have been reported in Acinetobacter baumannii TCDC-AB071527 and Acinetobacter baumannii AYE13. If the sequence similarity is seen in the light of the present findings, most likely the reported circular DNA as plasmids p1ABTCDC0715 and p2ABAYE can be considered to be genomes of phage particles.
Methods
Bacterial culture and gene manipulations
Bacterial strains and plasmids used in this study are listed in Table 1. The primer sequences are given in Supplementary Table S1. Escherichia coli cells were grown at 37°C in LB medium. Acinetobacter sp. DS002 was grown at 30°C either in LB medium or in minimal medium prepared by dissolving 4.8 g of K2HPO4, 1.2 g of KH2PO4, 1 g of NH4NO3, 0.2 g of MgSO4.7H20, 0.04 g of Ca(NO3)2.4H20 and 0.001 g of Fe2(SO4)3 in 1000 ml of distilled water. Succinate (10 mM) was supplemented as a carbon source. When necessary, antibiotics ampicillin (100 μg/ml), kanamycin (30 μg/ml), streptomycin (20 μg/ml) and chloramphenicol (30 μg/ml) were added to the growth medium. Indigenous plasmids were isolated from Acinetobacter sp. DS002 essentially by following the Currier-Nester protocol45. Molecular cloning, blotting techniques and electroporation of E. coli and Acinetobacter sp. DS002 were performed following procedures described elsewhere46. Detection of a single-stranded intermediate of pTS236 in Acinetobacter sp. DS002 was done by following procedures described elsewhere37. The 16 S rRNA gene cloning and phylogenetic tree construction was done as described previously47.
Table 1. Bacterial strains and plasmids.
| S.N. | Strain/Plasmid/Primer Name | Genotype/Phenotype/Sequence Description | Reference/Source/Remark |
|---|---|---|---|
| Strains | |||
| 1 | E. coli DH5α | fhuA2 Δ(argF-lacZ)U169 phoA glnV44 Φ80 Δ(lacZ)M15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | 58 |
| 2 | E coli BL21 DE3 | hsdS gal(λcIts857 ind1 sam7 nin5lac UV5 T7 gene 1 | 59 |
| 3 | E. coli pir116 | F- mcrA Δ(mrr-hsdRMS-mcrBC) φ80dlacZΔM15 ΔlacX74 recA1 endA1 araD139 Δ(ara, leu)7697 galU galK λ- rpsL (StrR) nupG pir-116(DHFRv) | EPICENTRE Biotechnologies |
| 4 | Acinetobacter sp. DS002 | Smr and Cmr | This work |
| Plasmids | |||
| 5 | pET23b | Apr, Expression vector | Novogen |
| 6 | pTRW1 | Apr, repA cloned in pET23b from predicted start codon TTG | This work |
| 7 | pTRW2 | Apr, repA cloned in pET23b from its first ATG codon | This work |
| 8 | pTRM | Expression plasmid coding for RepA Y265F | This work |
| 9 | pTS236K | Rescued cloned pTS236 having minitransposon in the intergenic region of repA and orf96 | This work |
| 10 | pTS236K1 | pTS236 having kanamycin cassette at unique Mlu I site found in the intergenic region of repA and orf106 | This work |
| 11 | pT106M | pTS236-K having termination codon immediately downstream to the start codon of orf106 | This work |
| 12 | pT96M | pTS236-K having termination codon immediately downstream to the start codon of orf96 | This work |
Rescue-cloning of plasmids from Acinetobacter sp. DS002
Plasmids were rescue-cloned by tagging the mini-transposon with a R6Kγori replicative origin. The isolated plasmids were treated with plasmid safe (EPICENTRE Biotechnologies, USA) to remove linear DNA and were used to tag with EZ-Tn5™ <R6Kγori/KAN-2> using the Transposon Insertion Kit (EPICENTRE Biotechnologies, USA). A 10 μl reaction mixture contained 1 μl of 10 × buffer, 1 μg of plasmid DNA, an equimolar concentration of transposon and 1 U of transposase. The reaction mixture was incubated for 2 hours at 37°C and the reaction was stopped by adding 1 μl of stop solution followed by incubation of the reaction mixture at 70°C for 10 min. A 1 μl aliquot of the transposition mixture was then electroporated into E. coli EC100D pir-116 cells. The KanR colonies were independently subcultured in LB medium supplemented with arabinose (1 mM) and the rescued plasmids were isolated to obtain their restriction profile and those which gave an unique restriction profile were selected for sequence determination.
Direct sequencing of rescue-cloned plasmid pTS236
After isolating mini-transposon-tagged Acinetobacter sp.DS002 plasmids, the rescue-cloned plasmids were sequenced using transposon-specific primers followed by primer walking. The sequence was analyzed using bioinformatic tools available online. The NCBI GLIMMER programme48 was used to predict open reading frames (ORFs) and for promoter prediction, BPROM software (www.softberry.com/berry.phtml?topic=bprom) was used. NCBI BLAST49, EBI ClustalW250 and REPFIND software51 were used for sequence similarity search, multiple sequence alignment and for finding repeat sequences, respectively. The double-stranded origin (DSO) of replication was identified by aligning the pTS236 DNA sequence with known sequences of other rolling-circle plasmids available in the database52. DNA secondary structure was predicted using the Mfold programme53.
Expression and purification of RepA
The strategy followed for expression of RepA from the predicted start codon UUG and from its first conventional AUG as shown in Fig. 2b. While expressing RepA from the predicted start codon UUG, the repA gene was amplified from plasmid pTS236 using the forward (DS00109) and reverse (DS00110) primers carrying EcoRI and XhoI restriction recognition sequences, respectively. The repA amplicon was then digested with EcoRI and XhoI and ligated into pET23b digested with the same enzymes. The cloning strategy places repA in-frame with the vector-encoded His-tag of pET23b and the recombinant plasmid pTRW1, which encodes RepA with a C-terminal His-tag. The RepA6HIS induced in E. coli BL21 (pTRW1) was affinity-purified by using a Ni-Sepharose High Performance column (XK16/20, GE Healthcare) following procedures described elsewhere54. A similar strategy was followed while cloning of repA from its first AUG except that the primers DS00111 and DS00110 were used as forward and reverse primers and the resulting expression plasmid is named as pTRW2. In both the cases start and stop codons were modified to facilitate incorporation of EcoRI and XhoI sites at 5′ and 3′ ends of repA sequence.
Site-directed mutagenesis
Site-directed mutagenesis was performed following the protocol described elsewhere55. To replace the codon specifying tyrosine in repA with the codon specifying phenylalanine, oligos DS00103 and DS00104 were used. Similarly, oligos DS00105/DS00106 and DS00107/DS00108 were used to introduce termination codons in all frames immediately downstream of the start codon AUG of orf106 and orf96, respectively. In all PCR reactions plasmid pTS236-K was used as template. After performing standard PCR using Pfu-polymerase the amplicons were digested with DpnI and the DpnI-resistant plasmids were transformed into E. coli pir116 cells and the presence of the desired mutation was confirmed by determining the sequence of the entire gene.
Determination of RepA/DSO interactions
Both electrophoretic mobility shift assay (EMSA) and RepA-dependent conversion of supercoiled (SC) pTS236 to the open circular (OC) form were used to determine RepA-DSO interactions. The gel extracted supercoiled pTS236-K was used as substrate to determine RepA-dependent nick generation. About 0.8 μg of supercoiled pTS236 was incubated at 32°C with 170 ng of RepA in a reaction mixture (30 μl) containing 10 mM Tris-HCl (pH 8.0), 100 mM KCl, 10 mM Mg(OAc)2, 1 mM EDTA, and 10% (w/v) ethylene glycol. The reactions were stopped by adding 4 μl of 20% (w/v) EDTA at different time intervals and samples were analyzed on 0.8% (w/v) agarose gels. A similar analysis was made by incubating the supercoiled pTS236-K with increasing concentrations of RepA. The RepA-dependent conversion of pTS236-K (SC) to pTS236-K (OC) was identified based on changes observed in the band intensities of the SC and OC forms of pTS236-K in dependence on time and protein concentration.
EMSA was performed as described previously56. A 100 bp DNA fragment containing the DSO region was amplified using the primer pair DS00101 and DS00102 and the amplicon was end-labeled following the standard protocols46. The following amounts (0 ng, 100 ng, 250 ng, 500 ng and 1000 ng) of purified RepA6His were incubated with 2 picomoles of the labeled probe in 20 μl of binding buffer (20 mM Tris-HCl [pH 8.0], 1.0 mM EDTA, 6 mM MgCl2, 50 mM KCl, 50 μg/ml bovine serum albumin [BSA], and 5% (w/v) glycerol) containing 5 μg/ml of herring sperm DNA for 20 min at 25°C. The DNA-protein mixture was resolved on a 5% (w/v) native polyacrylamide gel and was analyzed by autoradiography.
Phage immuno-purification (PIP)
Polyclonal antibodies against recombinant Orf966His and Orf1066His were raised in male New Zealand (O.B) rabbits following standard procedures. The polyclonal antibodies raised against these two recombinant proteins were affinity purified and used for performing western blots and immuno-purification (IP) experiments. After establishing the specific reactivity of the antibodies with their respective antigens they were independently conjugated to a protein-A column. The culture supernatant collected from Acinetobacter sp. DS002 cultures grown for 24 hrs were PEG-precipitated by bringing the culture supernatant to a final concentration of 10% PEG and 1 M sodium chloride. After leaving the mixture overnight at 4°C it was centrifuged at 15, 000 rpm for 20 min to collect the precipitate. The precipitate was then re-dissolved in 20 mM sodium phosphate buffer, pH 7 and passed through protein-A columns linked to either polyclonal antibodies of Orf966His or Orf1066His. The columns were washed thoroughly with 20 mM sodium phosphate buffer, pH 7 and the particles bound to columns were eluted by passing Glycine- HCl, pH 2.7. The bound material collected was then used to perform western blots using both Orf96 and Orf106 antibodies and to amplify ORFs encoded by particles bound to the affinity column.
TEM images of phage AbDs1
The sucrose gradient-purified virions were adsorbed to a freshly glow-discharged 200 mesh carbon coated copper grid (Ted Pella Inc. USA) and stained with an aqueous solution of uranyl acetate (2% w/v). Electron micrographs of these virions were recorded on a FEI Technai G2 SuperTwin 200 instrument operating at an acceleration voltage of 120 kV. A magnification of 19,500 × with an effective magnification of 100 nm at the specimen level was used to record the micrographs. The images were recorded using a Gatan Orius 803 digital camera (2 k × 2 k) corresponding to 3.422 Å/pix at the specimen level as calibrated with the help of the calibration grid (Electron Microscopy Sciences, Hatfield, PA 19440, USA). A total of about 50 isolated particles (some selected virions are shown in Fig. 6, panel Ic) were taken for generating a class average using the EMAN software tool38 and to see the icosahedral features, typically seen during the formation of virions.
Infection assay
The pTS236 has unique Mlu I site in the intergenic region of repA and orf106. The kanamycin cassette amplified from pUC4K57 was amplified using primers, DS00112 and DS00113, appended with a Mlu I recognition site. This amplicon was then cloned into the Sphinx 2.36 homologue, pTS236 digested with the same enzyme. The recombinant circular DNA, named as pTS236-K1 (Fig. 7a) was then electroporated into Acinetobacter sp. DS002 cells and selected on kanamycin-containing agar plates. The supernatant collected from the kanamycin-resistant cultures of Acinetobacter sp. DS002 (pTS236-K1) was PEG-precipitated and passed through a protein-A column conjugated with Orf966His specific polyclonal antibodies. After repeated washings, the bound material was eluted as described in earlier sections and used to seed mid-log phase Acinetobacter sp. DS002 cells. A portion of culture was withdrawn every hour and serially diluted culture was then plated on kanamycin-containing agar plates to observe the generation of kanamycin-resistant colonies. A graph was plotted overtime on the X-axis against the number of kanamycin-resistant CFUs on the Y-axis (Fig. 7b). The affinity-purified phage particles from Acinetobacter sp. DS002 (pTS236-K1) were used as template to amplify the kanamycin-resistance gene (Fig. 7c).
Author Contributions
D.S. conceived and designed the study. T.L., S.K., G.P. executed the work. V.R.M., S.A., G.K. and M.A. purified the virions and obtained TEM. images. D.S., T.L., S.K. and G.K. wrote the manuscript.
Supplementary Material
Acinetobacter phage genome is similar to Sphinx236, the circular DNA copurified with TSE infected particles
Acknowledgments
We thank Dr. Gary Sawers for reading the manuscript. TL is the recipient of a Senior Research Fellowship from CSIR, New Delhi. SK and VRM received financial support from UGC-SAP and CSIR. SA and GP received financial support from DRDO and DBT-CREBB. The Centre for Nanotechnology is greatly acknowledged for permitting the use of their TEM facility. The research in the laboratory of DS is supported by CSIR, DBT and DRDO.
References
- Minas W. & Gutnick D. L. Isolation, characterization, and sequence analysis of cryptic plasmids from Acinetobacter calcoaceticus and their use in the construction of Escherichia coli shuttle plasmids. Appl Environ Microbiol 59, 2807–2816 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reams A. B. & Neidle E. L. Genome plasticity in Acinetobacter: new degradative capabilities acquired by the spontaneous amplification of large chromosomal segments. Mol Microbiol 47, 1291–1304 (2003). [DOI] [PubMed] [Google Scholar]
- Fischer R., Bleichrodt F. S. & Gerischer U. C. Aromatic degradative pathways in Acinetobacter baylyi underlie carbon catabolite repression. Microbiology 154, 3095–3103 (2008). [DOI] [PubMed] [Google Scholar]
- Towner K. J. Acinetobacter: an old friend, but a new enemy. J Hosp Infect 73, 355–363 (2009). [DOI] [PubMed] [Google Scholar]
- Bergogne-Berezin E. & Towner K. J. Acinetobacter spp. as nosocomial pathogens: microbiological, clinical, and epidemiological features. Clin Microbiol Rev 9, 148–165 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodford N., Turton J. F. & Livermore D. M. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35, 736–755 (2011). [DOI] [PubMed] [Google Scholar]
- Valenzuela J. K. et al. Horizontal gene transfer in a polyclonal outbreak of carbapenem-resistant Acinetobacter baumannii. J Clin Microbiol 45, 453–460 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec A. et al. Emergence of carbapenem resistance in Acinetobacter baumannii in the Czech Republic is associated with the spread of multidrug-resistant strains of European clone II. J Antimicrob Chemother 62, 484–489 (2008). [DOI] [PubMed] [Google Scholar]
- Durante-Mangoni E. & Zarrilli R. Global spread of drug-resistant Acinetobacter baumannii: molecular epidemiology and management of antimicrobial resistance. Future Microbiol 6, 407–422 (2011). [DOI] [PubMed] [Google Scholar]
- Rumbo C. et al. Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob Agents Chemother 55, 3084–3090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett P. M. Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br J Pharmacol 153 Suppl 1, S347–357 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T. L., Wu R. C., Shaio M. F., Fung C. P. & Cho W. L. Acquisition of a plasmid-borne blaOXA-58 gene with an upstream IS1008 insertion conferring a high level of carbapenem resistance to Acinetobacter baumannii. Antimicrob Agents Chemother 52, 2573–2580 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fondi M. et al. Exploring the evolutionary dynamics of plasmids: the Acinetobacter pan-plasmidome. BMC Evol Biol 10, 59 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. G. et al. New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev 21, 601–614 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams M. D. et al. Comparative genome sequence analysis of multidrug-resistant Acinetobacter baumannii. J Bacteriol 190, 8053–8064 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacono M. et al. Whole-genome pyrosequencing of an epidemic multidrug-resistant Acinetobacter baumannii strain belonging to the European clone II group. Antimicrob Agents Chemother 52, 2616–2625 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallenet D. et al. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PLoS One 3, e1805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner S. B. Prions. Proc Natl Acad Sci U S A 95, 13363–13383 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colby D. W. & Prusiner S. B. Prions. Cold Spring Harb Perspect Biol 3, a006833 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L., Chakrabarty T., Miyazawa K., Nduom N. A. & Emmerling K. The kuru infectious agent is a unique geographic isolate distinct from Creutzfeldt-Jakob disease and scrapie agents. Proc Natl Acad Sci U S A 106, 13529–13534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L., Liu Y. & Mullins B. Strain-specific viral properties of variant Creutzfeldt-Jakob disease (vCJD) are encoded by the agent and not by host prion protein. J Cell Biochem 106, 220–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. Transmissible encephalopathy agents: virulence, geography and clockwork. Virulence 1, 101–104 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. Dementias, neurodegeneration, and viral mechanisms of disease from the perspective of human transmissible encephalopathies. Ann N Y Acad Sci 724, 259–281 (1994). [DOI] [PubMed] [Google Scholar]
- Shlomchik M. J., Radebold K., Duclos N. & Manuelidis L. Neuroinvasion by a Creutzfeldt-Jakob disease agent in the absence of B cells and follicular dendritic cells. Proc Natl Acad Sci U S A 98, 9289–9294 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. Transmissible encephalopathies: speculations and realities. Viral Immunol 16, 123–139 (2003). [DOI] [PubMed] [Google Scholar]
- Manuelidis L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 17, 131–145 (2011). [DOI] [PubMed] [Google Scholar]
- Chen C. C. et al. Genome sequence of a dominant, multidrug-resistant Acinetobacter baumannii strain, TCDC-AB0715. J Bacteriol 193, 2361–2362 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaneechoutte M. et al. Naturally transformable Acinetobacter sp. strain ADP1 belongs to the newly described species Acinetobacter baylyi. Appl Environ Microbiol 72, 932–936 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddavattam D., Khajamohiddin S., Manavathi B., Pakala S. B. & Merrick M. Transposon-like organization of the plasmid-borne organophosphate degradation (opd) gene cluster found in Flavobacterium sp. Appl Environ Microbiol 69, 2533–2539 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandeeti E. V., Chakka D., Pandey J. P. & Siddavattam D. Indigenous organophosphate-degrading (opd) plasmid pCMS1 of Brevundimonas diminuta is self-transmissible and plays a key role in horizontal mobility of the opd gene. Plasmid 65, 226–231 (2011). [DOI] [PubMed] [Google Scholar]
- Holtwick R., von Wallbrunn A., Keweloh H. & Meinhardt F. A novel rolling-circle-replicating plasmid from Pseudomonas putida P8: molecular characterization and use as vector. Microbiology 147, 337–344 (2001). [DOI] [PubMed] [Google Scholar]
- Chang T. L., Kramer M. G., Ansari R. A. & Khan S. A. Role of individual monomers of a dimeric initiator protein in the initiation and termination of plasmid rolling circle replication. J Biol Chem 275, 13529–13534 (2000). [DOI] [PubMed] [Google Scholar]
- Khan S. A. & Novick R. P. Complete nucleotide sequence of pT181, a tetracycline-resistance plasmid from Staphylococcus aureus. Plasmid 10, 251–259 (1983). [DOI] [PubMed] [Google Scholar]
- Novick R. P. Staphylococcal plasmids and their replication. Annu Rev Microbiol 43, 537–565 (1989). [DOI] [PubMed] [Google Scholar]
- Josson K., Soetaert P., Michiels F., Joos H. & Mahillon J. Lactobacillus hilgardii plasmid pLAB1000 consists of two functional cassettes commonly found in other gram-positive organisms. J Bacteriol 172, 3089–3099 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S. A. Plasmid rolling-circle replication: highlights of two decades of research. Plasmid 53, 126–136 (2005). [DOI] [PubMed] [Google Scholar]
- te Riele H., Michel B. & Ehrlich S. D. Single-stranded plasmid DNA in Bacillus subtilis and Staphylococcus aureus. Proc Natl Acad Sci U S A 83, 2541–2545 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke S. J., Baldwin P. R. & Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128, 82–97 (1999). [DOI] [PubMed] [Google Scholar]
- Wilson C., Hughes L. E., Rashid T., Ebringer A. & Bansal S. Antibodies to Acinetobacter bacteria and bovine brain peptides, measured in bovine spongiform encephalopathy (BSE) in an attempt to develop an ante-mortem test. J Clin Lab Immunol 52, 23–40 (2003). [PubMed] [Google Scholar]
- Wilson C. et al. Antibodies to prion and Acinetobacter peptide sequences in bovine spongiform encephalopathy. Vet Immunol Immunopathol 98, 1–7 (2004). [DOI] [PubMed] [Google Scholar]
- Akowitz A., Sklaviadis T. & Manuelidis L. Endogenous viral complexes with long RNA cosediment with the agent of Creutzfeldt-Jakob disease. Nucleic Acids Res 22, 1101–1107 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geoghegan J. C. et al. Selective incorporation of polyanionic molecules into hamster prions. J Biol Chem 282, 36341–36353 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L. A 25 nm virion is the likely cause of transmissible spongiform encephalopathies. J Cell Biochem 100, 897–915 (2007). [DOI] [PubMed] [Google Scholar]
- Deleault N. R., Lucassen R. W. & Supattapone S. RNA molecules stimulate prion protein conversion. Nature 425, 717–720 (2003). [DOI] [PubMed] [Google Scholar]
- Currier T. C. & Nester E. W. Isolation of covalently closed circular DNA of high molecular weight from bacteria. Anal Biochem 76, 431–441 (1976). [DOI] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E. F. & Maniatis T. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press (1989). [Google Scholar]
- Pinjari A. B. et al. Mineralization of acephate, a recalcitrant organophosphate insecticide is initiated by a pseudomonad in environmental samples. PLoS One 7, e31963 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A. L., Harmon D., Kasif S., White O. & Salzberg S. L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res 27, 4636–4641 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic local alignment search tool. J Mol Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- Thompson J. D., Higgins D. G. & Gibson T. J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betley J. N., Frith M. C., Graber J. H., Choo S. & Deshler J. O. A ubiquitous and conserved signal for RNA localization in chordates. Curr Biol 12, 1756–1761 (2002). [DOI] [PubMed] [Google Scholar]
- Khan S. A. Rolling-circle replication of bacterial plasmids. Microbiol Mol Biol Rev 61, 442–455 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31, 3406–3415 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey J. P., Gorla P., Manavathi B. & Siddavattam D. mRNA secondary structure modulates the translation of organophosphate hydrolase (OPH) in E. coli. Mol Biol Rep 36, 449–454 (2009). [DOI] [PubMed] [Google Scholar]
- Fisher C. L. & Pei G. K. Modification of a PCR-based site-directed mutagenesis method. Biotechniques 23, 570–571, 574 (1997). [DOI] [PubMed] [Google Scholar]
- Hellman L. M. & Fried M. G. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc 2, 1849–1861 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller W., Keppner W. & Rasched I. Versatile kanamycin-resistance cartridges for vector construction in Escherichia coli. Gene 46, 131–133 (1986). [DOI] [PubMed] [Google Scholar]
- Hanahan D. Studies on transformation of Escherichia coli with plasmids. J Mol Biol 166, 557–580 (1983). [DOI] [PubMed] [Google Scholar]
- Studier F. W. & Moffatt B. A. Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189, 113–130 (1986). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Acinetobacter phage genome is similar to Sphinx236, the circular DNA copurified with TSE infected particles