

Abstract
Background
Angiotensin II (Ang II) is a critical mediator vascular inflammation and remodeling in a number of diseases including hypertension and atherosclerosis. The purpose of this study was to evaluate the role of the epithelium-specific ETS transcription factor-1 (ESE-1), a member of E26 transformation-specific sequence (ETS) transcription factors, as a mediator of Ang II–mediated vascular responses.
Methods
ESE-1 knockout mice were used to evaluate the role of ESE-1 in regulating Ang II–mediated vascular inflammation and remodeling.
Results
ESE-1 levels are low to undetectable under basal conditions but rapidly increase in response to Ang II. Intimal medial thickness and perivascular fibrosis of the aorta were significantly greater in ESE-1 knockout mice compared with the wild-type littermate controls. Proliferating cell nuclear antigen (PCNA) staining was also greater in the aorta of the Ang II–infused ESE-1 knockout mice compared with the controls. The infiltration of T cells and macrophage into the vessel wall of the aorta was dramatically enhanced in the ESE-1 knockout mice compared with the controls. Finally, Ang II–induced expression of a known downstream target of ESE-1, nitric oxide synthase 2 (NOS2), was significantly blunted in ESE-1 knockout mice compared to littermate controls. The alterations in vascular inflammation and remodeling were associated with an exaggerated systolic blood pressure response to Ang II in ESE-1 knockout mice.
Conclusions
ESE-1 is an Ang II–inducible transcription factor that plays an important counter-regulatory role in the setting of vascular inflammation and remodeling.
Keywords: blood pressure, ETS factors, gene transcription, hypertension
E26 transformation-specific sequence (ETS) factors are a family of transcription factors that share a highly conserved DNA-binding domain. The name ETS originates from a sequence that was detected in an avian erythroblastosis virus, E26, where it formed a transforming gene together with Δgag and c-myb.1,2 There are ~25–30 ETS family members in a variety of species from drosophila to man. ETS factors are involved in regulating a wide variety of biological processes including normal development and differentiation.3 As proto-oncogenes they have also been implicated in the pathogenesis of several different types of cancer.4,5
The epithelium-specific ETS transcription factor-1 (ESE-1) was originally identified as an epithelial-restricted ETS factor.6 Under noninflammatory conditions, ESE-1 is only expressed in cells of epithelial origin. However, in response to inflammatory stimuli such as endotoxin or proinflammatory cytokines including interleukin-1β (IL-1β) or tumor necrosis factor-α (TNF-α), this transcription factor is highly induced in cultured primary endothelial cells or vascular smooth muscle cells (VSMCs).7 In a mouse model of endotoxemia, ESE-1 is rapidly induced in the endothelium and first medial layer of VSMC of the mouse aorta.7 Target genes regulated by ESE-1 include nitric oxide synthase 2 (NOS2) and cyclooxygenase 2 (COX2).7,8 ESE-1 has also recently been shown to function in the regulation of TNF-α-mediated expression of angiopoietin-1 in the setting of inflammatory arthritis.9 The transcriptional activity of ESE-1 can be positively and negatively modified by its interaction with other proteins. Whereas binding of ESE-1 to CBP and p300 is associated with an increase in the transcriptional activity of ESE-1, interaction with Ku proteins represses ESE-1 function.10
ETS factors have also been shown to play an important role in angiotensin II (Ang II)–mediated vascular inflammation and remodeling. In particular, we have previously shown that the prototypic member of the ETS family, Ets-1, is a critical transcriptional mediator of Ang II–mediated vascular inflammatory responses.11 The participation of other selected ETS factors in vascular inflammation was recently reviewed.12 However, the role of ESE-1 in Ang II–mediated vascular inflammation and remodeling has not previously been explored. The primary purpose of this study was to determine whether ESE-1 expression is similarly induced in vivo, in the vasculature, in response to Ang II stimulation, and secondarily to determine whether or not ESE-1 plays an important modulatory role by positively or negatively regulating vascular inflammatory responses to Ang II.
Methods
Cell culture. Primary human aortic smooth muscle cells were obtained from Lonza (Basel, Switzerland) and grown in smooth muscle growth medium-2 (cat. no. CC-3182; Lonza). Rat aortic smooth muscle cells (RASMCs) were grown in Dulbecco's modified Eagle's medium with 10% of fetal bovine serum. RASMCs were isolated from the thoracic aorta of male Sprague–Dawley rats (Charles River, Wilmington, MA) by using the collagenase digestion method we have previously described.11 For all experiments, primary VSMCs from passages 4–7 were used. VSMCs were grown to 70–80% confluence and then made quiescent by starvation for 48h. The VSMCs were collected at 0, 1, 2, 4, 6, and 24h after stimulation with 100nmol/l of Ang II.
Animals and Ang II infusion. Animals were housed in accordance with the guidelines from the American Association for Laboratory Animal Care. The ESE-1−/− mice were generated and bred on a C57BL/6 background.5 ESE-1−/− mice were kindly provided by Ismail Kola, Monash University, Melbourne, Australia. Genotyping was performed by polymerase chain reaction (PCR) of genomic DNA isolated from tails of mice. All experiments were performed using 10-week-old male ESE-1−/− mice with littermate ESE-1+/+ controls. Mice were anesthetized by intraperitoneal injection of xylazine (5mg/kg) and ketamine (80mg/kg). An ALZET minipump (Duret, Cupertino, CA) containing angiotensin II (Sigma, St Louis, MO) 1.4mg/kg/day dissolved in saline or saline alone (vehicle) was implanted subcutaneously and infused continuously. Aortic tissue samples were collected at 0, 3, 7, and 14 days after initiation of Ang II infusion. For immunohistochemical studies, mice were killed and perfused with 4% paraformaldehyde under 100mmHg of perfusion pressure with a microperfusion pump after 0.1mol/l phosphate-buffered saline perfusion via a catheter inserted into the left ventricle. Fixed aortic tissue samples were embedded in paraffin.
Morphometric analysis. After the animals were euthanized, the thorax was opened, a 21-gauge needle was placed into the left ventricle, and the inferior vena cava was severed. The animals were perfused with normal saline at 100mmHg until the perfusate cleared and then pressure fixed at 100mmHg with 4% formalin. Tissues were paraffin embedded and cut into 5µmol/l sections. Sections were stained with Masson's trichrome stain to permit detection of perivascular fibrosis. Arterial thickening and perivascular fibrosis were assessed as previously described.13,14,15 The media/luminal area ratio was used as the index of arterial thickening, which is the ratio of cross-sectional medial area to luminal area. Measurements were performed using image-analyzing software (NIH Image). Three independent sections per artery of five pairs of mice were counted.
Immunohistochemistry. 5µmol/l sections cut from paraffin-embedded aorta were used for immunohistochemical staining. The primary antibodies used in the study were as follows: rabbit antihuman ESE-1 (Orbigen, San Diego, CA), rabbit antihuman CD3, rabbit IgG fraction as negative control for CD3 (Dako, Carpinteria, CA), rat anti-mouse Mac-3, rat IgG1 as negative control for Mac-3 (BD Pharmingen, San Jose, CA), rabbit antihuman vascular cell adhesion molecule (VCAM)-1 (Santa Cruz, Santa Cruz, CA), rabbit antihuman proliferating cell nuclear antigen (PCNA) (Santa Cruz), and rabbit IgG as negative control for rabbit Ab (Santa Cruz). Vectastain Elite ABC kits (Vector Labs, Burlingame, CA) were used for the immunohistochemical staining. The sections were counterstained with methyl green (Dako). CD3+ leukocytes and Mac-3+ macrophages in three independent sections per artery of five pairs of mice were counted. The number of cells were expressed as a percentage of the total cells.
Blood pressure measurement. BP-2000 blood pressure analysis system (Visitech Systems, Cincinnati, OH) was used to measure the systolic blood pressure of the mice. Systolic blood pressure was measured on 0, 3, 7, and 14 days after angiotensin infusion. Mice were weighed at 3 and 8 weeks, and every time before blood pressure measurements were made. No significant differences in weight were observed between the ESE-1 knockout and wild-type controls at the different time points.
RNA isolation, quantitative RT-PCR, and genotyping. RNA from aorta of C57BL mice was homogenized using Tissue-Tearor (Biospec Products, Bartlesville, OK). Human aortic smooth muscle cells were treated by QIAshredder (Qiagen, Valencia, CA) before mRNA extraction. mRNA was isolated using RNeasy Mini Kit (Qiagen) with RNase-free DNase set (Qiagen) to remove the genomic DNA. RNA was reverse transcribed to cDNA using oligo dT primer and SuperScript II RT-polymerase (Invitrogen, Carlsbad, CA). Real-time quantitative PCR was performed using the ABI PRISM 7700 machine (Applied Biosystems, Carlsbad, CA). Mouse tail DNA was isolated, and PCR for genotyping was performed for 30 cycles of 1min at 94°C, 1min at 55°C, and 1min at 72°C followed by 7min at 72°C. Five microliters of the amplification product were analyzed on a 2% agarose gel.
Measurement of DNA synthesis. VSMCs were stimulated by Ang II for 18h and pulsed with 1µCi/ml [3H]thymidine for 5h. Then cells were washed twice with phosphate-buffered saline, incubated for 5min in 5% trichloroacetic acid, washed by methanol, and dissolved in 99% formic acid. The incorporation of [3H]thymidine into trichloroacetic acid–insoluble material was measured by liquid scintillation spectrophotometer.
Statistical analysis. The data are presented as mean ± s.e.m. The statistical significance of differences was analyzed by analysis of variance with a subsequent Dunnett test in multiple comparisons.
Results
We have previously shown that ESE-1 is induced in vascular endothelial cells and VSMCs in response to proinflammatory cytokines including IL-1β and TNF-α.7 Because Ang II is known to function as a critical mediator of vascular inflammation as it relates to atherosclerosis, hypertension, and aging, we were also interested in determining whether Ang II could induce the expression of ESE-1in VSMCs.16 We first evaluated ESE-1 expression by semiquantitative RT-PCR in human aortic smooth muscle cells. Although low levels of ESE-1 were detectable under basal conditions, there was a rapid and transient induction of ESE-1 (Figure 1a). Using quantitative RT-PCR, the increase in ESE-1 was about twofold at 2h (Figure 1c). ESE-1 was similarly measured in the mouse aorta at different time points after the infusion of Ang II (1.4mg/kg/day). Minimal expression of ESE-1 was observed at baseline, but the levels of ESE-1 increased significantly by 3 days and were persistently elevated throughout the course of the infusion (Figure 1b). Robust ESE-1 expression was similarly detected by immunohistochemistry in the VSMCs of the mouse aorta after 2 weeks of Ang II infusion (Figure 1d). ESE-1 staining was also observed in the endothelium and adventitia.
Figure 1. ESE-1 induction in response to Ang II in vitro and in vivo. (a) ESE-1 expression as measured by PCR in HASMCs after Ang II stimulation (100nmol/l). (b) ESE-1 expression in HASMCs under Ang II stimulation by real-time Q-PCR. (c) ESE-1 expression in thoracic aorta under Ang II infusion (1.4mg/kg/day) for 3, 7, and 14 days by PCR. (d) Immunohistochemical analysis of ESE-1 expression after infusion of Ang II for 2 weeks in C57BL/6 compared to vehicle control. Original magnification ×250. HASMC, human aortic smooth muscle cell. Ang II, angiotensin II; ESE-1, epithelium-specific ETS transcription factor-1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; PCR, polymerase chain reaction.

In addition to the induction of inflammatory genes, systemic administration of Ang II also promotes the influx of inflammatory cells.17 These are first detected within the first week of infusion. To determine the role of ESE-1 in the regulation of the influx of inflammatory cells, Ang II was infused into ESE-1 knockout mice and littermate controls. We observed an increase in the number of T cells (CD3) and macrophages (Mac-3) by immunohistochemical staining (Figure 2a,c). This was particularly strong in the adventitia. We then counted the number of CD3 and Mac-3 staining cells per high power field magnification. The absence of ESE-1 was associated with over a tenfold increase in the number of inflammatory cells (Figure 2b,d). Interestingly, the levels of the vascular adhesion molecule VCAM-1 were exaggerated in the ESE-1−/− mice compared to controls (Figure 5).
Figure 2. Increased recruitment of inflammatory cells in ESE-1−/− mice in response to Ang II. (a,c) Immunohistochemical staining and (b,d) quantitative analysis of infiltration of CD3(T cells, original magnification ×250) and Mac-3 (macrophages, original magnification ×400) in the adventitia of thoracic aorta of ESE-1+/+ vs. ESE-1−/− mice after infusion of Ang II (1.4mg/kg/day) for 1 week. *P < 0.01 ESE-1−/− vs. ESE-1+/+. Values represent mean ± s.e.m., n = 5. Ang II, angiotensin II; ESE-1, epithelium-specific ETS transcription factor-1; IgG, immunoglobulin G.

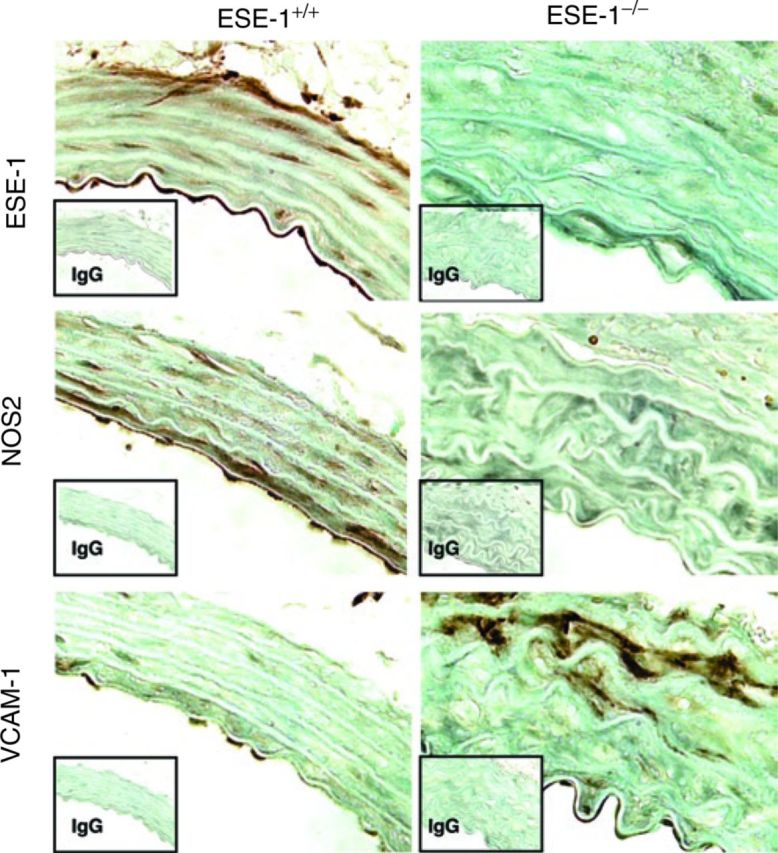
Figure 5. Immunohistochemical staining of ESE-1, NOS2, and VCAM-1 in the thoracic aorta of ESE-1+/+ and ESE-1−/− mice after Ang II infusion. Evaluation of expressions of ESE-1, NOS2, and COX2 in the thoracic aorta of ESE-1+/+ and ESE-1−/− mice, after infusion of Ang II (1.4mg/kg/day) for 1 week. Original magnification ×400. Isotype-matched controls are shown below each panel. Ang II, angiotensin II; COX2, cyclooxygenase 2; ESE-1, epithelium-specific ETS transcription factor-1; IgG, immunoglobulin G; NOS2, nitric oxide synthase 2; VCAM-1, vascular cell adhesion molecule-1.
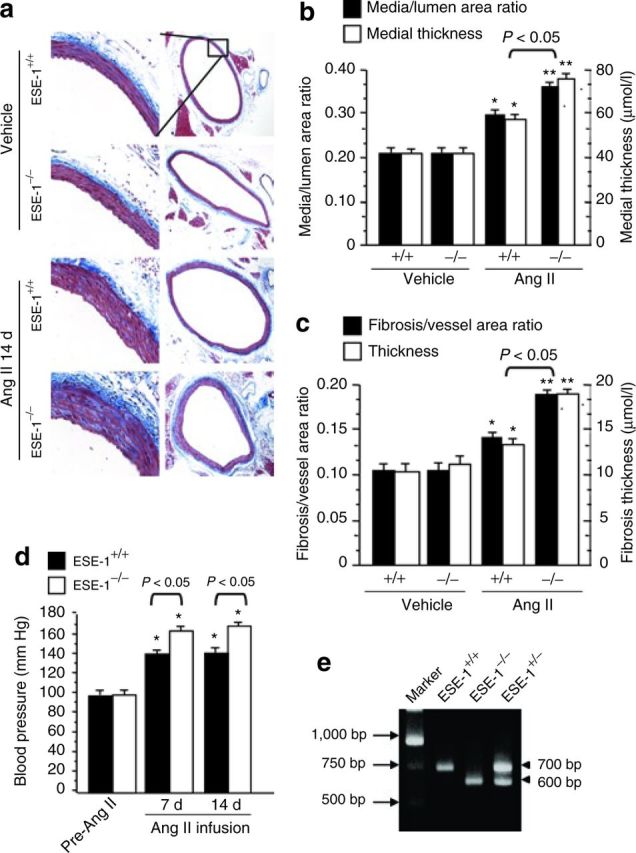
Chronic exposure of blood vessels to Ang II is associated with increased thickness and perivascular fibrosis.11 We also evaluated changes in vascular remodeling in ESE-1−/− mice and littermate controls (ESE-1+/+). There did not appear to be any significant morphological difference in vessel thickness prior to Ang II stimulation (Figure 3a, vehicle). However, after 2 weeks of infusion, there was a marked increase in thickness of the aortas and the extent of perivascular fibrosis in the ESE-1−/− compared to the littermate ESE-1+/+ control (Figure 3a; Ang II 14 days). Quantitation of the medial thickness and extent of perivascular fibrosis before and after stimulation confirmed these observations (Figure 3b,c). Interestingly, this was also associated with an exaggerated systolic blood pressure response to Ang II in the ESE-1−/− compared to the littermate ESE-1+/+ controls (Figure 3d). Genotyping of the mice was performed by PCR (Figure 3e).
Figure 3. Increase in Ang II–induced vascular remodeling and high blood pressure in ESE-1−/− vs. ESE-1+/+ mice. (a) Comparison of the effect of Ang II infusion vs. vehicle controls on perivascular fibrosis and arterial thickening in the aorta of ESE-1+/+ vs. ESE-1−/− mice. Aortic sections were stained with Masson's trichrome stain. Original magnification on left panel ×250 and right panel ×20. (b,c) Statistical analysis of aortic thickness (b) and perivascular fibrosis (c) in ESE-1+/+ vs. ESE-1−/− mice compared to vehicle controls. *P < 0.05 vs. vehicle of mice. **P < 0.01 vs. vehicle of mice (n = 5 per group). (d) The systolic blood pressure of mice was measured by using a Visitech BP-2000 system (see Methods) before (pre-Ang II) and after Ang II infusion (1.4mg/kg/day) for 1 week (7 days) and 2 weeks (14 days). *P < 0.01 vs. pre-Ang II of mice (n = 5 per group). (e) Representative PCR result for genotype of the ESE-1+/+, ESE-1+/−, and ESE-1−/−. Ang II, angiotensin II; ESE-1, epithelium-specific ETS transcription factor-1; PCR polymerase chain reaction.
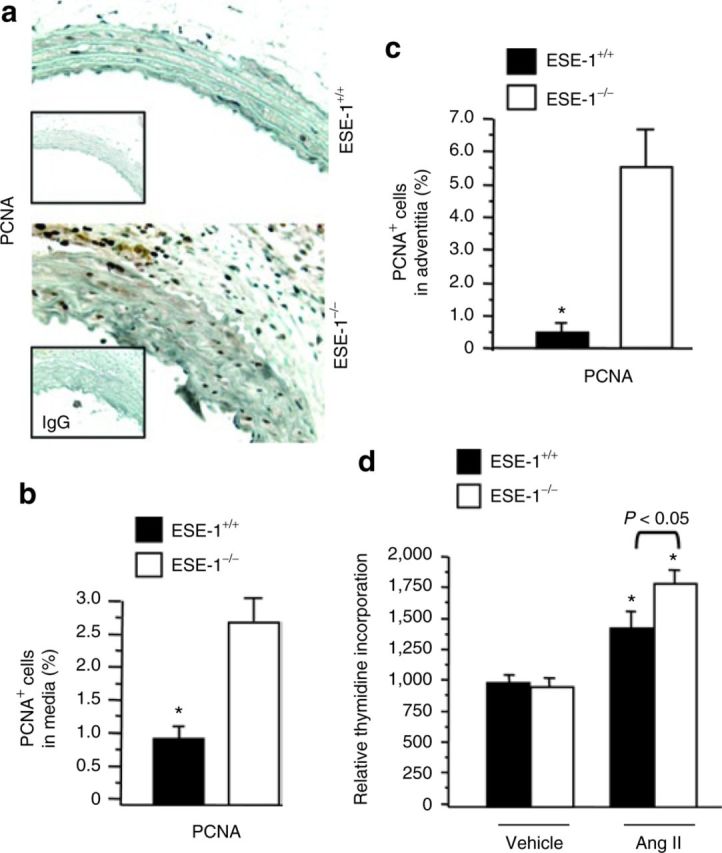
We hypothesized that one potential mechanism by which the increase in thickness could occur was through an increase in the proliferative capacity of the VSMCs from ESE-1−/− mice compared to control mice. PCNA staining of the media of ESE-1−/− mice was compared to littermate ESE-1+/+ controls. In addition to cells within the media of the aorta, we also noted an increase in the PCNA staining of cells within the adventitia (Figure 4a). The number of PCNA+ cells in the media (Figure 4b) and the adventitia (Figure 4c) were significantly higher in the ESE-1−/− mice compared to control mice. To further define whether this effect was Ang II–dependent, we isolated VSMCs from the aorta of ESE-1−/− and littermate ESE-1+/+ controls and assessed their relative uptake of tritiated thymidine. Interestingly, at baseline, there was no difference in proliferation. However, in the VSMCs obtained from ESE-1−/− mice exhibited and exaggerated response to Ang II compared to VSMCs from littermate ESE-1+/+ controls (Figure 4d).
Figure 4. Immunohistochemical staining of PCNA in the thoracic aorta of ESE-1+/+ and ESE-1−/− mice after Ang II infusion. (a) Evaluation of cell proliferation by PCNA staining after 2 weeks of Ang II infusion (1.4mg/kg/day) in the thoracic aorta of ESE-1+/+ compared to ESE-1−/− mice. Original magnification ×250. Isotype-matched controls are shown below each panel. (b,c) Statistical analysis of the positive cell staining with PCNA in media (b) and in adventitia (c). (d) Effect of Ang II on [3H]thymidine uptake. Quiescent human aortic smooth muscle cells were stimulated with Ang II (100nmol/l) for 18h and pulsed with [3H]thymidine for 5h. Incorporation of [3H]thymidine was measured by liquid scintillation spectrophotometer (n = 3). Values represent mean ± s.e.m. *P < 0.01 vs. vehicle, respectively. PCNA, proliferating cell nuclear antigen. Ang II, angiotensin II; ESE-1, epithelium-specific ETS transcription factor-1; IgG, immunoglobulin G.
We have previously shown that one of the direct targets of ESE-1 in response to proinflammatory cytokines in VSMCs and EC is NOS2 (ref. 7). We next evaluated whether NOS2 was also regulated by ESE-1 in response to Ang II. In wild-type mice, we observed a robust induction of ESE-1 and NOS2, that was significantly blunted in the ESE-1−/− mice (Figure 5).
Discussion
Until recently, very little was known about the role of ETS factors in regulating vascular inflammation. Over the past few years, several studies have been completed that support a role for selected members of the ETS transcription factor family in the regulation of vascular inflammation, including endothelial activation in response to inflammatory mediators, the recruitment of inflammatory cells to the vessel wall, and proliferation and migration of VSMCs.12 We and others have observed that Ets-1 is induced in VSMCs and endothelial cells in response to a variety of stimuli including Ang II, PDGF-BB, thrombin, IL-1β, and TNF-α.11,18,19,20,21,22,23 Target genes identified to be downstream of Ets-1 in the setting of acute vascular inflammation include the chemokine MCP-1 and the adhesion molecule VCAM-1. Systemic administration of the vasoactive peptide Ang II via continuous infusion is not only associated with increases in systolic blood pressure but also promotes the recruitment of inflammatory cells, including T cells and monocytic cells, to the vessel wall.24 One of the major mediators of vascular inflammation within the vessel wall is reactive oxygen species. Ang II, for example, promotes the generation of superoxide anions in VSMCs largely via the activity of NAD(P)H oxidases, that can be converted to hydrogen peroxide by superoxide dismutase.25
The results of our study support apparent opposing or counter-regulatory effects of different Ets factors to the same inflammatory stimuli, Ang II. One explanation for these opposite effects could be that they simply provide a mechanism for providing balance so that inflammatory responses do not go unopposed. The lack of ESE-1 expression in the ESE-1 knockout mice, for example, could lead to an exaggerated Ang II–mediated inflammatory response via an associated augmentation in the expression of Ets-1. The production of reactive oxygen species, such as hydrogen peroxide, is known to induce the expression of the ETS factor Ets-1 (ref. 26). When ESE-1 is normally expressed, the production of NO by NOS2 acts to counterbalance this increased inflammatory response leading to a reduction in adverse or pathologic remodeling. The ESE-1-dependent NO production can therefore be viewed as directly contributing to the regulation Ets-1 gene transcription by modifying the level of reactive oxygen species.
We also observed an exaggerated blood pressure response to Ang II in the ESE-1 knockout mice. We have previously identified NOS2 as a downstream target of ESE-1 in the setting of endotoxemia and in VSMC in response to proinflammatory cytokines such as TNF-α.7 ESE-1 synergizes with NF-κB to induce the expression of NOS2 in VSMC. The results of the current study also support a role for ESE-1 in the regulation of NOS2 downstream of Ang II. However, previous studies have demonstrated that Ang II does not directly induce NOS2 in cultured VSMC.27 We postulate that one possible mechanism by which Ang II might lead to increase expression of NOS2 in vivo is through the recruitment of inflammatory cells such as T cells that are known to secrete proinflammatory cytokines such as TNF-α.28 Therefore, although the primary vascular effect of Ang II on VSMC is to promote vasoconstriction, it is possible that the recruitment of inflammatory cells and the paracrine release of proinflammatory cytokines could explain the increased NOS2 expression we observed 1 week after Ang II infusion.
Prostaglandins may also play an important role in the regulation of blood pressure in response to Ang II. For example, it is known that COX2 synthesizes vasodilator prostaglandins. Inhibition of COX2 has been shown to augment the pressor effects of Ang II.29 Furthermore, COX2 is a downstream target of ESE-1 in the setting of inflammation.8 Therefore, another possible explanation for the increased blood pressure observed in the ESE-1 knockout mice at 7 and 14 days after Ang II infusion is a reduction in the production of vasodilator prostaglandins by COX2.
Leukotrienes, the products of the lipoxygenase enzymes have also been implicated in the pathogenesis of hypertension.30 For example, the lipoxygenase derived 12-hydroxyeicosatetraenoic acid (12-HETE) is associated with the development of hypertension and is a mediator of Ang II–mediated mesangial injury in diabetic nephropathy. Similarly, the P450 hydroxylase–derived 20-HETE is also a potent vasoconstrictor. 20-HETE has also been shown to promote the development of hypertension. We have previously shown that ESE-1 can also function as a transcriptional repressor.31 ESE-1 could potentially function as a repressor of these enzymes and thereby inhibit the production of selected leukotrienes known to play a role in the pathogenesis of hypertension.
In addition to the role of ETS factors as positive regulators of gene transcription, two members of the ETS factor family have been shown to function as transcriptional repressors of selected genes under noninflammatory conditions. For example, Elk-3 regulates the expression of the heme oxygenase-1 (HO-1) gene.32 HO-1 is a cytoprotective enzyme that is rapidly induced in monocytic cells in response to inflammatory stimuli such as endotoxin.33 Elk-3 functions as a potent transcriptional repressor that binds to regulatory sites within the HO-1 promoter and thereby inhibits the transcriptional activity of this promoter. In response to endotoxin, levels of Elk-3 rapidly diminish in cultured primary macrophages, which is associated with increased HO-1 levels.32 Under basal conditions, Elk-3 functions as a potent repressor of HO-1 expression, thereby contributing to transcriptional regulation of HO-1 gene under inflammatory and noninflammatory conditions. Elk-3 similarly functions as a repressor of NOS2 gene expression under noninflammatory conditions.34 More recently, we have shown that the ETS-related gene (ERG) functions as a transcriptional repressor of the IL-8 gene in endothelial cells under basal conditions.35 In response to proinflammatory cytokines such as TNF-α, a reduction in ERG is associated with a marked increase in IL-8 production and neutrophil attachment. These studies suggest that selected members of the ETS transcription factor family function to promote vascular quiescence in the absence of inflammation. In the presence of inflammatory stimuli, these transcriptional repressors are suppressed, thereby allowing the expression of selected inflammatory genes.
In summary, the results of our studies as well as those of others provide strong evidence for an important regulatory role of several different members of the ETS transcription in the setting of vascular inflammation remodeling. Whereas Ets-1 generally promotes inflammation downstream of Ang II, we have identified a counter-regulatory role for ESE-1 that may, at least in part, be mediated through the induction of the NOS2.
Acknowledgements
This work was supported by NIH grants R01-HL-67219 (P.O.) and R01-HL-82717 (P.O.).
Disclosure
The authors declared no conflict of interest.
References
- 1.Nunn MF, Seeburg PH, Moscovici C, Duesberg PH: Tripartite structure of the avian erythroblastosis virus E26 transforming gene. Nature 1983;306:391–395 [DOI] [PubMed] [Google Scholar]
- 2.Lepage A, Uzan G, Touche N, Morales M, Cazenave JP, Lanza F, de La Salle C: Functional characterization of the human platelet glycoprotein V gene promoter: a specific marker of late megakaryocytic differentiation. Blood 1999;94:3366–3380 [PubMed] [Google Scholar]
- 3.Wasylyk B, Hahn SL, Giovane A: The Ets family of transcription factors. Eur J Biochem 1993;211:7–18 [DOI] [PubMed] [Google Scholar]
- 4.Seth A, Watson DK: ETS transcription factors and their emerging roles in human cancer. Eur J Cancer 2005;41:2462–2478 [DOI] [PubMed] [Google Scholar]
- 5.Oikawa T, Yamada T: Molecular biology of the Ets family of transcription factors. Gene 2003;303:11–34 [DOI] [PubMed] [Google Scholar]
- 6.Oettgen P, Alani RM, Barcinski MA, Brown L, Akbarali Y, Boltax J, Kunsch C, Munger K, Libermann TA: Isolation and characterization of a novel epithelium-specific transcription factor, ESE-1, a member of the ets family. Mol Cell Biol 1997;17:4419–4433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rudders S, Gaspar J, Madore R, Voland C, Grall F, Patel A, Pellacani A, Perrella MA, Libermann TA, Oettgen P: ESE-1 is a novel transcriptional mediator of inflammation that interacts with NF-kappa B to regulate the inducible nitric oxide synthase gene. J Biol Chem 2001;276:3302–3309 [DOI] [PubMed] [Google Scholar]
- 8.Grall FT, Prall WC, Wei W, Gu X, Cho JY, Choy BK, Zerbini LF, Inan MS, Goldring SR, Gravallese EM, Goldring MB, Oettgen P, Libermann TA: The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J 2005;272:1676–1687 [DOI] [PubMed] [Google Scholar]
- 9.Brown C, Gaspar J, Pettit A, Lee R, Gu X, Wang H, Manning C, Voland C, Goldring SR, Goldring MB, Libermann TA, Gravallese EM, Oettgen P: ESE-1 is a novel transcriptional mediator of angiopoietin-1 expression in the setting of inflammation. J Biol Chem 2004;279:12794–12803 [DOI] [PubMed] [Google Scholar]
- 10.Wang H, Fang R, Cho JY, Libermann TA, Oettgen P: Positive and negative modulation of the transcriptional activity of the ETS factor ESE-1 through interaction with p300, CREB-binding protein, and Ku 70/86. J Biol Chem 2004;279:25241–25250 [DOI] [PubMed] [Google Scholar]
- 11.Zhan Y, Brown C, Maynard E, Anshelevich A, Ni W, Ho IC, Oettgen P: Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J Clin Invest 2005;115:2508–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oettgen P: Regulation of vascular inflammation and remodeling by ETS factors. Circ Res 2006;99:1159–1166 [DOI] [PubMed] [Google Scholar]
- 13.Takemoto M, Egashira K, Usui M, Numaguchi K, Tomita H, Tsutsui H, Shimokawa H, Sueishi K, Takeshita A: Important role of tissue angiotensin-converting enzyme activity in the pathogenesis of coronary vascular and myocardial structural changes induced by long-term blockade of nitric oxide synthesis in rats. J Clin Invest 1997;99:278–287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu L, Iwai M, Nakagami H, Chen R, Suzuki J, Akishita M, de Gasparo M, Horiuchi M: Effect of angiotensin II type 1 receptor blockade on cardiac remodeling in angiotensin II type 2 receptor null mice. Arterioscler Thromb Vasc Biol 2002;22:49–54 [DOI] [PubMed] [Google Scholar]
- 15.Ishibashi M, Egashira K, Zhao Q, Hiasa K, Ohtani K, Ihara Y, Charo IF, Kura S, Tsuzuki T, Takeshita A, Sunagawa K: Bone marrow-derived monocyte chemoattractant protein-1 receptor CCR2 is critical in angiotensin II-induced acceleration of atherosclerosis and aneurysm formation in hypercholesterolemic mice. Arterioscler Thromb Vasc Biol 2004;24:e174–e178 [DOI] [PubMed] [Google Scholar]
- 16.Phillips MI, Kagiyama S: Angiotensin II as a pro-inflammatory mediator. Curr Opin Investig Drugs 2002;3:569–577 [PubMed] [Google Scholar]
- 17.Piqueras L, Kubes P, Alvarez A, O'Connor E, Issekutz AC, Esplugues JV, Sanz MJ: Angiotensin II induces leukocyte-endothelial cell interactions in vivo via AT(1) and AT(2) receptor-mediated P-selectin upregulation. Circulation 2000;102:2118–2123 [DOI] [PubMed] [Google Scholar]
- 18.Hultgårdh-Nilsson A, Cercek B, Wang JW, Naito S, Lövdahl C, Sharifi B, Forrester JS, Fagin JA: Regulated expression of the ets-1 transcription factor in vascular smooth muscle cells in vivo and in vitro. Circ Res 1996;78:589–595 [DOI] [PubMed] [Google Scholar]
- 19.Goetze S, Kintscher U, Kaneshiro K, Meehan WP, Collins A, Fleck E, Hsueh WA, Law RE: TNFalpha induces expression of transcription factors c-fos, Egr-1, and Ets-1 in vascular lesions through extracellular signal-regulated kinases ½. Atherosclerosis 2001;159:93–101 [DOI] [PubMed] [Google Scholar]
- 20.Naito S, Shimizu S, Maeda S, Wang J, Paul R, Fagin JA: Ets-1 is an early response gene activated by ET-1 and PDGF-BB in vascular smooth muscle cells. Am J Physiol 1998;274:C472–C480 [DOI] [PubMed] [Google Scholar]
- 21.Redlich K, Kiener HP, Schett G, Tohidast-Akrad M, Selzer E, Radda I, Stummvoll GH, Steiner CW, Gröger M, Bitzan P, Zenz P, Smolen JS, Steiner G: Overexpression of transcription factor Ets-1 in rheumatoid arthritis synovial membrane: regulation of expression and activation by interleukin-1 and tumor necrosis factor alpha. Arthritis Rheum 2001;44:266–274 [DOI] [PubMed] [Google Scholar]
- 22.Liu AY, Corey E, Vessella RL, Lange PH, True LD, Huang GM, Nelson PS, Hood L: Identification of differentially expressed prostate genes: increased expression of transcription factor ETS-2 in prostate cancer. Prostate 1997;30:145–153 [DOI] [PubMed] [Google Scholar]
- 23.Santiago FS, Khachigian LM: Ets-1 stimulates platelet-derived growth factor A-chain gene transcription and vascular smooth muscle cell growth via cooperative interactions with Sp1. Circ Res 2004;95:479–487 [DOI] [PubMed] [Google Scholar]
- 24.Dzau VJ: Theodore Cooper Lecture: tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension 2001;37:1047–1052 [DOI] [PubMed] [Google Scholar]
- 25.Griendling KK, Sorescu D, Ushio-Fukai M: NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res 2000;86:494–501 [DOI] [PubMed] [Google Scholar]
- 26.Bonello MR, Bobryshev YV, Khachigian LM: Peroxide-inducible Ets-1 mediates platelet-derived growth factor receptor-alpha gene transcription in vascular smooth muscle cells. Am J Pathol 2005;167:1149–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakayama I, Kawahara Y, Tsuda T, Okuda M, Yokoyama M: Angiotensin II inhibits cytokine-stimulated inducible nitric oxide synthase expression in vascular smooth muscle cells. J Biol Chem 1994;269:11628–11633 [PubMed] [Google Scholar]
- 28.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG: Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD: Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest 2002;110:61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hao CM, Breyer MD: Physiologic and pathophysiologic roles of lipid mediators in the kidney. Kidney Int 2007;71:1105–1115 [DOI] [PubMed] [Google Scholar]
- 31.Peng H, Tan L, Osaki M, Zhan Y, Ijiri K, Tsuchimochi K, Otero M, Wang H, Choy BK, Grall FT, Gu X, Libermann TA, Oettgen P, Goldring MB: ESE-1 is a potent repressor of type II collagen gene (COL2A1) transcription in human chondrocytes. J Cell Physiol 2008;215:562–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chung SW, Chen YH, Yet SF, Layne MD, Perrella MA: Endotoxin-induced down-regulation of Elk-3 facilitates heme oxygenase-1 induction in macrophages. J Immunol 2006;176:2414–2420 [DOI] [PubMed] [Google Scholar]
- 33.Chen YH, Yet SF, Perrella MA: Role of heme oxygenase-1 in the regulation of blood pressure and cardiac function. Exp Biol Med (Maywood) 2003;228:447–453 [DOI] [PubMed] [Google Scholar]
- 34.Chen YH, Layne MD, Chung SW, Ejima K, Baron RM, Yet SF, Perrella MA: Elk-3 is a transcriptional repressor of nitric-oxide synthase 2. J Biol Chem 2003;278:39572–39577 [DOI] [PubMed] [Google Scholar]
- 35.Yuan L, Nikolova-Krstevski V, Zhan Y, Kondo M, Bhasin M, Varghese L, Yano K, Carman CV, Aird WC, Oettgen P: Antiinflammatory effects of the ETS factor ERG in endothelial cells are mediated through transcriptional repression of the interleukin-8 gene. Circ Res 2009;104:1049–1057 [DOI] [PMC free article] [PubMed] [Google Scholar]