

Abstract
The A53T genetic missense mutation of the wild-type α-synuclein (αS) protein was initially identified in Greek and Italian families with familial Parkinson’s disease. Detailed understanding of the structures and the changes induced in the wild-type αS structure by the A53T mutation, as well as establishing the direct relationships between the rapid conformational changes and free energy landscapes of these intrinsically disordered fibrillogenic proteins, helps to enhance our fundamental knowledge and to gain insights into the pathogenic mechanism of Parkinson’s disease. We employed extensive parallel tempering molecular dynamics simulations along with thermodynamic calculations to determine the secondary and tertiary structural properties as well as the conformational free energy surfaces of the wild-type and A53T mutant-type αS proteins in an aqueous solution medium using both implicit and explicit water models. The confined aqueous volume effect in the simulations of disordered proteins using an explicit model for water is addressed for a model disordered protein. We also assessed the stabilities of the residual secondary structure component interconversions in αS based on free energy calculations at the atomic level with dynamics using our recently developed theoretical strategy. To the best of our knowledge, this study presents the first detailed comparison of the structural properties linked directly to the conformational free energy landscapes of the monomeric wild-type and A53T mutant-type α-synuclein proteins in an aqueous solution environment. Results demonstrate that the β-sheet structure is significantly more altered than the helical structure upon A53T mutation of the monomeric wild-type αS protein in aqueous solution. The β-sheet content close to the mutation site in the N-terminal region is more abundant while the non-amyloid-β component (NAC) and C-terminal regions show a decrease in β-sheet abundance upon A53T mutation. Obtained results utilizing our new theoretical strategy show that the residual secondary structure conversion stabilities resulting in α-helix formation are not significantly affected by the mutation. Interestingly, the residual secondary structure conversion stabilities show that secondary structure conversions resulting in β-sheet formation are influenced by the A53T mutation and the most stable residual transition yielding β-sheet occurs directly from the coil structure. Long-range interactions detected between the NAC region and the N- or C-terminal regions of the wild-type αS disappear upon A53T mutation. The A53T mutant-type αS structures are thermodynamically more stable than those of the wild-type αS protein structures in aqueous solution. Overall, the higher propensity of the A53T mutant-type αS protein to aggregate in comparison to the wild-type αS protein is related to the increased β-sheet formation and lack of strong intramolecular long-range interactions in the N-terminal region in comparison to its wild-type form. The specific residual secondary structure component stabilities reported herein provide information helpful for designing and synthesizing small organic molecules that can block the β-sheet forming residues, which are reactive toward aggregation.
Keywords: α-Synuclein, genetic missense mutation, free energy landscape, molecular dynamics simulations
Parkinson’s disease (PD) results in the loss of the dopaminergic neurons in the substantia nigra of the brain and is characterized pathologically by the presence of large proteinaceous aggregates termed Lewy bodies.1,2 Lewy bodies are mainly composed of the α-synuclein (αS) protein, an intrinsically disordered fibrillogenic protein consisting of 140 amino acid residues.3 Furthermore, several cases of familial PD are associated with genetic missense mutations of the αS protein.4−6 The first of these genetic missense mutations of the αS protein identified in Italian and Greek families with early onset PD was the A53T mutation, which results in the conversion of the 53rd amino acid residue from alanine to threonine.4 Due to its role in the formation of Lewy bodies, the aggregation of the αS protein is proposed to play a role in the pathogenic mechanism of PD. Specifically, the αS monomers and oligomers are reported to be the neurotoxic species.7,8 Furthermore, the disordered structures of the monomeric and oligomeric αS are purported to determine the aggregation mechanism.9,10
A53T mutation has been shown to alter the neurotoxicity and aggregation of the wild-type αS protein. Different trends in the neurotoxicity of the wild-type and A53T mutant-type αS proteins have been reported in the current literature; Choong et al. reported that the A53T mutant-type αS is neurotoxic whereas the wild-type αS is neuroprotective toward the SH-SY5Y cells in the presence of rotenone and maneb.11 Furthermore, Wersinger et al. detected an increase in neurotoxicity of the A53T mutant-type αS in comparison to the wild-type αS for human SK-N-MC cells in the vicinity of peroxide.12 However, others presented that the A53T mutant-type αS is less toxic than the wild-type via rat models and SH-SY5Y cells in the presence of dopamine.7,13 Membrane interaction investigations reported that the A53T mutation does not affect the binding affinity with vesicles and cell membranes in comparison to the wild-type αS protein.14−18 Other experiments proposed an increased membrane permeabilizing ability upon A53T mutation of the wild-type αS.19,20 It was reported that the wild-type αS fibrils display a straight filament structure whereas the A53T mutant-type αS forms filaments with a twisted structure.15,21−23 In addition, numerous measurements reported an increased rate of aggregation for both the fibrillization and oligomerization of the A53T mutant-type αS in comparison to the wild-type αS protein.9,10,15,21−28 Due to these measured differences in the aggregation rates and the proposed direct link between the aggregation mechanism and the monomeric protein structures, it is crucial to possess the knowledge of monomeric A53T mutant-type and wild-type αS structures. However, the exact changes induced by the A53T mutation at the monomeric or oligomeric level and the structural and thermodynamic relationships with the aggregation mechanisms and pathology remain poorly understood due to challenges in measuring the properties of the monomeric and oligomeric species, such as fast aggregation, rapid conformational changes, and solvent effects.
Circular dichroism (CD) measurements presented identical spectra that predicted random coil for both the wild-type and A53T mutant-type αS proteins.9,21,26,27 Nuclear magnetic resonance (NMR) measurements by Bussell and Eliezer reported that the α-helical character of the αS protein is unaffected by the A53T mutation.29 However, various single molecular force (SMF) and Fourier transform infrared (FTIR) and NMR spectroscopy measurements presented an increase in β-sheet content in the A53T mutant-type in comparison to the wild-type αS protein.9,10,29−31 Furthermore, Bertoncini et al. presented that long-range interactions between the C-terminal (Lys96–Ala140) and the non-amyloid-β component (NAC; Lys60–Val95) regions are released upon A53T mutation of the wild-type αS protein.32
Although experimental studies have provided valuable insights into the effect of the A53T mutation on the wild-type αS protein, atomic level information with dynamics can be gained from theoretical studies of the wild-type and A53T mutant-type αS proteins at the monomeric level in solution that are not easily observable using conventional experimental tools. The current literature includes several theoretical studies, but none of the existing theoretical investigations have provided a complete picture of the impact of the A53T mutation on the wild-type αS structures via assessing the structure and free energy landscape relationships. For example, Kumar et al. reported an increase in the aggregation propensity of the A53T mutant-type αS protein due to similar secondary structure contents of their simulated A53T mutant-type αS structure to NMR measurements of the αS fibril structure and a sequence analysis via classical molecular dynamics (MD) simulations in explicit water.33 Detailed information regarding the secondary and tertiary structure contents as well as the thermodynamic properties was not shown in this study. The effect of the A53T mutation on the phospholipid bilayer or sodium dodecyl sulfate (SDS)-bound structures of the wild-type αS were determined by Perlmutter et al. via classical MD simulations.34 They reported that the A53T mutation has little affect on the structure of wild-type αS.34 Balesh et al. performed classical MD and annealing MD (AMD) simulations of the wild- and mutant-type αS proteins.35 They reported similar helical and β-sheet contents for the wild-type and A53T mutant-type αS proteins. Furthermore, they presented a more compact structure for the A53T mutant-type αS than the wild-type. However, detailed residual information regarding the secondary and tertiary structure properties, as well as the relationships of the structural properties with free energy landscapes, have not been presented. MD simulations of the wild-type and A53T mutant-type αS, as well as the mouse αS protein, by Carloni and co-workers reported interesting trends regarding the impact of the A53T mutation on the wild-type αS structures.36 Specifically, they found that intramolecular interactions of the C-terminal region with either the N-terminal (Met1–Lys60) or NAC regions of the α-synuclein protein are lost upon A53T mutation. Furthermore, they reported that the average radius of gyration (Rg) of the A53T mutant-type αS protein is larger than that of the wild-type αS for most of the protein ensembles investigated. Even though important insights are gained from this successful study, detailed secondary structure and thermodynamic properties were not presented. We also should mention here that MD simulations without the usage of special sampling techniques have the potential to suffer from the multiple minima problem and may result in poor sampling of intrinsically disordered proteins.
We performed all-atom replica exchange molecular dynamics (REMD) simulations on the full-length monomeric wild-type and A53T mutant-type αS proteins in aqueous solution utilizing implicit and explicit water models. The resulting structures obtained from these simulations were utilized in calculations of the secondary and tertiary structural properties, as well as free energy landscapes, of the wild-type and A53T mutant-type αS. Furthermore, we applied our recently developed in house theoretical strategy to provide insight into the thermodynamic preference of transitions between different secondary structure components for each residue at the atomic level with dynamics based on free energy change calculations. In addition, we address the determined structural variations in aqueous intrinsically disordered proteins based on the confined aqueous volume effect using a smaller model protein and an explicit model for water. In this study, we report the structures and free energy landscapes of the A53T mutant-type αS protein as well as the impact of A53T mutation on the structures and free energy landscapes of the wild-type αS protein in aqueous solution. We relate our results to the proposed toxicity of these proteins and to the pathology.
Results and Discussion
Figure 1 presents the abundance of secondary structure components per residue for the wild-type and A53T mutant-type αS proteins. Specific secondary structure components, that is, α-helix and β-sheet structures, are proposed to play important roles in the physiological function and aggregation mechanism of the αS protein. Overall, the α-helix content of the wild-type and A53T mutant-type αS proteins shows only minor deviations in abundance (<5%) with a few notable exceptions. Specifically, residues Val3–Lys12 in the N-terminal region, Ala89–Ala91 in the NAC region, and Asn103–Glu105 in the C-terminal region adopt up to 10% more abundant α-helix structure upon A53T mutation of the wild-type αS. However, residues Phe94–Gly101 present an opposite trend with an up to 15% less abundant α-helix structure formation in the A53T mutant-type αS than in the wild-type αS. In general, this finding agrees with circular dichroism (CD) measurements that reported similar α-helix contents for the wild-type and A53T mutant-type αS.9,10,27,28 In addition, our results support the findings of Bussell and Eliezer who reported that the α-helical character of Ala18–Gly31 is unperturbed by the A53T mutation via NMR measurements.29 Furthermore, we observe that the abundance of α-helix structure is greater in the N-terminal and NAC regions than in the C-terminal region, especially for the last 38 residues, for both the wild-type and A53T mutant-type αS in aqueous solution. This finding agrees with the observed helical tendency of the first 100 residues of the wild-type and A53T mutant-type αS proteins via NMR measurements. Jónsson et al. detected the same trend for the wild-type αS protein structures via Monte Carlo simulations.37 We should mention here that the formation of the α-helix structure in the N-terminal and NAC regions of the αS protein has been proposed to be a key factor for vesicle and membrane binding.14,17,18,20,38−40 Therefore, the overall similarity in the α-helix contents in the N-terminal and NAC regions of the wild-type and A53T mutant-type αS proteins indicates that the binding of αS to vesicles and membranes would not be significantly influenced by the A53T mutation. In fact several in vitro and in vivo experiments reported that the binding affinity of αS with cell membranes and phospholipid vesicles is unaffected by the A53T mutation.14−18 Furthermore, our results present that previously reported differences for the neurotoxicity of the mutant- and wild-type αS proteins when interacting with cell membranes might be related to changes in the oligomeric rather than monomeric structures. Despite the minimal changes observed in the α-helix content, large differences in the abundance of 310-helix formation occur between the wild-type and A53T mutant-type αS proteins. Residues Val16–Ala18, Glu20–Thr22, Glu28–Gly31, Val74–Val77, and Glu131–Gln134 form more abundant (up to 10%) 310-helix in the wild-type protein in comparison to the same residues in the A53T mutant-type αS protein structures. On the other side, Val52–Val55, Lys102–Glu105, and Gln109–Leu113 adopt larger abundant 310-helix structure (up to 10%) upon A53T mutation of the wild-type αS protein.
Figure 1.

Wild-type and A53T mutant-type αS secondary structure components. Secondary structure component abundances per residue for the wild-type (black) and A53T mutant-type (red) αS structures obtained after convergence. The abundances for the π-helix and coil structures are not displayed.
The most abundant β-sheet structure (up to 20%) occurs at Val70, Val71, Val82, Glu83, Ala89–Ala91, Lys102, Asn103, and Pro108, Gln109 in the NAC and C-terminal regions of the wild-type αS. Upon A53T mutation, the β-sheet formation at these residues is either less stable or disappears. The same trend is also observed for residues Phe4, Glu13, Val16, Gln24, Ala27, Thr33, and Lys34 located in the N-terminal region. Prominent β-sheet formation occurs at Leu8, Ala30, Glu35, Val37, Tyr39, Glu46, and His50 in the A53T mutant-type structures in comparison to those of the wild-type αS protein. Overall, these findings show an increase in β-sheet formation close to the mutation site in the N-terminal region upon A53T mutation of the wild-type αS. Our results at the atomic level with dynamics in aqueous solution support the findings of Bussell and Eliezer, who reported more likely β-sheet structure formation around the mutation site of the A53T mutant-type αS in comparison to the wild-type via NMR measurements,29 but we also present the specific residues along with the probabilities of the secondary structure components. Abundant β-sheet structure upon A53T mutation was also observed for αS via single molecule force (SMF) and Fourier transform infrared (FTIR) spectroscopy measurements.9,10,31 Interestingly, Balesh et al. did not detect an increase in β-sheet propensity around the mutation site of the A53T mutant-type αS in comparison to the wild-type via annealing MD simulations.35 The formation of β-sheet structure in αS has been linked to its aggregation process.9,10,15,26−28,41 Therefore, our results demonstrate that these specific residues located in the N-terminal region around the mutation site (Ala30–His50) play an important role in the aggregation mechanism of αS due to the increase in β-sheet content upon A53T mutation.
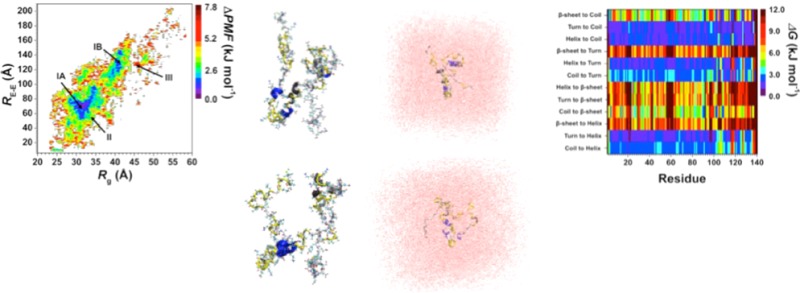
Using our recently developed theoretical strategy,42−44 we studied the change in free energy upon transition between two different secondary structure components at the atomic level with dynamics for each residue of the wild-type and A53T mutant-type αS proteins (Figure 2). The determined free energy values yield valuable insight into the most preferred secondary structure transitions for each residue, which provides the most likely secondary structure component that will precede the formation of the secondary structure component of interest. Transitions to helix and β-sheet structures are of special interest because of the proposed role of these secondary structure formations in the physiological function and aggregation process. Regarding the formation of helical structures, we find that the transition from either a coil or turn structure to helix structure is the most stable conversion for the wild-type and A53T mutant-type αS proteins. Furthermore, the conversion stability yielding helix is not significantly affected by the A53T mutation. This finding supports the similar α-helix content of the wild-type and A53T mutant-type αS proteins.9,10,27−29 Stable coil to β-sheet conversions occur at Phe4, Glu13, Ala90, Ala91, Lys102, Asn103, Pro108 ,and Gln109 in the wild-type αS protein structures. Upon A53T mutation, we note that this thermodynamic stability significantly decreases (>1kBT) at Phe4, Glu13, Ala90, and Gln109. Furthermore, only Tyr39 displays a favorable thermodynamic preference for conversion to a β-sheet structure from a coil structure in the structures of the mutant-type protein. Interestingly, the stability decreases for residual secondary structure conversions resulting in β-sheet structure from either a turn, coil, or helix structure at the mutation site (Ala53/Thr53) in the A53T mutant-type αS protein in comparison to the wild-type αS protein.
Figure 2.
Wild-type and A53T mutant-type αS secondary structure transition stabilities. The stability of secondary structure transitions between two specific secondary structure components per residue for the wild-type (WT) and A53T mutant-type (A53T) αS proteins based on free energy calculations performed using our recently developed theoretical strategy.44,46 The color scale corresponds to the free energy value associated with the specific secondary structure transition between two secondary structure components for a specific residue.
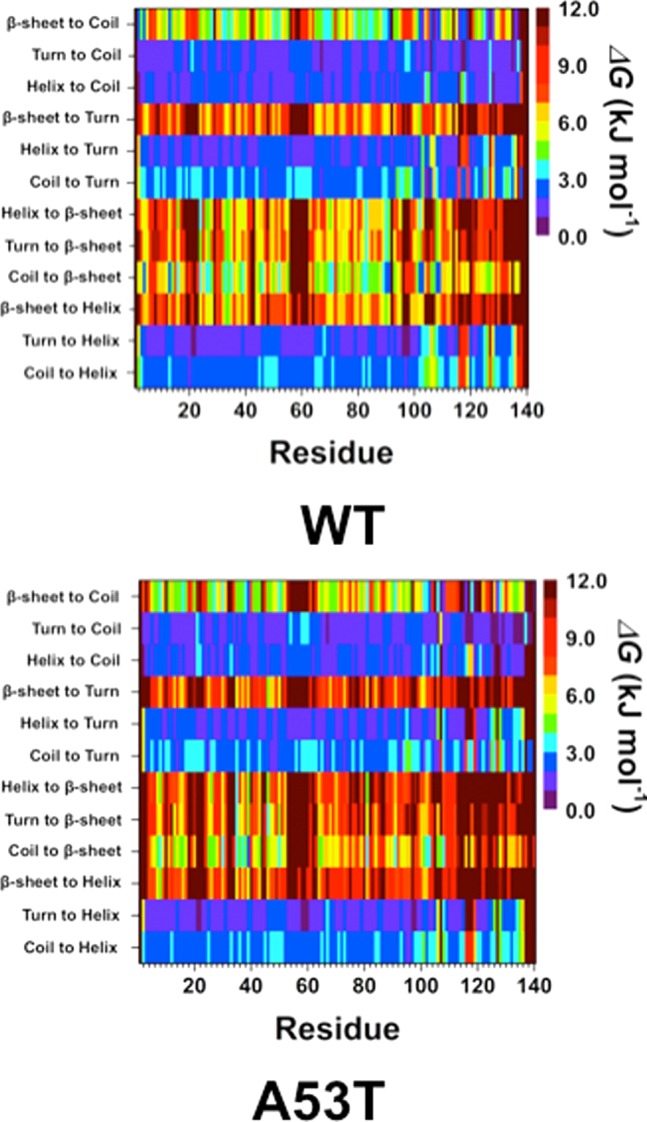
Figure 3 presents the intramolecular protein interactions in the structures of the wild-type and A53T mutant-type αS proteins, respectively. Gly86–Asn103 and Glu104–Asn122 located in the NAC and C-terminal regions present strong intramolecular interactions (>50%) in the wild-type αS structures in aqueous solution. Additionally, stable intramolecular interactions (up to 88%) within the NAC region of the wild-type αS are detected between Val70–Gly84 and Ala85–Leu100. Moreover, abundant intramolecular interactions occur between Ala56–Gly106 and Gly84–Gln134 (up to 42%). Upon A53T mutation, the abundance of intramolecular interactions between the NAC and C-terminal regions (Gly86–Asn103 with Glu104–Asn122) and within the NAC region (Val70–Gly84 with Ala85–Leu100) decreases. Furthermore, the intramolecular interactions between Ala56–Gly106 and Gly84–Gln134 are reduced (<20%) upon A53T mutation. The abundance of interactions between Glu28–Glu46 in the N-terminal region and Glu60–Lys80 in the NAC region, as well as within the C-terminal region between Gly86–Glu104 and Glu130–Ala140, of the A53T mutant-type αS increases slightly upon A53T mutation. Interestingly, we note that the intramolecular interactions of the C-terminal region with the N-terminal or NAC regions almost disappear upon A53T mutation. Carloni and co-workers reported a similar loss in intramolecular interactions between the wild-type and A53T mutant-type αS proteins using classical MD simulations in explicit water.36 Furthermore, NMR measurements of the wild-type and A53T mutant-type αS proteins performed by Bertoncini et al. also reported decreased long-range interactions upon A53T mutation, especially between the C-terminal and NAC regions.32 Therefore, these findings present reduced long-range interactions involving the NAC region and indicate that the NAC region is more solvent-exposed upon A53T mutation. This finding is supported by Hazy et al., who reported that the A53T mutant-type αS is more hydrated than the wild-type αS via differential scanning calorimeter measurements.45 Increased exposure of the NAC region is related to enhanced aggregation propensity of αS due to its proposed critical role in the aggregation process. Therefore, our tertiary structure findings indicate that the aggregation propensity of the αS protein is increased upon A53T mutation, which is in agreement with previous experiments.9,10,21−28
Figure 3.
Wild-type and A53T mutant-type αS tertiary structures. Calculated intramolecular interactions of the wild-type (WT) and the A53T mutant-type (A53T) αS. The color scale corresponds to the probability (P) of the distance between the heavy atoms (C, N, O, S) of a residue being ≤20 Å from each other.
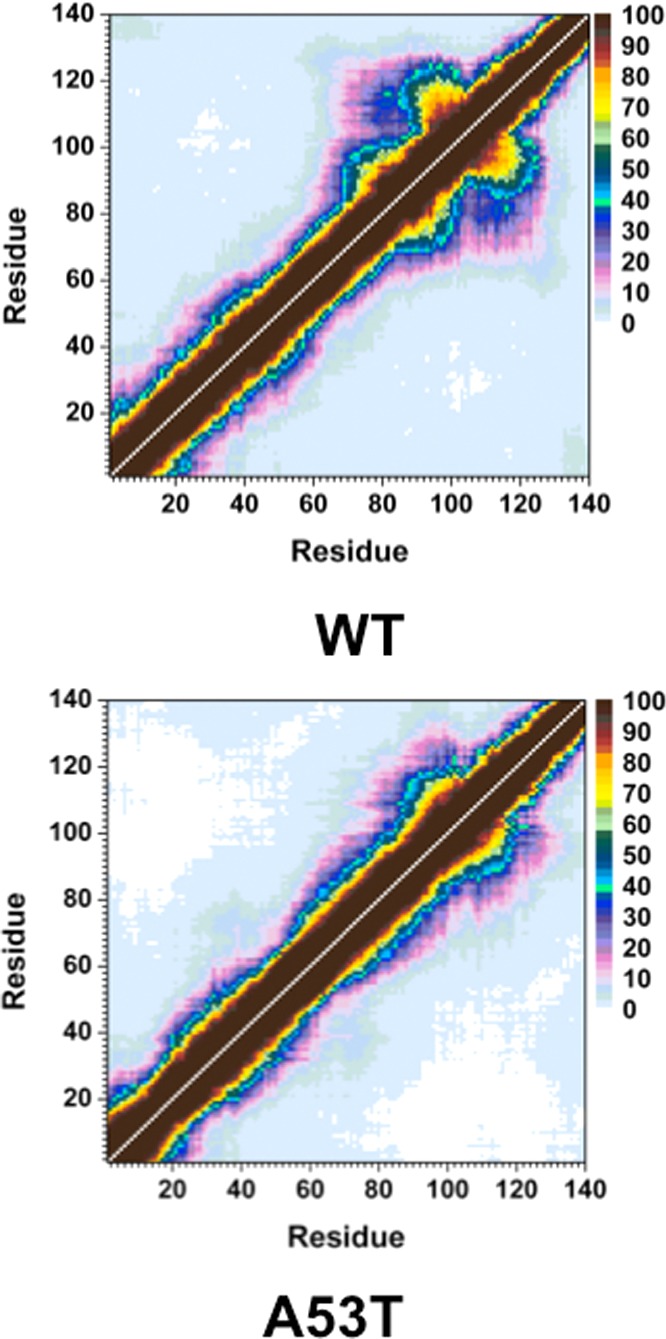
Different structures belonging to the wild-type and A53T mutant-type αS proteins simulated utilizing an implicit water model were selected (based on abundant α-helix and β-sheet structure and prominent long-range intramolecular interaction formations) as initial structures in additional sets of MD simulations utilizing an explicit model for water in order to address the impact of different water models on the simulated structures of the large-size disordered proteins. Figure 4 shows the specific initial structures (REMD; implicit water) and the obtained final structures (MD; explicit water). The secondary structures per residue with dynamics, which are in agreement with experiments and some previous theoretical studies, are not influenced significantly by the usage of an explicit water model (Figure 5). However, additional β-sheet formation occurs at Val40, Ser42, and Asn122 (structure I; Figure 5); Gly101 and Asn103 (structure III; Figure 5); and Gly101 and Met116 (structure V, Figure 5) in the conformations of the wild-type αS protein simulated using an explicit model for water. The β-sheet at Ala78 and Pro120 (structure I; Figure 5) and Gln24 and Glu28 (structure V; Figure 5) in the wild-type αS protein conformations disappear when an explicit water model is used. Interestingly, such differences in β-sheet formation do not occur for the A53T mutant-type αS protein (Figure 5). However, helical and turn structure formations at some residues of the mutant-type form are slightly influenced by the usage of an implicit or explicit water model (Figure 5). Moreover, for understanding whether the usage of an implicit and explicit water model affects the simulated intramolecular interactions, the distance variations between the heavy atoms of different residues were calculated utilizing the following relationship:
where r is the distance between heavy atoms (C, N, O, S) of different residues. We note differences demonstrated by the Δ⟨r⟩ values for the long-range intramolecular interactions of the wild-type αS (see Supporting Information). Even though the overall trend for the long-range intramolecular interactions is not significantly affected, the distances between the heavy atoms located at residues in the N- and C-terminal or NAC regions of the wild-type protein are different than the ones simulated using an implicit model for water. These findings show that intermolecular hydrogen-bonding interactions between the surrounding water molecules and the wild-type αS may influence the abundance of specific residual intramolecular interactions. As shown above, the long-range intramolecular interactions between the different regions either are less abundant or disappear upon A53T mutation of the wild-type αS. This trend does not change with the usage of an explicit model for water (see Supporting Information).
Figure 4.
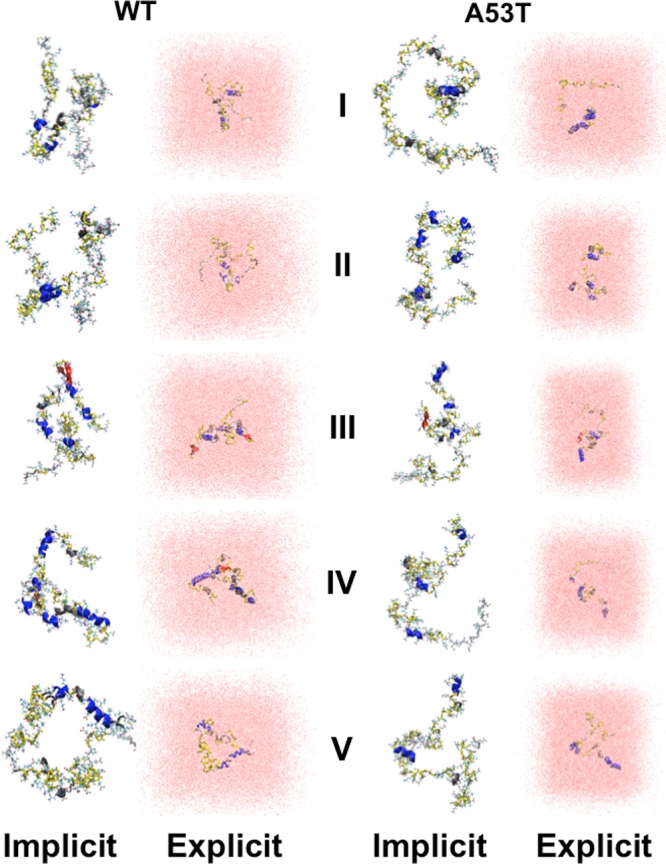
Wild-type and A53T mutant-type αS structures in implicit and explicit solvent. Pictorial representation of the wild-type (WT) and A53T mutant-type (A53T) αS structures obtained from REMD simulations in implicit solvent (Implicit) and from the classical MD simulations in explicit solvent (Explicit). The secondary structure of each residue is displayed according to the color of the protein back according to the following key: α-helix (blue), 310-helix (gray), π-helix (purple), β-sheet (red), β-bridge (black), turn (yellow), and coil (white).
Figure 5.
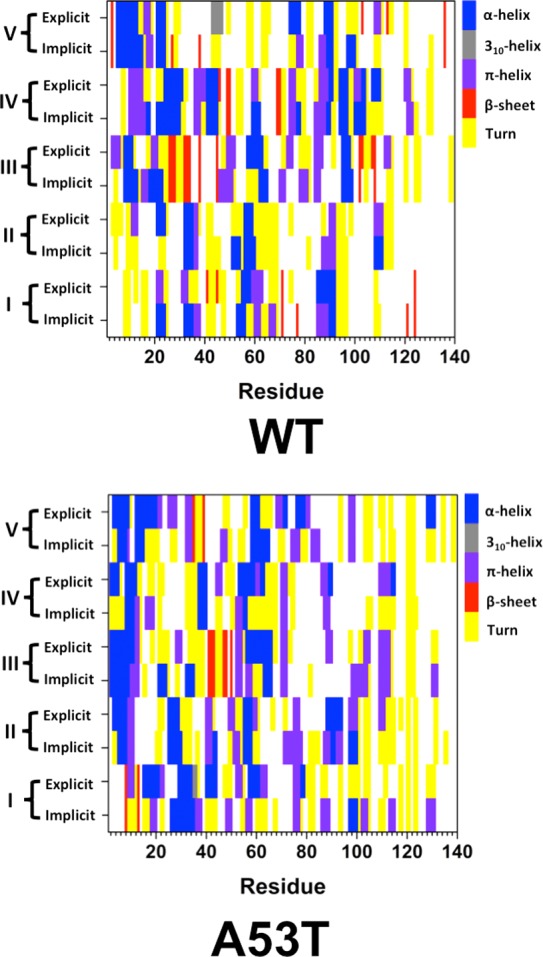
Secondary structures of the wild-type and A53T mutant-type αS structures in implicit and explicit solvent. Secondary structure components per residues of the wild-type (WT) and A53T mutant-type (A53T) αS conformations obtained from REMD simulations in implicit solvent (Implicit) and MD simulations in explicit solvent (Explicit). The color scale next to each figure displays the secondary structure component associated with a specific color.
Different than the simulations of native state proteins, which usually have a more stable structure than disordered proteins, one needs to consider the confined aqueous volume effect in the simulations of highly flexible intrinsically disordered large-size proteins. The current literature includes studies on intrinsically disordered proteins solvated in water utilizing explicit water models and small volumes, which in turn usually do not capture the many hydration shells around the protein, nor do such simulations provide the required space for the disordered protein to adopt specific conformations. For example, αS simulations were performed with small solvation shell thicknesses up to 5.0 Å.45,59 A smaller intrinsically disordered protein, amyloid-β (Aβ), which is at the center of neurodegenerative diseases, has been widely investigated using small aqueous volumes with hydration shell thicknesses usually ≤5.0 Å.62−64,68 The confined aqueous volume effect is visible when we compare the structures reported by different groups using varying hydration shell thicknesses for the same intrinsically disordered protein: Raffa and Rauk reported β-sheet formation in the N-terminal region of Aβ42 using a small confined aqueous volume that captures only the first hydration shell.64 However, Luttman and Fels showed no β-sheet formation in the N-terminal region of Aβ42 utilizing a larger hydration shell thickness of 22.0 Å in their simulations.67 To the best of our knowledge, the confined aqueous volume effect on the simulated structures of full-length intrinsically disordered proteins, which are at the center of neurodegenerative diseases, has not been addressed in detail. We performed additional sets of MD simulations on the full-length Aβ42 peptide using an explicit water model for addressing this effect. Our results (Supporting Information) clearly show that the confined aqueous volume using an explicit model for water significantly impacts the simulated structures. We should mention here again that simulations using an implicit water model do not face these challenges and limitations, but they ignore the intermolecular hydrogen bonding interactions between the disordered proteins and water molecules.
The intramolecular interaction maps that we obtain from our simulations are in excellent agreement with experiments and the simulation results of Carloni and co-workers,36 who used an explicit model for water. In agreement with their findings, our results show that intramolecular interactions of the C-terminal region with either the N-terminal (Met1–Lys60) or NAC regions of the α-synuclein protein are lost upon A53T mutation. This trend does not change using an implicit or explicit model for water. Furthermore, they reported that the average radius of gyration (Rg) of the A53T mutant-type αS protein is larger than that of the wild-type αS for most of the protein ensembles investigated, which is in agreement with our findings (see below). Possible variations in the results reported by Balesh et al.35 and between our studies, which are in agreement with experiments and the results presented by Carloni and co-workers,36 may occur due to various reasons: the initial structure chosen by Balesh et al. is the micelle bound αS structure instead of an extended structure or an experimentally determined free αS conformation; the impact of the confined aqueous volume on the simulations performed by Balesh et al. cannot be addressed because the solvation process is not described in detail (volume and number of water molecules are missing); decreasing the temperature to 0 K might influence the reported structures for the body temperature depending on the simulation time.35
The probability distribution of the Rg values for the wild-type and A53T mutant-type αS proteins are presented in Figure 6. For Rg values less than 35 Å, we observe that the probability is higher for the wild-type αS structures rather than the A53T mutant-type αS structures. At Rg values larger than 35 Å, we find that the A53T mutant-type αS structures have higher probabilities than the wild-type αS structures. Based on these distributions, the average Rg of the wild-type and A53T mutant-type α-synuclein proteins are 35.3 ± 6.1 Å and 38.1 ± 5.7 Å, respectively. Differential scanning calorimeter experiments reported a more hydrated structure for the A53T mutant-type rather than wild-type αS protein.45 Furthermore, classical MD simulations performed by Losasso et al. reported larger Rg for most conformers of the A53T mutant-type than the wild-type αS.36 However, small-angle X-ray scattering (SAXS) measurements by Li et al. conducted at a pH value of 7.5 in 100 mM NaCl yielded the same Rg value (40 ± 1 Å) for both the wild- and A53T mutant-type αS proteins.9,10 On the other hand, annealing MD simulations by Balesh et al. suggested that the A53T mutant-type αS protein is more compact than the wild-type αS protein.35 More compact protein structures are more likely to exhibit a higher abundance of intramolecular interactions between different regions of the protein. Therefore, the lower probability of A53T mutant-type structures in comparison to the wild-type αS protein with Rg values smaller than 35 Å agrees with the less probable long-range interactions in the A53T mutant-type αS structures described above. Furthermore, the αS structures with a higher Rg value are more solvent exposed, which as stated above may result in a higher aggregation propensity of these structures. Consequently, the higher probability of A53T mutant-type αS structures with Rg > 35 Å in comparison to the wild-type αS is related to the increased rate of aggregation of the αS protein upon A53T mutation.9,10,21−28
Figure 6.
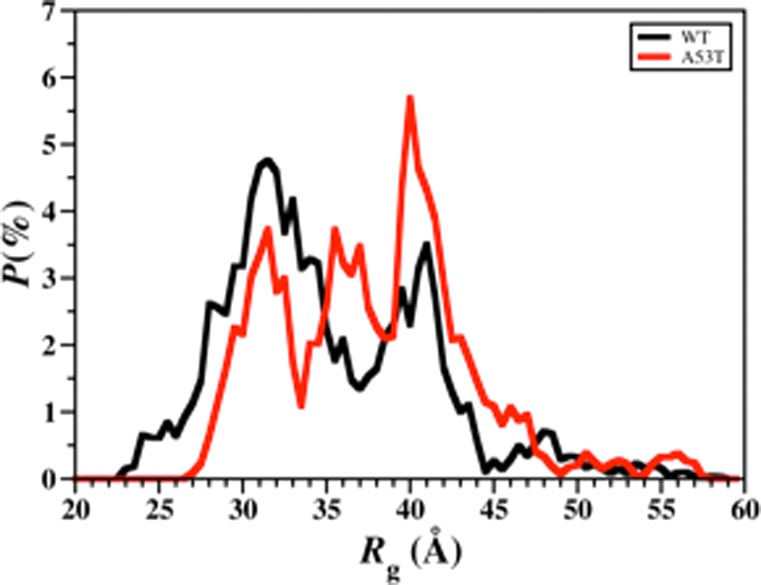
Wild-type and A53T mutant-type αS radius of gyration values. The probability distribution of the radius of gyration (Rg) values of the wild-type (black) and A53T mutant-type (red) αS for the structures obtained after convergence.
The average conformational Gibbs free energies along with their enthalpic and entropic contributions are presented in Table 1. The structures of the A53T mutant-type α-synuclein are thermodynamically more preferred than the structures of the wild-type α-synuclein by a ΔG value of 132.9 kJ mol–1. This result suggests that the A53T mutant-type αS structures are more stable than the wild-type αS protein structures in an aqueous solution medium. Specifically, the thermodynamic results suggest that the structures of the A53T mutant-type are more stable than its wild-type form largely due to the enthalpic contribution. Overall, these findings are attributed to the solvent exposure variations of the two intrinsically disordered proteins (see above).
Table 1. The Calculated Average Enthalpy (H), Solvation Free Energy (Gsol), H – Gsol, Entropy (TS), and Gibbs Free Energy (G) Values for the Wild-Type (WT) and A53T Mutant-Type (A53T) α-Synuclein Protein Structures in an Aqueous Solution Environment.
| peptide | ⟨Etotal⟩ (kJ mol–1) | ⟨Gsol⟩ (kJ mol–1) | ⟨H⟩ (kJ mol–1) | –T⟨S⟩ (kJ mol–1) | ⟨G⟩ (kJ mol–1) |
|---|---|---|---|---|---|
| WT | 7146.5 ± 174.3 | –16704.1 ± 169.7 | –9557.4 ± 18.4 | –7043.8 ± 8.4 | –16601.4 ± 13.0 |
| A53T | 7700.0 ± 188.8 | –17362.3 ± 185.2 | –9662.3 ± 21.0 | –7072.0 ± 9.6 | –16734.3 ± 10.2 |
Following our recent studies on intrinsically disordered proteins, we also assessed the conformational preferences of the wild-type and A53T mutant-type αS proteins using the potential of mean force (PMF) surfaces along the coordinates of the Rg and end-to-end distance (RE-E) as shown in Figure 7.42,44,46−48 The PMF surfaces provide information on whether these structural parameters are affected by the mutation via linking these characteristics to thermodynamics with dynamics. The presence of different basins with similar thermodynamic values along the PMF surface indicates that different conformations of the same protein have similar thermodynamic preferences. Evaluating the structures located in these different thermodynamically preferred basins yields insights into which conformational properties influence the favorability of the protein structures. The wild-type αS protein shows two favorable PMF basins: basins IA and IB. Basin IA occurs at Rg values varying between 28.0 and 35.5 Å and at RE-E values from 51 to 98 Å, whereas basin IB is located at Rg values varying between 37.4 and 42.0 Å and RE-E values varying between 108.0 and 148.1 Å. The PMF surface for the A53T mutant-type αS protein also presents two favorable basins located at Rg values varying between 29.0 and 33.0 Å for basin IA and 34.0 and 43.2 Å for basin IB and RE-E values varying between 45.2 and 73.1 Å for basin IA and 91.1 and 137.3 Å for basin IB. Moreover, the PMF surfaces indicate that transitions between the structures located in basin IA and basin IB require the overriding of larger energy barriers (>1kBT) for both the wild-type and A53T mutant-type αS proteins.
Figure 7.
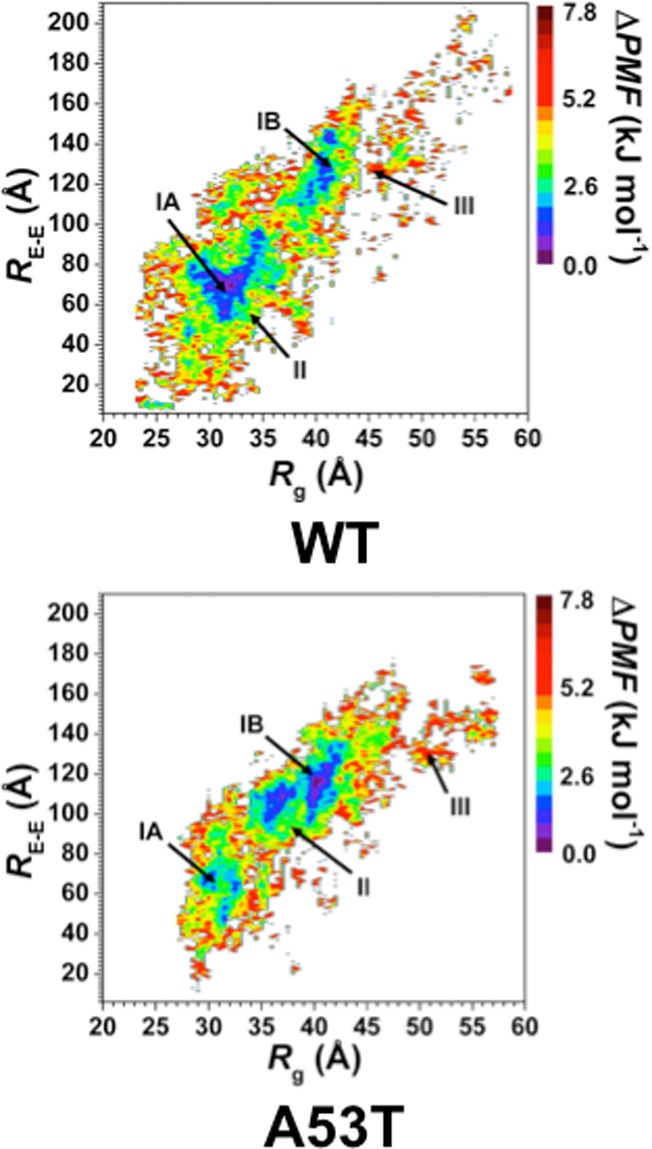
Wild-type and A53T mutant-type αS PMF surfaces. Change in the potential of mean force (ΔPMF) of the wild-type (WT) and A53T mutant-type (A53T) αS along the coordinates of radius of gyrations (Rg) and end-to-end distance (RE-E) in units of kJ mol–1. The reference values for the PMF surfaces are 6.73 kJ and 6.48 kJ mol–1 for the wild-type and A53T mutant-type αS, respectively.
For gaining insights into the relationships between the structural properties and PMF surfaces, we also calculated the secondary and tertiary structure components per residue of each PMF basin for the wild-type and A53T mutant-type αS proteins. As shown in Figure 8, the basin IA structures of the wild-type αS protein present the most abundant α-helical structure (up to 56%) in the C-terminal region at Phe94–Gly101. This abundance decreases by 30% for the basin IB structures (Figure 8). Instead, basin IB structures of wild-type αS present the highest abundance of α-helix formation in the N-terminal and NAC regions at Glu20–Val26, Lys32–Gly36, and Lys28–Thr64 (up to 46%), which decreases in the basin IA structures. In addition, α-helix content at Met5, Lys6, Glu28, Ala29, Leu38, Val40–Lys43, Glu61, Lys80–Glu83, and Ala89–Thr92 decreases from basin IA to basin IB. Along the PMF surface, only residues Glu20–Gln24 and Lys32–Glu35 present a decreasing abundance of α-helix structure with decreasing PMF values for structures located in basin IB (basin IB > basin II > basin III) of the wild-type αS. Differences in the 310-helix structure abundance of the structures in the favorable PMF basins of the wild-type αS protein are also noted (Figure 8). Specifically, residues Lys43–Lys45, Gln79–Ala85, and Asp121–Tyr125 present a more abundant 310-helix structure in the basin IA rather than basin IB structures, whereas the opposite trend is detected for residues Val15–Ala17, Gly25–Gly31, Gly36–Leu38, Val40–Ser42, Gly86–Ile88, Val95–Gln99, and Gly101. The β-sheet content of the wild-type αS structures also shows significant deviations between the conformations located in basins IA and IB. Residues Val16, Gln24, Glu35, Val70, Val71, Lys102, and Pro108 adopt β-sheet structure in the basin IA conformations (up to 22%) of the wild-type αS protein. On the other side, the most abundant β-sheet formation (up to 60%) occurs at Phe4, Glu13, Val82, Glu83, Ala90, Ala91 and Gln109 in the structures located in basin IB.
Figure 8.

Wild-type and A53T mutant-type αS secondary structure components of the PMF basin structures. Calculated secondary structure abundances per residue for the structures of the wild-type (WT) and A53T mutant-type (A53T) αS located in basin IA (purple), basin IB (blue), basin II (green), and basin III (red) along the PMF surface. The abundances for the π-helix and coil structures are not displayed.
For the A53T mutant-type αS protein, the most abundant α-helix formation occurs in the N-terminal region at Val3–Ala11 (up to 50%) for the structures located in basin IA but this stability decreases by up to 20% in the same region in the structures located in basin IB (Figure 8). Residues Lys96–Gln99 in the C-terminal region of the structures located in basin IB form abundant α-helix (up to 45%), which form less stable α-helix in the structures located in basin IA. Residues Lys21–Gln24 in the N-terminal region adopt more abundant α-helix in the structures located in basin IB rather than the same region in the structures located in basin IA of the A53T mutant-type αS. The opposite trend is detected for Lys32–Glu35 located in the N-terminal region. Regarding 310-helix structure formation, the most prominent formations occur in parts of the NAC and N-terminal regions (Val52–Gly61) and the C-terminal region (Gln109–Glu114) with up to 40% abundance for both basins IA and IB. Interestingly, more abundant 310-helix formation occurs in the structures located in basin IA rather than those in basin IB for the A53T mutant-type αS (Lys32–Glu35, Val40–Ser42, and Lys102–Glu105), whereas the opposite trend is detected at Lys43–Lys45, Phe94–Lys96, and Pro120–Ala124. However, helical structure abundance does not display a consistent trend with PMF values. Furthermore, we determined the noteworthy deviations in the β-sheet formation of the wild-type and the A53T mutant-type α-synuclein between the most preferred PMF basins. The N-terminal region of the basin IA structures of the A53T mutant-type αS presents the highest abundance of β-sheet structure at Glu35 and Tyr39 (up to 20%), but decreases in the basin IB conformations. Further significant β-sheet structure formations are detected at Gly7, Leu8, Glu13, Gly14, Gly101, Asn103, and Gly106 in the basin IA structures. On the other side, the basin IB structures form the highest β-sheet content at Thr81 and Ala91 in the NAC region (up to 12%), which is not present in the basin IA structures. In addition, slightly less abundant β-sheet formation is also observed at Leu8, Val15, Val26, Ala30, Glu46, His50, Lys102, and Pro108. However, distinct trends in the abundance of β-sheet formation along the PMF surface are not evident for the A53T mutant-type αS protein.
The intramolecular interactions of the structures located in the most favorable PMF basins for the wild-type and A53T mutant-type αS proteins display prominent differences (Figure 9). The NAC and C-terminal regions of the wild-type αS protein present abundant interactions (up to 90%) both between and within the regions Gly86–Glu104 and Val70–Glu83 or Glu105–Ala124 (Figure 9). Moderately abundant (up to 45%) interactions of the C-terminal regions are also observed with the N-terminal and NAC regions: Val55–Gly86 and Gly104–Tyr136. Furthermore, residues Leu8–Val16 of the N-terminal region and Gly132–Ala140 of the C-terminal region interact weakly (5%) in the conformations located in basin IA of the wild-type αS protein. Similar to the basin IA structures, the basin IB structures of the wild-type αS protein present abundant intramolecular interactions between the NAC and C-terminal regions via Thr72–Ile88 and Ile88–Val118 as well as Ala90–Asn103 and Glu104–Pro120 (Figure 9). Weak intramolecular interactions between Met1–Ala18 in the N-terminal region and the NAC or C-terminal regions in basin IA conformation are no longer present in the basin IB structures. Instead, we observe an increase in abundance of the intramolecular interactions within the N-terminal region between residues Met1–Lys10 and Gly14–Gly25. Deviations in the intramolecular interactions of the basin IA and basin IB structures of the A53T mutant-type αS are also observed. Strong intramolecular interactions (50–90%) occur between Glu83–Glu110 and Leu100–Pro128 in the NAC and C-terminal regions of the basin IA conformations of the A53T mutant-type αS protein. Interestingly, strong intramolecular interactions between the same regions are observed in the basin IB structures, as well as between Leu100–Pro120 and Ala85–Gly106. However, the moderately abundant interactions between Lys58–Ala78 and Gly84–Leu100, as well as those between Gln24–Gly47 and Lys60–Ala76, that are present in the basin IA structures are weaker by up to 20% in the basin IB structures of the A53T mutant-type αS protein. Furthermore, the weakly abundant intramolecular interactions of the N-terminal and NAC regions (Met1–Val66) with the C-terminal region (Ala124–Ala140) in the basin IA conformations disappear in the basin IB structures. These findings clearly demonstrate that the structure and thermodynamic property relationships are largely affected by the A53T mutation of the wild-type αS protein in an aqueous solution.
Figure 9.

Wild-type and A53T mutant-type αS tertiary structures of the favorable PMF basin structures. Calculated intramolecular interactions of the wild-type αS structures located in basin IA (WT IA) and basin IB (WT IB) of the PMF surface and of the A53T mutant-type αS structures located in basin IA (A53T IA) and basin IB (A53T IB) of the PMF surface. The color scale corresponds to the probability (P) of the distance between the heavy atoms (C, N, O, S) of a residue being ≤20 Å from each other.
The results and discussions presented in the Supporting Information section for the confined aqueous volume effect in the simulations of intrinsically disordered proteins clearly show that the chosen aqueous volume impacts the simulated disordered protein structures. Even though simulations using an implicit water model do not capture the intermolecular interactions between the protein and water molecules, such simulations do not suffer from the confined aqueous volume effect, which impacts the simulated structures of intrinsically disordered proteins enormously because these proteins lack a stable structure unlike native state proteins and require a larger confined volume than the ones presented in the current literature.
Summary
Overall, we report the structural and thermodynamic properties including the conformational Gibbs free energies and secondary structure conversion free energies at the atomic level with dynamics for the wild-type and A53T mutant-type αS proteins in aqueous solution, which reveals the impact of the A53T mutation at the monomeric level on the wild-type αS protein. Even though some structural properties have been presented by experiments and theoretical studies before, in this work, we present all structural properties in detail along with thermodynamic properties, and we provide details that are currently not available in the literature. The helical content of wild-type αS is minimally affected by the A53T mutation except for a few residues in the N-terminal and C-terminal regions. This result agrees with several reported experimental measurements as well as the similar binding affinities of the wild-type and A53T mutant-type αS proteins for phospholipid vesicles and membranes. Interestingly, we detect a decrease in the β-sheet content formed in the NAC and C-terminal regions, while the N-terminal region around the mutation site presents an increase in β-sheet abundance upon A53T mutation. This finding indicates that the N-terminal region close to the 53rd amino acid residue plays an active role in the aggregation mechanism of the A53T mutant-type αS protein. Furthermore, our newly developed strategy reveals that the free energy stability of secondary structure transitions resulting in a helical structure formation is unaffected by the A53T mutation. However, the preferred residual secondary structure component transitions to a β-sheet structure shift to the N-terminal region around the mutation site except at the 53rd amino acid residue itself, which shows a significant decrease in the stability for transitions yielding a β-sheet structure. Additionally, transitions from a coil to a β-sheet structure are the most favorable for the abundant β-sheet forming residues (see above) of the wild-type and A53T mutant-type αS proteins in aqueous solution. The intramolecular interactions occurring in the wild-type and A53T mutant-type αS protein structures reveal that the long-range interactions between the NAC region and the N- and C-terminal regions disappear upon A53T mutation. This result indicates that the NAC region is more solvent exposed in the A53T mutant-type rather than wild-type αS, which is related to an increase in the rate of aggregation. In addition to the overall secondary and tertiary structural characteristics of the wild-type and A53T mutant-type αS, the PMF surfaces of both proteins present two favorable conformational basins with different secondary and tertiary structural properties. However, transitions of structures between these two basins are predicted to occur more readily for the A53T rather than the wild-type αS due to the decreased energy barrier between the basins in the A53T mutant-type in comparison to the wild-type αS protein. Overall, our results show that the A53T mutation significantly impacts the structural properties of the wild-type αS protein. These resulting differences agree with previous observed experimental measurements and can provide further insight into the aggregation mechanism of these two proteins. The details yielded from this study aid in providing the currently missing fundamental knowledge about the A53T mutant-type αS protein and the impact of the A53T mutation on the structures of the wild-type αS protein. The results reported herein can help in the design and synthesis of more efficient drugs that can block the specific residues forming certain structures, that is, β-sheet structure, which are reactive toward aggregation.
Methods
Intrinsically disordered proteins can adopt a multitude of different conformations. As a result, the theoretical method for investigating intrinsically disordered proteins needs to be chosen carefully so that the different possible protein conformations are adequately sampled. REMD simulations utilize special sampling throughout the course of the simulation to overcome energy barriers between different minimum energy conformations.49,50 We performed extensive REMD simulations with the AMBER 11 software package on the wild-type and A53T mutant-type αS proteins utilizing the AMBER ff99SB force field parameters for proteins.51,52 The usage of an explicit solvent model in REMD simulations can result in errors due to variations in the heat capacity of water as well as conformational effects due to confined aqueous volume and therefore the Onufriev–Bashford–Case Generalized Born implicit solvent model was utilized.43,44,47,53,54 A total number of 56 replicas were employed for the wild-type and A53T mutant-type αS with temperature exponentially distributed between 283 and 400 K, yielding an exchange probability of 0.70.55 The initial fully extended structures of the wild-type and A53T mutant-type αS proteins were equilibrated for 500 ns per replica. REMD simulations were performed for a total of 40 ns with exchanges between replicas attempted every 5 ps with a time step of 2 fs. Trajectories were saved every 500 steps. Langevin dynamics was used to maintain the temperature of each replica with a collision frequency of 2 ps–1.56,57 Following our recent studies, the particle mesh Ewald (PME) method was used to treat long-range interactions.56,57 The bonds to hydrogen atoms were constrained using the SHAKE algorithm.58 Despite the confined aqueous volume effect in the simulations of highly flexible large-size intrinsically disordered proteins, usage of an implicit water model in the simulations of these species ignores the impact of intermolecular hydrogen bonding interactions as well as short- and long-range solvent structuring and local density effects on the determined disordered protein conformations. Therefore, sets of additional simulations were performed utilizing specific wild-type and A53T mutant-type conformations that we obtained from our REMD simulations using an implicit water model as the initial structures. Specifically, all structures were solvated using the modified TIP5P explicit model for water74,75 in a box where the closest distance between the protein and any box edge was 20 Å and simulated for additional 30 ns via separate classical MD simulation runs at the same temperature and pressure of interest (310 K temperature and a pressure of 0.1 MPa). In order to further address the confined aqueous volume effect on the simulations of a disordered protein using an explicit water model, we also performed all-atom MD simulations using a smaller size disordered protein in water, amyloid-β(1–42) (Aβ42). Aβ42 was solvated in cubic boxes with layers of water for 20 and 30 Å in two separate simulations (corresponding to 10 314 and 25 777 explicit water molecules), respectively. We should mention here that the current literature includes smaller solvation boxes used in the simulations of intrinsically disordered proteins, and the confined aqueous volume effect becomes more significant in such simulations.36,45,59−68 The integration time step was set to 2 fs. An isobaric–isothermal ensemble was applied using Langevin dynamics.56,57 The RATTLE algorithm was applied to restrain the bonds between heavy atoms and hydrogen atoms.69 The temperature was set to 310 K to correlate to the physiological temperature. The long-range interactions were treated using the particle mesh Ewald method and the cutoff value for nonbonded interactions was set to 12 Å.56,57 Counter ions (Na+) were used for neutralizing the charges.
The cumulative secondary structure abundance (see Supporting Information) was used to verify the convergence of the REMD simulations of the wild-type and A53T mutant-type αS proteins at 20 ns of simulation time as shown in our previous studies on intrinsically disordered proteins.42,44,46−48,70 The structural and thermodynamic properties of the wild-type and A53T mutant-type αS proteins were calculated from the structures obtained after convergence from the replica closest to physiological temperature (310 K). The abundances of the secondary structure components per residue for the wild-type and A53T mutant-type αS proteins were calculated via the DSSP program.71 In addition, we applied our recently developed theoretical strategy to calculate the free energy change associated with transitions between two different secondary structure components at the atomic level with dynamics.42,44,46 This method calculates the PMF of each transition via the conditional probability, defined as (P(ti→j|Stj)). Within this conditional probability, P(ti→j) is the probability of a transition between two different secondary structures, i and j, while P(Stj) identifies probability of a transition resulting in the formation of a specific secondary structure, j, for a certain residue. The free energy change of each secondary structure transition is then calculated using our ProtMet software package (eq 1):
| 1 |
where kB is the Boltzmann constant, T is the temperature, and Z(λ) is the conditional probability ratio of the specific secondary structure transition. More details can be found in refs (42, 44 and 46). The intramolecular interactions of the wild-type and A53T mutant-type αS proteins were determined by calculating the probability of interactions between two different residues. Intramolecular interactions between two different residues occur if a heavy atom (C, N, O, or S) of a residue is at least 20 Å from a heavy atom of any other residue, as shown in our previous studies regarding the αS protein.44,46 The thermodynamic preferences of the wild-type and A53T mutant-type αS proteins were determined using both the MM/PBSA and PMF methods as shown in our previous studies on intrinsically disordered fibrillogenic proteins.42,44,46−48,70 The MM/PBSA method utilizes the potential energy (Etot), solvation free energy (Gsol), and entropy (S) of each protein structure to calculate the estimated conformational Gibbs free energy (G) of the same protein structure at a specific temperature (T) via eq 2:
| 2 |
The Gsol is the summation of the electrostatic and nonpolar contributions of each protein structure to the G. The electrostatic contribution to the G is calculated using dielectric constant values of 1 and 80 for the protein and solvent environment, respectively. The entropy values were estimated using the normal-mode analysis method.72 Entropy value calculations using a quasi-harmonic method, namely, the Schlitter method,73 were also attempted; however, the conformational changes were too large for this method to be applied. The coordinates of Rg and RE-E were used to determine the PMF surfaces of both the wild-type and A53T mutant-type αS proteins, as shown in our previous studies of the αS and amyloid-β proteins.42,44,46−48
Acknowledgments
The calculations and simulations were performed on Kraken at the National Institute for Computational Sciences, Texas Advanced Computing Center (TACC), and the Computational Biology Initiative and The University of Texas at San Antonio. The authors thank Aquila Dunn, Isin Sakallioglu, Ahmet K. Aloglu for their help in technical assistance.
Glossary
Abbreviations
- αS
α-synuclein
- far-UV-CD
far-ultraviolet-circular dichroism
- SMF
single molecule force
- FTIR
Fourier transform infrared
- FRET
Förster resonance energy transfer
- NAC
non-amyloid-β component
- SDS
sodium dodecyl sulfate
- REMD
replica exchange molecular dynamics
- NMR
nuclear magnetic resonance
- Rg
radius of gyration
- RE-E
end-to-end distance
- PMF
potential of mean force
- MM/PBSA
molecular mechanics/Poisson–Boltzmann surface area
- NMA
normal-mode analysis
- Aβ42
amyloid-β(1–42)
Supporting Information Available
The convergence figures for the wild-type and A53T mutant-type αS, the convergence figures for the secondary structure transition stabilities and the PMF surfaces of both peptides, the structural properties of both proteins for the least favorable basins on the PMF surfaces, and the results from our classical MD simulations of the model disordered peptide (Aβ42) using an explicit model for water that demonstrate the impact of confined aqueous volume. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by an allocation and computing resources provided by the National Science Foundation (Grant No. TG-CHE110044).
The authors declare no competing financial interest.
Supplementary Material
References
- Goedert M. (2001) Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2, 492–501. [DOI] [PubMed] [Google Scholar]
- Braak H.; Del Tredici K.; Rub U.; de Vos R. A. I.; Steur E.; Braak E. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 24, 197–211. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G.; Schmidt M. L.; Lee V. M. Y.; Trojanowski J. Q.; Jakes R.; Goedert M. (1997) Alpha-synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Polymeropoulos M. H.; Lavedan C.; Leroy E.; Ide S. E.; Dehejia A.; Dutra A.; Pike B.; Root H.; Rubenstein J.; Boyer R.; Stenroos E. S.; Chandrasekharappa S.; Athanassiadou A.; Papapetropoulos T.; Johnson W. G.; Lazzarini A. M.; Duvoisin R. C.; DiIorio G.; Golbe L. I.; Nussbaum R. L. (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276, 2045–2047. [DOI] [PubMed] [Google Scholar]
- Zarranz J. J.; Alegre J.; Gomez-Esteban J. C.; Lezcano E.; Ros R.; Ampuero I.; Vidal L.; Hoenicka J.; Rodriguez O.; Atares B.; Llorens V.; Tortosa E. G.; del Ser T.; Munoz D. G.; de Yebenes J. G. (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 55, 164–173. [DOI] [PubMed] [Google Scholar]
- Kruger R.; Kuhn W.; Muller T.; Woitalla D.; Graeber M.; Kosel S.; Przuntek H.; Epplen J. T.; Schols L.; Riess O. (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 18, 106–108. [DOI] [PubMed] [Google Scholar]
- Winner B.; Jappelli R.; Maji S. K.; Desplats P. A.; Boyer L.; Aigner S.; Hetzer C.; Loher T.; Vilar M.; Campionic S.; Tzitzilonis C.; Soragni A.; Jessberger S.; Mira H.; Consiglio A.; Pham E.; Masliah E.; Gage F. H.; Riek R. (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. U.S.A. 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro T. F.; Putcha P.; Tetzlaff J. E.; Spoelgen R.; Koker M.; Carvalho F.; Hyman B. T.; McLean P. J. (2008) Formation of Toxic Oligomeric α-Synuclein Species in Living Cells. Plos One 3, e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Uversky V. N.; Fink A. L. (2001) Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human alpha-synuclein. Biochemistry 40, 11604–11613. [DOI] [PubMed] [Google Scholar]
- Li J.; Uversky V. N.; Fink A. L. (2002) Conformational behavior of human alpha-synuclein is modulated by familial Parkinson’s disease point mutations A30P and A53T. Neurotoxicology 23, 553–567. [DOI] [PubMed] [Google Scholar]
- Choong C. J.; Say Y. H. (2011) Neuroprotection of alpha-synuclein under acute and chronic rotenone and maneb treatment is abolished by its familial Parkinson’s disease mutations A30P, A53T and E46K. Neurotoxicology 32, 857–863. [DOI] [PubMed] [Google Scholar]
- Wersinger C.; Sidhu A. (2003) Differential cytotoxicity of dopamine and H2O2 in a human neuroblastoma divided cell line transfected with alpha-synuclein and its familial Parkinson’s disease-linked mutants. Neurosci. Lett. 342, 124–128. [DOI] [PubMed] [Google Scholar]
- Moussa C. E. H.; Wersinger C.; Tomita Y.; Sidhu A. (2004) Differential cytotoxicity of human wild type and mutant alpha-synuclein in human neuroblastoma SH-SY5Y cells in the presence of dopamine. Biochemistry 43, 5539–5550. [DOI] [PubMed] [Google Scholar]
- Bussell R.; Eliezer D. (2004) Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochemistry 43, 4810–4818. [DOI] [PubMed] [Google Scholar]
- Choi W.; Zibaee S.; Jakes R.; Serpell L. C.; Davletov B.; Crowther R. A.; Goedert M. (2004) Mutation E46K increases phospholipid binding and assembly into filaments of human alpha-synuclein. FEBS Lett. 576, 363–368. [DOI] [PubMed] [Google Scholar]
- Jensen P. H.; Nielsen M. S.; Jakes R.; Dotti G.; Goedert M. (1998) Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 273, 26292–26294. [DOI] [PubMed] [Google Scholar]
- Jo E. J.; Fuller N.; Rand R. P.; St George-Hyslop P.; Fraser P. E. (2002) Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 315, 799–807. [DOI] [PubMed] [Google Scholar]
- Perrin R. J.; Woods W. S.; Clayton D. F.; George J. M. (2000) Interaction of human alpha-synuclein and Parkinson’s disease variants with phospholipids - Structural analysis using site-directed mutagenesis. J. Biol. Chem. 275, 34393–34398. [DOI] [PubMed] [Google Scholar]
- Ding T. T.; Lee S. J.; Rochet J. C.; Lansbury P. T. (2002) Annular alpha-synuclein protofibrils are produced when spherical protofibrils are incubated in solution or bound to brain-derived membranes. Biochemistry 41, 10209–10217. [DOI] [PubMed] [Google Scholar]
- Volles M. J.; Lee S. J.; Rochet J. C.; Shtilerman M. D.; Ding T. T.; Kessler J. C.; Lansbury P. T. (2001) Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 40, 7812–7819. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Harper J. D.; Lansbury P. T. (1998) Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 4, 1318–1320. [DOI] [PubMed] [Google Scholar]
- Giasson B. I.; Uryu K.; Trojanowski J. Q.; Lee V. M. Y. (1999) Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 274, 7619–7622. [DOI] [PubMed] [Google Scholar]
- Serpell L. C.; Berriman J.; Jakes R.; Goedert M.; Crowther R. A. (2000) Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U. S. A. 97, 4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyoshihara T.; Kojima M.; Ueda K.; Tashiro M.; Shimotakahara S. (2007) Observation of multiple intermediates in alpha-synuclein fibril formation by singular value decomposition analysis. Biochem. Biophys. Res. Commun. 355, 398–403. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A.; Petre B. M.; Wall J.; Simon M.; Nowak R. J.; Walz T.; Lansbury P. T. (2002) alpha-synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 322, 1089–1102. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Lee S. J.; Rochet J. C.; Ding T. T.; Williamson R. E.; Lansbury P. T. (2000) Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. U. S. A. 97, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narhi L.; Wood S. J.; Steavenson S.; Jiang Y. J.; Wu G. M.; Anafi D.; Kaufman S. A.; Martin F.; Sitney K.; Denis P.; Louis J. C.; Wypych J.; Biere A. L.; Citron M. (1999) Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 274, 9843–9846. [DOI] [PubMed] [Google Scholar]
- Ono K.; Ikeda T.; Takasaki J.; Yamada M. (2011) Familial Parkinson disease mutations influence alpha-synuclein assembly. Neurobiol. Dis. 43, 715–724. [DOI] [PubMed] [Google Scholar]
- Bussell R.; Eliezer D. (2001) Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J. Biol. Chem. 276, 45996–46003. [DOI] [PubMed] [Google Scholar]
- Heise H.; Celej M. S.; Becker S.; Riede D.; Pelah A.; Kumar A.; Jovin T. M.; Baldus M. (2008) Solid-state NMR reveals structural differences between fibrils of wild-type and disease-related A53T mutant alpha-synuclein. J. Mol. Biol. 380, 444–450. [DOI] [PubMed] [Google Scholar]
- Brucale M.; Sandal M.; Di Maio S.; Rampioni A.; Tessari I.; Tosatto L.; Bisaglia M.; Bubacco L.; Samori B. (2009) Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers. ChemBioChem 10, 176–183. [DOI] [PubMed] [Google Scholar]
- Bertoncini C. W.; Jung Y. S.; Fernandez C. O.; Hoyer W.; Griesinger C.; Jovin T. M.; Zweckstetter M. (2005) Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl. Acad. Sci. U. S. A. 102, 1430–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S.; Sarkar A.; Sundar D. (2009) Controlling aggregation propensity in A53T mutant of alpha-synuclein causing Parkinson’s disease. Biochem. Biophys. Res. Commun. 387, 305–309. [DOI] [PubMed] [Google Scholar]
- Perlmutter J. D.; Braun A. R.; Sachs J. N. (2009) Curvature dynamics of alpha-synuclein familial parkinson disease mutants molecular simulations of the micelle- and bilayer-bound forms. J. Biol. Chem. 284, 7177–7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balesh D.; Ramjan Z.; Floriano W. B. (2011) Unfolded annealing molecular dynamics conformers for wild-type and disease-associated variants of alpha-synuclein show no propensity for beta-sheet formation. J. Biophys. Chem. 2, 124–134. [Google Scholar]
- Losasso V.; Pietropaolo A.; Zannoni C.; Gustincich S.; Carloni P. (2011) Structural role of compensatory amino acid replacements in the alpha-synuclein protein. Biochemistry 50, 6994–7001. [DOI] [PubMed] [Google Scholar]
- Jonsson S. A.; Mohanty S.; Irback A. (2012) Distinct phases of free alpha-synuclein-A Monte Carlo study. Proteins 80, 2169–2177. [DOI] [PubMed] [Google Scholar]
- Clayton D. F.; George J. M. (1998) The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 21, 249–254. [DOI] [PubMed] [Google Scholar]
- Davidson W. S.; Jonas A.; Clayton D. F.; George J. M. (1998) Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- Jo E. J.; McLaurin J.; Yip C. M.; St George-Hyslop P.; Fraser P. E. (2000) Alpha-synuclein membrane interactions and lipid specificity. J. Biol. Chem. 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- Conway K. A.; Harper J. D.; Lansbury P. T. (2000) Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloi. Biochemistry 39, 2552–2563. [DOI] [PubMed] [Google Scholar]
- Coskuner O.; Wise-Scira O.; Perry G.; Kitahara T. (2013) The structure of the E22Δ mutant-type amyloid-β alloforms and the impact of E22Δ mutation on the structures of the wild-type amyloid-β alloforms. ACS Chem. Neurosci. 4, 310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. (2007) Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 103, 17–37. [DOI] [PubMed] [Google Scholar]
- Wise-Scira O.; Dunn A.; Aloglu A. K.; Sakallioglu I. T.; Coskuner O. (2013) Structures of the E46K mutant-type α-synuclein protein and impact of E46K mutation on the structures of the wild-type α-synuclein protein. ACS Chem. Neurosci. 4, 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazy E.; Bokor M.; Kalmar L.; Gelencser A.; Kamasa P.; Han K. H.; Tompa K.; Tompa P. (2011) Distinct hydration properties of wild-type and familial point mutant A53T of alpha-synuclein associated with Parkinson’s disease. Biophys. J. 101, 2260–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Aloglu A. K.; Dunn A.; Sakallioglu I. T.; Coskuner O. (2013) Structures and free energy landscapes of the wild-type and A30P mutant-type α-synuclein proteins with dynamics. ACS Chem. Neurosci. 4, 486–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Kitahara T.; Perry G.; Coskuner O. (2011) Amyloid-beta peptide structure in aqueous solution varies with fragment size. J. Chem. Phys. 135, 205101. [DOI] [PubMed] [Google Scholar]
- Wise-Scira O.; Xu L.; Perry G.; Coskuner O. (2012) Structures and free energy landscapes of aqueous zinc(II)-bound amyloid-beta(1–40) and zinc(II)-bound amyloid-beta(1–42) with dynamics. J. Biol. Inorg. Chem. 17, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugita Y.; Okamoto Y. (1999) Replica-exchange molecular dynamics method for protein folding. Chem. Phys. Lett. 314, 141–151. [Google Scholar]
- Zhang W.; Wu C.; Duan Y. (2005) Convergence of replica exchange molecular dynamics. J. Chem. Phys. 123, 154105. [DOI] [PubMed] [Google Scholar]
- Case D., Darden T., Cheatham T., Simmerling C., Wang J., Duke R., Luo R., Walker R., Zhang W., and Merz K. (2010) AMBER 11, University of California, San Francisco.
- Hornak V.; Abel R.; Okur A.; Strockbine B.; Roitberg A.; Simmerling C. (2006) Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Onufriev A.; Bashford D. (2004) Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 55, 383–394. [DOI] [PubMed] [Google Scholar]
- Windisch M.; Wolf H.; Hutter-Paier B.; Wronski R. (2008) The role of alpha-synuclein in neurodegenerative diseases: A potential target for new treatment strategies?. Neurodegener. Dis. 5, 218–221. [DOI] [PubMed] [Google Scholar]
- van der Spoel D.; Patriksson A. (2008) A temperature predictor for parallel tempering simulations. Phys. Chem. Chem. Phys. 10, 2073–2077. [DOI] [PubMed] [Google Scholar]
- Allen M. P., and Tildesley D. J. (1999) Computer Simulation of Liquids; Clarendon Press, Oxford, U.K.. [Google Scholar]
- Frenkel D., and Smit B. (2002) Understanding Molecular Simulation: From Algorithms to Applications, Vol. 4, Academic Press, San Diego, CA. [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341. [Google Scholar]
- Chatterjee P.; Sengupta N. (2012) Effect of the A30P mutation on the structural dynamics of micelle-bound alpha Synuclein released in water: a molecular dynamics study. Eur. Biophys. J. Biophys. Lett. 41, 483–489. [DOI] [PubMed] [Google Scholar]
- Baumketner A.; Shea J. E. (2006) Folding landscapes of the Alzheimer amyloid-beta(12–28) peptide. J. Mol. Biol. 362, 567–579. [DOI] [PubMed] [Google Scholar]
- Xu Y. C.; Shen J. J.; Luo X. M.; Zhu W. L.; Chen K. X.; Ma J. P.; Jiang H. L. (2005) Conformational transition of amyloid beta-peptide. Proc. Natl. Acad. Sci. U. S. A. 102, 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Ham S. (2011) Characterizing amyloid-beta protein misfolding from molecular dynamics simulations with explicit water. J. Comput. Chem. 32, 349–355. [DOI] [PubMed] [Google Scholar]
- Brovchenko I.; Burri R. R.; Krukau A.; Oleinikova A.; Winter R. (2008) Intrinsic thermal expansivity and hydrational properties of amyloid peptide A beta(42) in liquid water. J. Chem. Phys. 129, 195101. [DOI] [PubMed] [Google Scholar]
- Raffa D. F.; Rauk A. (2007) Molecular dynamics study of the beta amyloid peptide of Alzheimer’s disease and its divalent copper complexes. J. Phys. Chem. B 111, 3789–3799. [DOI] [PubMed] [Google Scholar]
- Triguero L.; Singh R.; Prabhakar R. (2008) Comparative molecular dynamics studies of wild-type and oxidized forms of full-length Alzheimer amyloid beta-peptides A beta(1–40) and A beta(1–42). J. Phys. Chem. B 112, 7123–7131. [DOI] [PubMed] [Google Scholar]
- Cruz L.; Urbanc B.; Borreguero J. M.; Lazo N. D.; Teplow D. B.; Stanley H. E. (2005) Solvent and mutation effects on the nucleation of amyloid beta-protein folding. Proc. Natl. Acad. Sci. U. S. A. 102, 18258–18263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttmann E.; Fels G. (2006) All-atom molecular dynamics studies of the full-length beta-amyloid peptides. Chem. Phys. 323, 138–147. [Google Scholar]
- Sgourakis N. G.; Yan Y. L.; McCallum S. A.; Wang C. Y.; Garcia A. E. (2007) The Alzheimer’s peptides A beta 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol. 368, 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen H. C. (1983) RATTLE: A “Velocity” version of the SHAKE algorithm for molecular dynamics calculations. J. Comput. Phys. 52, 24–34. [Google Scholar]
- Fawver J. N.; Duong K. T.; Wise-Scira O.; Schall H. E.; Coskuner O.; Zhu X.; Colom L. V.; Murray I. V. J. (2012) Probing and trapping a sensitive conformation: Amyloid-β fibrils, oligomers, and dimers. J. Alzheimer’s Dis. 32, 197–215. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. (1983) Dictionary of protein secondary structure - pattern-recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Case D. A. (1994) Normal-mode analysis of protein dynamics. Curr. Opin. Struct. Biol. 4, 285–290. [Google Scholar]
- Schlitter J. (1993) Estimation of absolute and relative entropies of macromolecules using the covariance-matrix. Chem. Phys. Lett. 215, 617–621. [Google Scholar]
- Coskuner O.; Deiters U. K. (2006) Hydrophobic interactions by Monte Carlo simulations. Z. Phys. Chem. 220, 349–369. [Google Scholar]
- Coskuner O.; Deiters U. K. (2007) Hydrophobic interactions of xenon by Monte Carlo simulations. Z. Phys. Chem. 221, 785–799. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.