

Abstract
Although β-lactams have been the most effective class of antibacterial agents used in clinical practice for the past half century, their effectiveness on Gram-negative bacteria has been eroded due to the emergence and spread of β-lactamase enzymes that are not affected by currently marketed β-lactam/β-lactamase inhibitor combinations. Avibactam is a novel, covalent, non-β-lactam β-lactamase inhibitor presently in clinical development in combination with either ceftaroline or ceftazidime. In vitro studies show that avibactam may restore the broad-spectrum activity of cephalosporins against class A, class C, and some class D β-lactamases. Here we describe the structures of two clinically important β-lactamase enzymes bound to avibactam, the class A CTX-M-15 extended-spectrum β-lactamase and the class C Pseudomonas aeruginosa AmpC β-lactamase, which together provide insight into the binding modes for the respective enzyme classes. The structures reveal similar binding modes in both enzymes and thus provide a rationale for the broad-spectrum inhibitory activity of avibactam. Identification of the key residues surrounding the binding pocket allows for a better understanding of the potency of this scaffold. Finally, avibactam has recently been shown to be a reversible inhibitor, and the structures provide insights into the mechanism of avibactam recyclization. Analysis of the ultra-high-resolution CTX-M-15 structure suggests how the deacylation mechanism favors recyclization over hydrolysis.
INTRODUCTION
Antibiotic resistance currently represents a highly relevant global public health issue. The β-lactam antibiotics, discovered over 80 years ago, still represent the most widely used class of antibacterial agents due to their high level of activity, especially in serious Gram-negative infections, and their good tolerability profiles. A worrisome issue is represented by the widespread diffusion of KPC-producing Klebsiella pneumoniae isolates, which were responsible for many outbreaks worldwide, and infections caused by such strains are associated with significant mortality rate (1–3). Thus, resistance to β-lactam antibiotics is a clinical issue of considerable concern because of the global spread of multidrug-resistant strains (4–6).
Resistance to β-lactams is commonly mediated by the production of one or more β-lactamases, which inactivate the antibiotic by hydrolyzing the amide bond of the β-lactam ring (7). β-Lactamases have rapidly evolved in response to the introduction of new β-lactams and increasingly represent an important challenge to the efficacy of these agents, including the newest cephalosporins and carbapenems (8). The primary strategy for maintaining the potency of β-lactam drugs in the face of emerging resistance from β-lactamase enzymes has been to coadminister the β-lactam drug in combination with a β-lactamase enzyme inhibitor that will protect it from hydrolysis (9, 10). Currently, there are only three β-lactamase inhibitors approved for use in the clinic: clavulanic acid, sulbactam, and tazobactam (Fig. 1a to c) (11). All of these inhibitors are derived from β-lactam scaffolds. Besides their propensity to be hydrolyzed at low levels, these inhibitors cover only the class A β-lactamases and are clinically ineffective against class C and class D β-lactamase enzymes (9, 12). Therefore, there is an urgent need to develop β-lactamase inhibitors with a broader spectrum of inhibition that can restore the effectiveness of the β-lactam antibiotics (13, 14).
Fig 1.
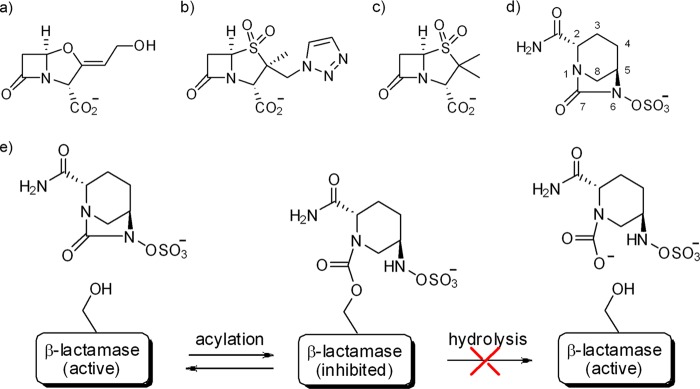
Chemical structures of β-lactamase inhibitors. (a) Clavulanic acid; (b) tazobactam; (c) sulbactam; (d) avibactam; (e) schematic representation of the proposed pathway for avibactam inhibition.
Avibactam, previously known as AVE1330A or NXL104, is a novel non-β-lactam inhibitor of β-lactamases that is currently in clinical development (Fig. 1d) (15–17). Avibactam is a representative of a bridged bicyclic ((2S,5R)-7-oxo-6-(sulfooxy)-1,6-diazabicyclo[3,2,1]octane-2-carboxamide) (DBO) scaffold that was rationally designed by incorporating the existing knowledge about β-lactams into the properties of a novel scaffold (18). Despite the fact that avibactam exploits the same rationale of previous β-lactamase inhibitor/β-lactam combinations (e.g., amoxicillin-clavulanate and piperacillin-tazobactam), by inhibiting the β-lactamase and thereby restoring the susceptibility of β-lactamase-producing strains, it has unique properties that account for its broad spectrum of activity and in vitro/in vivo efficacy (19–22). These properties include the low molecular weight of the molecule, its reactivity, its polarity, and its potent inhibition of a wide range of class A and class C β-lactamases, including extended-spectrum β-lactamases (ESBLs) (CTX-M-15), class A carbapenemases (KPC-2), and chromosomal and acquired AmpC-type class C enzymes with potencies in the nanomolar range (16, 23). In recent in vitro studies, avibactam paired with ceftazidime was effective against a wide range of β-lactamase-producing Gram-negative strains, and this combination was superior to clavulanate- or tazobactam-based combinations (21, 24).
Another remarkable feature of avibactam is its unusual covalent reversible mechanism of inhibition with β-lactamases (25) (Fig. 1e). This mechanism is very different from that of clinically used β-lactam-based β-lactamase inhibitors, with which the acyl-enzyme formation is practically irreversible and the acyl-enzyme intermediate can decompose through hydrolysis or further chemical rearrangements (26). The observed reversible ring closure of the strained and highly reactive avibactam ring system remains unexpected and intriguing. To understand the rationale for the potent broad-spectrum activity of avibactam across class A and class C enzymes, the structures of two clinically important β-lactamases were solved in complex with avibactam: (i) the CTX-M-15 ESBL, the most prevalent member of the CTX-M family, which is able to hydrolyze both cefotaxime and ceftazidime, and (ii) the Pseudomonas aeruginosa chromosomal AmpC, whose induction/deregulation can confer resistance to oxyimino-cephalosporins and which is not susceptible to currently available conventional β-lactamase inhibitors. In particular, the ultrahigh resolution of a native and an avibactam-bound CTX-M-15 structure allowed us to examine closely the interactions within the acyl-enzyme binding pocket and put forward a structure-based explanation of the avibactam mechanism of reversible inhibition.
MATERIALS AND METHODS
Protein production and purification.
The CTX-M-15 and P. aeruginosa AmpC β-lactamases were obtained from a culture of Escherichia coli BL21(DE3) carrying the plasmid vectors pET-CTX-M-15 and pET-AmpC, respectively, obtained by cloning the β-lactamase-encoding genes in the NdeI-BamHI restriction sites of plasmid vector pET-29a or pET-9a (Stratagene), as previously described (27). One liter of the expression strain containing CTX-M-15 [BL21(DE3)/pET29a-CTX-M-15 clone] was grown in the presence of 50 μg/ml kanamycin at 25°C for 24 h. The cell pellet was suspended in 400 ml of 0.5 M Tris-HCl (pH 8.0) with 0.5 mM MgCl2 and 1 mM EDTA, centrifuged for 30 min (14,000 × g), and resuspended in 400 ml ice-cold osmotic shock buffer (0.5 mM MgCl2, 1 mM EDTA, 10 mM Tris-HCl, pH 9.0) on ice for 30 min. This was then loaded at 3 ml/min on a 60-ml Q Sepharose column previously equilibrated with the osmotic shock buffer at 4°C. The flowthrough was concentrated using Amicon Ultra 15-10K down to 25 ml and injected at 2 ml/min (system pump; Akta), followed by buffer A (20 mM MES [morpholineethanesulfonic acid], pH 6). The protein were eluted using an NaCl gradient (buffer B) (20 mM MES [pH 6], 1 M NaCl) at 2 ml/min and then loaded onto a gel filtration column (Superdex 75 16/60). Elution was analyzed by SDS-PAGE, and the protein concentration (∼32 mg/ml) was assessed using the Bradford assay.
P. aeruginosa AmpC was purified from the culture supernatant of E. coli BL21(DE3)/pET-AmpC grown in the autoinducing medium ZYP-5052 (28) at 37°C for 48 h. The supernatant was clarified by centrifugation (10,000 × g, 10°C, 30 min), concentrated by ultrafiltration (YM10 membrane [Millipore, Billerica, MA] and using an Amicon 2000A high-performance ultrafiltration cell), and desalted using a HiPrep desalting 26/10 column (GE Healthcare, Uppsala, Sweden) with 10 mM HEPES buffer, pH 7.5 (buffer H). The sample was then loaded on an XK 16/20 column packed with 25 ml of CM Sepharose FF (GE Healthcare) previously equilibrated with buffer H. Bound proteins were eluted using a linear gradient (flow rate, 4 ml/min) of NaCl in buffer H (up to 1 M NaCl in 75 ml). Active fractions were pooled, concentrated by ultrafiltration using a Millipore Ultra-15 (Ultracel-10K; molecular mass, cutoff, 10 kDa), and loaded onto an XK 16/100 column packed with 125 ml of Superdex 75 preparative-grade gel. Proteins were eluted (flow rate, 1 ml/min) with buffer H supplemented with 0.15 M NaCl. Purified β-lactamases (final concentration, 8 to 10 mg/ml) were stored at −20°C.
Crystallization and structure determination.
Crystals of native CTX-M-15 β-lactamase and of its complex with avibactam were obtained by vapor diffusion in a hanging-drop setup. An 8-mg/ml solution of CTX-M-15 in 0.1 M Tris-HCl at pH 8.0 was incubated with a 3-mg/ml solution of avibactam (provided by Novexel SA, Romainville, France). Ammonium sulfate at 2.2 to 2.4 M was used as a precipitant. Crystallization of P. aeruginosa AmpC (10 mg/ml) was similarly obtained using 0.1 M Tris (pH 8.0), 20% polyethylene glycol (PEG) 8000, and 0.1 M K2HPO4 as the precipitant solution. Crystals were optimized by means of a microseeding procedure, as described previously (29). Crystals of AmpC complexed with avibactam were obtained from soaks of wild-type AmpC crystals grown by vapor diffusion in 2-μl hanging drops in 19% (wt/vol) PEG 3350, 10% (wt/vol) 2-propanol, and 0.1 M imidazole (pH 7.0) using the hanging-drop vapor diffusion method. AmpC crystals were harvested and soaked for 30 min in a 200 mM solution of avibactam in crystallization buffer.
Crystals were cryoprotected using 20% glycerol in the crystallization buffer and flash cooled in liquid nitrogen. X-ray diffraction data were collected on beamlines BM14U (CTX-M-15) or ID29 (native AmpC) of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) or, with avibactam-AmpC crystals, using an FRE+ X-ray source (Rigaku Corporation, Tokyo, Japan). Data processing was done with MOSFLM and scaled with Scala (CCP4 suite) (30). The autoBUSTER (31) software suite was used to index, integrate, and scale AmpC-avibactam data. The data collection parameters are reported in Table 1.
Table 1.
Data collection statistics
| Parameter | Valuea for: |
|||
|---|---|---|---|---|
| Native CTX-M-15 | CTX-M-15–avibactam complex | Native AmpC | AmpC-avibactam complex | |
| X-ray source | ESRF BM14U | ESRF BM14U | ESRF ID29 | FRE+ |
| Wavelength (Å) | 0.918 | 0.918 | 0.936 | 1.54178 |
| Data collection temp (K) | 100 | 100 | 100 | 100 |
| Space group | P212121 | P212121 | P3221 | P212121 |
| Cell dimensions (Å) | a = 44.698, b = 45.730, c = 117.086 | a = 44.483, b = 45.69, c = 117.721 | a = b = 83.06, c = 122.42 | a = 44.8814, b = 71.2045, c = 106.2468 |
| Subunits/asu | 1 | 1 | 1 | 1 |
| Matthews coefficient (Å3Da-1) | 2.08 | 2.08 | 3.05 | 2.44 |
| Solvent concn (%) | 41.01 | 41.01 | 59.67 | 49.66 |
| Resolution limits (Å) | 29.27–1.10 (1.16–1.10) | 23.54–1.10 (1.16–1.10) | 46.617–1.79 (1.89–1.79) | 106.29–1.86 (1.96–1.86) |
| Reflections measured | 767,395 (109,593) | 596,968 (85,566) | 79,535 (9,784) | 126,025 (11,566) |
| Unique reflections | 97,955 (14,022) | 98,124 (14,148) | 38,317 (5,537) | 28,813 (3,828) |
| Completeness (%) | 99.8 (99.2) | 99.9 (100.0) | 83.3 (82.7) | 97.9 (90.5) |
| Rmerge (%) | 7.5 (43.8) | 5.1 (14.5) | 6.2 (37.4) | 12.0 (62.7) |
| Multiplicity | 7.8 (7.8) | 6.1 (6.0) | 2.1 (1.8) | 4.9 (3.0) |
| I/σ(I) | 14.9 (4.3) | 20.3 (11.2) | 8.7 (1.8) | 22.8 (1.8) |
Data in parentheses are for the highest-resolution shell.
The CTX-M-15 native and cocomplex structures were obtained by molecular replacement using Protein Data Bank (PDB) code 1IYS as a model. Both structures were refined at 1.1 Å resolutions with REFMAC5 (CCP4 suite) using anisotropic temperature factors and hydrogen atoms in calculated positions. The AmpC structure was determined by molecular replacement using the P. aeruginosa AmpC wild-type structure (PDB code 2WZZ). The model was refined to 1.9 Å, and σ-weighted electron density maps were also calculated with ccp4i for both (30). Manual rebuilding and placement of water molecules were performed with the program Coot (32) and alternated with rounds of positional and B-factor refinement with REFMAC5. The final refinement statistics are reported in Tables 2 and 3. The structural alignments for comparison and the figures for protein structures were prepared using the program PyMOL (33) or CCP4mg. The model for the closed form of avibactam was made in Corina using smiles file and manually docked on the acylated CTX-M-15 using PyMOL.
Table 2.
Refinement statistics for native CTX-M-15 and avibactam complex
| Parameter | Valuea for: |
|
|---|---|---|
| Native CTX-M-15 | CTX-M-15–avibactam complex | |
| Resolution range (Å) | 29.27–1.10 (1.13–1.10) | 22.85–1.10 (1.13–1.10) |
| No. of reflections | 92994 (6381) | 93132 (6786) |
| Refinement type | Anisotropic + calc hydrogens | Anisotropic + calc hydrogens |
| Rcryst (%) | 11.69 (17.10) | 10.44 (7.60) |
| Rfree set size (%) | 5 | 5 |
| Rfree (%) | 13.64 (19.20) | 12.24 (10.60) |
| Protein atoms | 1,941 | 1,969 |
| Ligand atoms | - | 17 (avibactam) |
| Water molecules | 299 | 289 |
| Ethylene glycol | 4 | 6 |
| Chloride ions | 2 | 1 |
| Sulfate ions | 4 | 3 |
| Average B factor (Å2) | 6.14 | 6.024 |
| RMSD bond lengths (Å) | 0.005 | 0.023 |
| RMSD bond angles (°) | 1.128 | 2.092 |
| RMSD planes (Å) | 0.004 | 0.015 |
| RMSD chiral centers (Å3) | 0.063 | 0.126 |
| RMSD atomic positions (Å) | 0.015 | 0.011 |
Data in parentheses are for the highest-resolution shell.
Table 3.
Refinement statistics for native AmpC and avibactam complex
| Parameter | Value for: |
|
|---|---|---|
| Native AmpC | AmpC-avibactam complex | |
| Resolution limit (Å) | 1.79 | 1.9 |
| Resolution range (Å) | 34.51–1.79 | 34.27 1.92 |
| Completeness overall (%) | 82.07 | 84.07 |
| Reflections, unique | 36,340 | 23,950 |
| Roverall (%)a | 19.30 | 20.95 |
| Rfree (%)b | 24.38 | 24.19 |
| Nonhydrogen protein atoms | 2,843 | 2,776 |
| Nonhydrogen ligand atoms | 4 | 17 |
| Solvent molecules | 231 | 115 |
| RMSD from ideal values | ||
| Bond lengths (Å) | 0.023 | 0.009 |
| Bond angles (°) | 2.029 | 1.303 |
| Avg B values (Å2) | ||
| Protein main-chain atoms | 18.654 | 19.226 |
| All protein atoms | 20.414 | 19.98 |
| Ligand | 29.203 | 38.048 |
| Solvent | 27.807 | 21.015 |
| Φ, Ψ angle distribution for residues inc: | ||
| Most favored regions (%) | 96 | 92 |
| Additional allowed regions (%) | 4 | 8 |
| Generously allowed regions (%) | 0 | 0 |
| Disallowed regions (%) | 0 | 0 |
Roverall = Σhkl ||Fobs| − |Fcalc||/Σhkl |Fobs|.
Rfree is the cross-validation R factor computed for the test set of 5% of unique reflections.
Ramachandran statistics as defined by PROCHECK.
Protein structure accession numbers.
Coordinates and structure factors have been deposited in the Protein Data Bank under accession numbers 4HBT (native CTX-M-15), 4HBU (CTX-M-15–avibactam complex), 4GZB (native AmpC), and 4HEF (AmpC-avibactam complex).
RESULTS
Native and avibactam-bound structures of CTX-M-15 at ultrahigh resolution.
The structure of CTX-M-15 β-lactamase was obtained at a resolution of 1.1 Å, offering a detailed view of the enzyme active site in both the native form and the inhibitor-bound enzyme (statistics are listed in Tables 1 and 2) and thus allowing unambiguous determination of both the binding mode and any changes in the pocket upon binding.
This is the first view of the native CTX-M-15 structure, which maintains the αβα-sandwich fold typical of other class A β-lactamases and is identical (root mean square deviation [RMSD], 0.25 Å) to that of other members of the CTX-M type II class A β-lactamases (e.g., CTX-M-9). The high resolution of the electron density maps of both native and avibactam cocomplex CTX-M-15 show some interesting features of the structures. Avibactam was observed to form a covalent bond to Ser70-O (numberings for CTX-M-15 and AmpC residues are based on conventional β-lactamase numbering for the respective classes), with all its atoms clearly visible in the electron density, indicating that the inhibitor was bound tightly with little flexibility and no conformational variability (Fig. 2a). The positions of catalytically relevant residues are conserved, with very little conformational rearrangement upon avibactam binding (Fig. 2b), with an RMSD of only 0.15 Å upon least-squares superposition of all atoms between native and complexed structures. The six-membered ring is observed in a chair conformation, with the hydrogen atoms on C3, C5, and N6 and the axial one on C4 visible in the density (Fig. 2c and d). Avibactam binding occurred upon cleavage of the C7-N6 bond. The DBO scaffold, containing the urea moiety, offers two possible C-N bonds for cleavage, 7-1 or 7-6 (see Fig. S1a in the supplemental material). The observation of the crystal structure excludes the possibility of cleavage of the C7-N1 bond, similar to the case for the β-lactams (see Fig. S1b in the supplemental material), which was initially expected from early scaffold design (18). Three key interactions are observed between the inhibitor and the protein: the acyl-enzyme covalent bond, the highly polar sulfate recognition, and the carboxamide group. Besides the covalent carbamoyl-ester linkage, avibactam also forms a hydrogen bond to Ser70-O through the N6 proton that can be observed by the connectivity in the electron density when contoured to lower sigma. The C7 carbonyl of the newly formed carbamate is well positioned in the oxyanion hole in a conformation similar to that observed for the acylated β-lactam molecules, such as ceftazidime and tazobactam (34, 35), and forms hydrogen bonds with the amido-nitrogens of Ser70 (2.7 Å) and Ser237 (2.9 Å). The residues around the vicinity of the Ser70-O linkage are Lys73, Ser130, and a water molecule positioned by Glu166 and Asn170.
Fig 2.

Active site of CTX-M-15 in native avibactam and avibactam-enzyme complex. (a) Avibactam interactions in the CTX-M-15 pocket. Residues around the binding pocket of the avibactam–CTX-M-15 crystal structure are depicted as yellow sticks, and that from adjacent crystal contact (Arg42) is colored green. Avibactam is depicted in cyan. Polar interactions within 4 Å are shown in red dashed lines. Backbones from different secondary structural elements are colored differently. Deacylating water is depicted as a red sphere. (b) Overlay of the native CTX-M-15 active site (magenta) on CTX-M-15 covalently bound to avibactam (green). The residues within 4 Å around avibactam are shown in sticks. Avibactam is depicted in cyan, while the water molecules from each structure are shown as spheres with the respective colors. (c) Electron density map of CTX-M-15 in the native structure. The 2Fo-Fc map in blue is contoured at 1.5 sigma, and the fo-fc map in red is drawn around Glu166 for clarity. Residues are depicted as yellow sticks, while water is shown as red spheres. (d) Electron density map of the CTX-M-15 active site bound to avibactam. The 2Fo-Fc map (contoured at 1.5 sigma) is shown in blue, while the Fo-Fc map is shown in red, around Glu166. Residues and avibactam are depicted as green sticks, while water is shown as a red sphere.
While the inhibitor binding did not cause significant changes in the conformation of the active-site residues, the high resolution of the two structures allowed two subtle but functionally significant observations: (i) the side chain of Lys73 is oriented toward Ser130 rather than Glu166 upon ligand binding (Fig. 2b), and (ii) Glu166 is clearly protonated in the inhibitor-bound form, while it appears to be deprotonated in the native form (Fig. 2c and d). These two changes are associated with a significant modification of the hydrogen bond interactions between catalytically important residues and the position and orientation of the “deacylating” water molecule, as described in further detail below.
The sulfate group of avibactam is bound to the positively charged pocket that is involved in stabilizing the carboxylate moieties of β-lactams. The sulfate group makes polar contacts with Thr235, Lys234, Ser237, and Ser130 and via a water molecule with Tyr105 (not shown in Fig. 2). An intermolecular interaction with Arg42 of the adjacent molecule in the crystal contact (green sticks in Fig. 2a) is observed, which is a crystallographic artifact since the protein is a monomer in solution. The carbonyl oxygen of the carboxamide group forms hydrogen bonds with the side chains of Asn132 (2.8 Å) and Asn104 (3.0 Å). The amide group of the carboxamide also forms a hydrogen bond with Wat97 (3.0 Å), which in turn interacts with the Ser237 side chain. Thus, Ser237 interacts with both ends of the molecule, stabilizing the activating sulfate group as well as providing a hydrogen bond to the carboxamide, as observed from the alternate conformations in the crystal structure. In addition to these interactions, the piperidine ring forms a hydrophobic stack with the Tyr105 side chain which is conserved across all class A β-lactamase enzymes. The binding of avibactam displaces five water molecules from the active site, including one displaced due to the change in orientation of Lys73. However, the catalytic water molecule predicted to be involved in the deacylation of β-lactams (36) is retained in the pocket of the acyl-enzyme intermediate (Wat2, Fig. 2d).
Structure of avibactam bound to Pseudomonas aeruginosa AmpC.
The structure of avibactam was also solved in complex with P. aeruginosa AmpC to 1.8 Å, by soaking native AmpC crystals with the compound, and was compared with the native structure solved to 1.9 Å (statistics are listed in Table 3). The binding pockets of class C enzymes are more open than the class A pockets, reflecting their greater ability to accommodate the bulkier side chains of cephalosporins (37, 38). The electron density of avibactam bound to AmpC shows a covalent linkage to the catalytic Ser64 (residues are numbered based on conventional AmpC numbering, and a sequence alignment to P. aeruginosa AmpC is provided in Fig. S4 in the supplemental material), with very little movement between the bound and native structures (Fig. 3a to d). The largest change upon avibactam binding is the movement of the Tyr150 ring by 50 degrees planar rotation and movement of the hydroxyl group by 2 Å (Fig. 3b). The carbonyl oxygen of the acylated compound is located in the oxyanion hole, at a distance of 2.8 Å from the backbone amides of both Ser64 and Ser318. Interactions with the surrounding residues include amino acids that are conserved among all AmpC β-lactamase enzymes, such as Lys67, Tyr150, Gln120, Asn152, and Lys315. Substitutions at any of these residues have been previously reported to dramatically lower the enzymatic activity (39). The piperidine ring interacts with the hydrophobic side chains of Leu119, Leu320, and Ala293, compared with only Tyr105 in CTX-M-15. The two additional residues, Ala293 and Leu119, are unique to class C β-lactamases, as they are located on the extra domain characteristic of this class. The carboxamide group of avibactam forms hydrogen bonds with the Asn152 and Gln120 side chains, while the sulfate group is stabilized by the polar groups of Arg349, Lys315, Thr316, Asn346, and Ser318. The sulfate pocket observed here has previously been shown to be occupied by the carboxylate moiety of β-lactams such as ceftazidime in their acyl-enzyme complexes (see Fig. S2 in the supplemental material) (37, 40). There are fewer water molecules observed in close proximity to avibactam in AmpC than in CTX-M-15. The only water near the vicinity of Ser64-O interacts with the carboxamide group and the carbonyl group of Ser318. Multiple water-mediated interactions with the sulfate moiety are observed, however (Fig. 3a).
Fig 3.

Avibactam binding pocket on AmpC. (a) Interactions of avibactam in the pocket. Residues within 4 Å are shown as sticks, avibactam is colored magenta, polar interactions are shown as blue dashed lines, and water molecules are depicted as red spheres. (b) Overlay of AmpC bound to avibactam (orange) on the native AmpC structure (green). Avibactam is depicted in magenta sticks, while the water molecules observed close to the pocket are shown as spheres of respective colors. (c) Electron density map of the active site of the P. aeruginosa AmpC native structure. The 2Fo-Fc electron density map of the active-site residues contoured at 1.5 σ level is shown in pink mesh; the residues in the pocket are depicted as yellow sticks, while a cryoprotectant ethylene glycol molecule observed in the pocket has been colored green. (d) Electron density map of the active site of the P. aeruginosa AmpC structure bound to avibactam. The 2Fo-Fc electron density map of the active-site residues contoured at the 1.5 σ level is shown in pink mesh; the residues in the pocket are depicted as green sticks, while avibactam is shown as a cyan stick. Water molecules a, c, and d are depicted as red spheres.
DISCUSSION
Spectrum and potency.
The structures presented here, along with the previously published structure of avibactam bound to Mycobacterium tuberculosis BlaC (41), allow a view of the avibactam binding mode in both class A and C β-lactamases. Although the residues interacting with avibactam are not identical in AmpC and CTX-M-15, the overall conformations of avibactam in the two structures are very similar (see Fig. S3 in the supplemental material). The four residues that lie in similar tertiary space in the two classes are Ser64, Lys67, Lys315, and Tyr150 in AmpC, which overlay Ser70, Lys73, Lys234, and Ser130 in CTX-M-15, respectively. The observed binding mode in both CTX-M-15 and AmpC shows that each functional group on avibactam makes efficient interactions with key and conserved residues in the active-site pocket, supporting a potent binding conformation of the acyl-enzyme complex against these enzymes. In particular, the interaction of the activating groups, i.e., the sulfate moiety, is stronger than that of the carboxylates in β-lactam inhibitors due to their ability to form more polar contacts in the pocket. In both CTX-M-15 and AmpC, the sulfate group makes multiple hydrogen bonds to the polar residues such as Ser130, Thr235, and Arg276 in CTX-M-15 and Asn346, Thr316, and Lys315 in AmpC. The remaining elements of interaction, such as stabilization of the carboxamide group, and the placement of the carbonyl group in the oxyanion hole or the plane of the hexocyclic ring are conserved between CTX-M-15 and AmpC. Preservation of the avibactam binding mode and conservation of its inhibitory properties across the two classes can be attributed to the limited flexibility of the avibactam molecule and the lack of additional rearrangements. This mitigates the need for additional, potentially variable, catalytic residues in the binding pocket and is in contrast to the observations with clavulanic acid and tazobactam between class A and class C β-lactamases, where the large differences in activity have been attributed to the positions of certain residues and their accessibility to the inhibitor, such as the presence of Tyr150 versus Ser130 and the absence or presence of Glu166 in AmpC versus CTX-M-15.
Mechanism of recyclization.
The mechanism of recyclization can be divided into two parts, the carbamylation of the catalytic serine and reversible decarbamylation via compound recyclization (25). The ultrahigh resolution of the CTX-M-15 structure provides a glimpse of the protonation states and thus a clearer view of the mechanistic interplay in this enzyme between the two states. Acylation in all serine β-lactamases occurs by the nucleophilic attack of the conserved serine hydroxyl on the carbonyl group to form a high-energy tetrahedral intermediate that subsequently collapses to a carbamoyl enzyme intermediate in the case of avibactam. The position of the carbamate bond in avibactam suggests that the residues that are most likely to act as the conjugated general bases in class A enzymes are Glu166, Lys73, and Ser130 (Fig. 2a and b). A comparison of the native and the avibactam-bound CTX-M-15 structures shows a difference in hydrogen bonding patterns and protonation states of these residues (Fig. 4a and b). Glu166 in the native structure is deprotonated or anionic, while in the avibactam covalent complex it is protonated and neutral (Fig. 2c and d). The conformation of Lys73 appears to be coupled to the protonation state of Glu166: when Glu166 is charged, Lys73 interacts with both Glu166 and Ser70, and when Glu166 is protonated, Lys73 flips away from Glu166 and interacts with Ser130 (Fig. 2b). This is consistent with the previously described proton shuttle pathway from Lys73 to Ser130 (42) and may reflect a concomitant change in the protonation state of Lys73. Based on this observation, the deprotonated Glu166 is most likely the general base for acylation that activates the conserved water molecule to pull the proton from Ser70, while the general acid to donate a proton to avibactam N6 is likely to be either Lys73 or Ser130.
Fig 4.
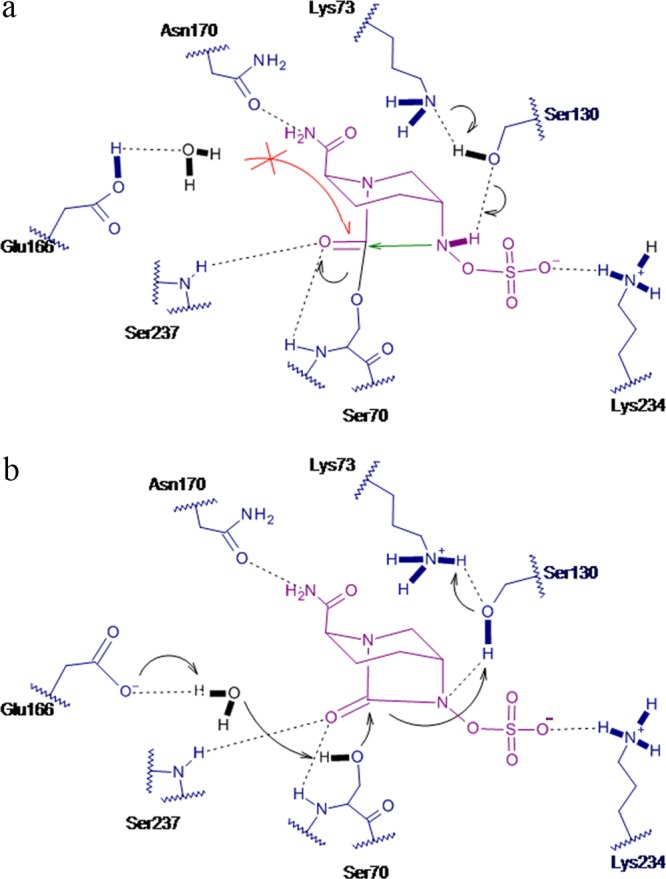
Proposed mechanism of avibactam inhibition in CTX-M-15 during acylation (a) and during deacylation (b).
Deacylation of avibactam occurs by recyclization in spite of the presence of a water molecule in the deacylating position. In case of CTX-M-15, the lack of hydrolysis can be explained by the protonation state of Glu166. While in the acylation stage, Glu166 is anionic and acts as a general base; in the deacylating stage, it is protonated, where the orientation of the interacting water is flipped, making it a weaker nucleophile. Disfavored hydrolysis is further helped by the greater inherent stability of the carbamoyl linkage of the avibactam-enzyme complex. A plausible explanation for the difference in protonation states of Glu166 with avibactam binding could be the resultant charge neutralization in the vicinity by the newly formed hydrogen bonds between the carboxamide groups of avibactam and the nearby polar residues, Asn132 and Asn104. In the case of AmpC, no water molecule in the vicinity of carbamoyl linkage that could potentially compete for deacylation via hydrolysis was observed.
Recyclization can be explained by the geometry of the open-ring conformation. In the active-site pocket, the open form of avibactam retains the substrate-like all-axial/pseudoaxial conformation upon cleavage (Fig. 5a). This is a result of multiple factors. First, just one atom distance between the sessile bond atoms and the nearest strong anchors of the ligand to the active-site pocket (i.e., C7 to covalent Ser70 linkage and N6 to the sulfate) restrains significant displacement and further repositioning upon cleavage. Furthermore, the cleaved product contains a centrally located rigid piperidine linker that makes it significantly less flexible than the carbam/cephem rings in β-lactams (see Fig. S2 in the supplemental material). The newly formed carbamate linker also makes the axial orientation of the C2 carboxamide and consequently the one of the C5 substituent more energetically favored (43). Finally, the electron density of avibactam suggests a hydrogen bond between N6 and Ser70-O, which further stabilizes the constrained conformation within the binding pocket. As a result of this conformation, the two ends of the cleaved bond, i.e., the C7 and N6 atoms, are displaced by only 1.4 Å to a final distance of 2.8 Å. This puts the N6 of avibactam closer to the carbamoyl group than the deacylating water (Fig. 5b). The angle between the carbamoyl plane and N6 is 113.4°, making it within the Burgi-Dunitz trajectory (44) for a nucleophilic attack. Similar intramolecular geometry was also observed in the crystal structures of 5,5-trans-lactone inhibitors, which show reversible acylation for porcine pancreatic elastase acyl-enzyme (45) as well as predicted reversible acylation by cyclic acyl phosphates (46). The disposition of residues around the N6 nucleophile in the two acylated structures suggests that Ser130 and Tyr150 are likely to participate as general bases for recyclization in class A and class C, respectively (Fig. 2a and Fig. 3a).
Fig 5.
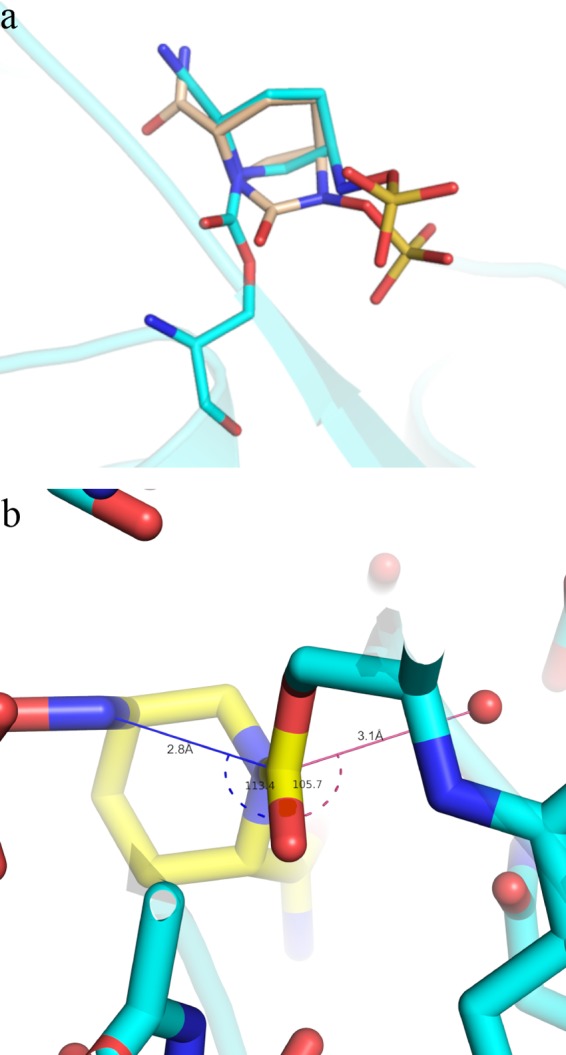
Substrate-like conformation of avibactam in the enzyme pocket. (a) Overlay of covalently modified avibactam on CTX-M-15 (cyan) on a closed avibactam model (light brown). The protein backbone is shown in cartoon form. (b) Bond distance and angle near recyclization event in CTX-M-15. Avibactam is depicted as a yellow stick, while Ser70 is in cyan. The distance between deacylating water (shown as red spheres) and carbonyl C7 is shown as a pink line, while that between attacking N6 and carbonyl C7 is a blue line. The angles are depicted in the same color with dashed lines.
Conclusion.
Overall, the results described here reveal that avibactam inhibits serine β-lactamases by forming a covalently bound enzyme complex that has similar characteristics with class A and class C β-lactamase enzymes, thus providing a structural basis for the broad-spectrum inhibition by avibactam. This is an important difference with respect to inhibitors such as clavulanic acid. Various positional disorders have been observed previously with other larger inhibitors, such as the phosphonates, where the differences in the active-site volume available for the specific ligand binding change the position of the attacking group and, in turn, the activity of these inhibitors against different enzyme classes. In the case of avibactam, its small size and efficient interacting groups allow it to bind to nearly all β-lactamases and make strong interactions with the key catalytic residues near the active sites. This should have a positive implication for susceptibility against preexisting resistance as well as in suppression of emergence of new resistance in the clinic. In addition, the structural data show that upon covalent bonding of avibactam to the catalytic serine, the avibactam ring opens but retains the conformation close to that of the unreacted form. This conformation explained the surprising reversible mechanism observed with such a strained ring inhibitor. While the stability of the avibactam–β-lactamase complex against water-mediated hydrolysis has allowed a longer half-life in the periplasmic space, the reversibility of the scaffold allows for good coverage of most β-lactamases in the milieu where these are expressed.
Supplementary Material
ACKNOWLEDGMENTS
We thank Richard Alm, Dave Ehmann, Thomas Keating, and Wright Nichols for careful reading and constructive suggestions. We also acknowledge the technical contribution of Jean-Michel Bruneau (formerly with Novexel S.A., Romainville, France). Finally, we thank Christine Miossec and Michael T. Black (formerly with Novexel S.A., Romainville, France), who initially commissioned part of the present study, for their constant interest in this work and for helpful discussion of the data.
Footnotes
Published ahead of print 25 February 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02247-12.
REFERENCES
- 1. Nordmann P, Naas T, Poirel L. 2011. Global spread of carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 17:1791–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Borer A, Saidel-Odes L, Riesenberg K, Eskira S, Peled N, Nativ R, Schlaeffer F, Sherf M. 2009. Attributable mortality rate for carbapenem-resistant Klebsiella pneumoniae bacteremia. Infect. Control Hosp. Epidemiol. 30:972–976 [DOI] [PubMed] [Google Scholar]
- 3. Snitkin ES, Zelazny AM, Thomas PJ, Stock F, NISC Comparative Sequencing Program, Henderson DK, Palmore TN, Segre JA. 2012. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci. Transl. Med. 4:148ra116 doi:10.1126/scitranslmed.3004129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kanoksil M, Jatapai A, Peacock SJ, Limmathurotsakul D. 2013. Epidemiology, microbiology and mortality associated with community-acquired bacteremia in northeast Thailand: a multicenter surveillance study. PLoS One 8:e54714 doi:10.1371/journal.pone.0054714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel G, Bonomo RA. 2011. Status report on carbapenemases: challenges and prospects. Expert Rev. Anti Infect. Ther. 9:555–570 [DOI] [PubMed] [Google Scholar]
- 6. Neuner EA, Yeh JY, Hall GS, Sekeres J, Endimiani A, Bonomo RA, Shrestha NK, Fraser TG, van Duin D. 2011. Treatment and outcomes in carbapenem-resistant Klebsiella pneumoniae bloodstream infections. Diagn. Microbiol. Infect. Dis. 69:357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bush K. 2010. The coming of age of antibiotics: discovery and therapeutic value. Ann. N. Y. Acad. Sci. 1213:1–4 [DOI] [PubMed] [Google Scholar]
- 8. Bush K, Fisher JF. 2011. Epidemiological expansion, structural studies, and clinical challenges of new beta-lactamases from gram-negative bacteria. Annu. Rev. Microbiol. 65:455–478 [DOI] [PubMed] [Google Scholar]
- 9. Bebrone C, Lassaux P, Vercheval L, Sohier J, Jehaes A, Sauvage E, Galleni M. 2010. Current challenges in antimicrobial chemotherapy: focus on beta-lactamase inhibition. Drugs 70:651–679 [DOI] [PubMed] [Google Scholar]
- 10. Bush K. 2010. Bench-to-bedside review: The role of beta-lactamases in antibiotic-resistant Gram-negative infections. Crit. Care 14:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Biondi S, Long S, Panunzio M, Qin WL. 2011. Current trends in beta-lactam based beta-lactamases inhibitors. Curr. Med. Chem. 18:4223–4236 [DOI] [PubMed] [Google Scholar]
- 12. Totir MA, Cha J, Ishiwata A, Wang B, Sheri A, Anderson VE, Buynak J, Mobashery S, Carey PR. 2008. Why clinically used tazobactam and sulbactam are poor inhibitors of OXA-10 beta-lactamase: Raman crystallographic evidence. Biochemistry 47:4094–4101 [DOI] [PubMed] [Google Scholar]
- 13. Bush K. 2012. Improving known classes of antibiotics: an optimistic approach for the future. Curr. Opin. Pharmacol. 12:527–534 [DOI] [PubMed] [Google Scholar]
- 14. Bush K, Macielag MJ. 2010. New beta-lactam antibiotics and beta-lactamase inhibitors. Expert Opin. Ther. Pat. 20:1277–1293 [DOI] [PubMed] [Google Scholar]
- 15. Bonnefoy A, Dupuis-Hamelin C, Steier V, Delachaume C, Seys C, Stachyra T, Fairley M, Guitton M, Lampilas M. 2004. In vitro activity of AVE1330A, an innovative broad-spectrum non-beta-lactam beta-lactamase inhibitor. J. Antimicrob. Chemother. 54:410–417 [DOI] [PubMed] [Google Scholar]
- 16. Stachyra T, Levasseur P, Pechereau MC, Girard AM, Claudon M, Miossec C, Black MT. 2009. In vitro activity of the β-lactamase inhibitor NXL104 against KPC-2 carbapenemase and Enterobacteriaceae expressing KPC carbapenemases. J. Antimicrob. Chemother. 64:326–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coleman K. 2011. Diazabicyclooctanes (DBOs): a potent new class of non-beta-lactam beta-lactamase inhibitors. Curr. Opin. Microbiol. 14:550–555 [DOI] [PubMed] [Google Scholar]
- 18. Aszodi J, Rowlands DA, Mauvais P, Collette P, Bonnefoy A, Lampilas M. 2004. Design and synthesis of bridged gamma-lactams as analogues of beta-lactam antibiotics. Bioorg. Med. Chem. Lett. 14:2489–2492 [DOI] [PubMed] [Google Scholar]
- 19. Wiskirchen DE, Crandon JL, Furtado GH, Williams G, Nicolau DP. 2011. In vivo efficacy of a human-simulated regimen of ceftaroline combined with NXL104 against extended-spectrum-beta-lactamase (ESBL)-producing and non-ESBL-producing Enterobacteriaceae. Antimicrob. Agents Chemother. 55:3220–3225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cattoir V. 2011. NXL-104, a novel beta-lactamase inhibitor with broad-spectrum activity. J. Anti-Infect. 13:20–24 [Google Scholar]
- 21. Aktas Z, Kayacan C, Oncul O. 2012. In vitro activity of avibactam (NXL104) in combination with beta-lactams against Gram-negative bacteria, including OXA-48 beta-lactamase-producing Klebsiella pneumoniae. Int. J. Antimicrob. Agents 39:86–89 [DOI] [PubMed] [Google Scholar]
- 22. Endimiani A, Hujer KM, Hujer AM, Pulse ME, Weiss WJ, Bonomo RA. 2011. Evaluation of ceftazidime and NXL104 in two murine models of infection due to KPC-producing Klebsiella pneumoniae. Antimicrob. Agents Chemother. 55:82–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stachyra T, Pechereau MC, Bruneau JM, Claudon M, Frere JM, Miossec C, Coleman K, Black MT. 2010. Mechanistic studies of the inactivation of TEM-1 and P99 by NXL104, a novel non-beta-lactam beta-lactamase inhibitor. Antimicrob. Agents Chemother. 54:5132–5138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Citron DM, Tyrrell KL, Merriam V, Goldstein EJ. 2011. In vitro activity of ceftazidime-NXL104 against 396 strains of beta-lactamase-producing anaerobes. Antimicrob. Agents Chemother. 55:3616–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ehmann DE, Jahic H, Ross PL, Gu RF, Hu J, Kern G, Walkup GK, Fisher SL. 2012. Avibactam is a covalent, reversible, non-beta-lactam beta-lactamase inhibitor. Proc. Natl. Acad. Sci. U. S. A. 109:11663–11668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Drawz SM, Bonomo RA. 2010. Three decades of beta-lactamase inhibitors. Clin. Microbiol. Rev. 23:160–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Docquier JD, Lamotte-Brasseur J, Galleni M, Amicosante G, Frere JM, Rossolini GM. 2003. On functional and structural heterogeneity of VIM-type metallo-beta-lactamases. J. Antimicrob. Chemother. 51:257–266 [DOI] [PubMed] [Google Scholar]
- 28. Studier FW. 2005. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 41:207–234 [DOI] [PubMed] [Google Scholar]
- 29. Benvenuti M, Mangani S. 2007. Crystallization of soluble proteins in vapor diffusion for X-ray crystallography. Nat. Protoc. 2:1633–1651 [DOI] [PubMed] [Google Scholar]
- 30. Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. 2011. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67:235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smart OS, Womack TO, Flensburg C, Keller P, Paciorek W, Sharff A, Vonrhein C, Bricogne G. 2012. Exploiting structure similarity in refinement: automated NCS and target-structure restraints in BUSTER. Acta Crystallogr. D Biol. Crystallogr. 68:368–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 33. DeLano WL. 2002. The PyMOL molecular graphics system. DeLano Scientific, San Carlos, CA [Google Scholar]
- 34. Frase H, Smith CA, Toth M, Champion MM, Mobashery S, Vakulenko SB. 2011. Identification of products of inhibition of GES-2 beta-lactamase by tazobactam by X-ray crystallography and spectrometry. J. Biol. Chem. 286:14396–14409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Delmas J, Chen Y, Prati F, Robin F, Shoichet BK, Bonnet R. 2008. Structure and dynamics of CTX-M enzymes reveal insights into substrate accommodation by extended-spectrum beta-lactamases. J. Mol. Biol. 375:192–201 [DOI] [PubMed] [Google Scholar]
- 36. Minasov G, Wang X, Shoichet BK. 2002. An ultrahigh resolution structure of TEM-1 beta-lactamase suggests a role for Glu166 as the general base in acylation. J. Am. Chem. Soc. 124:5333–5340 [DOI] [PubMed] [Google Scholar]
- 37. Beadle BM, Trehan I, Focia PJ, Shoichet BK. 2002. Structural milestones in the reaction pathway of an amide hydrolase: substrate, acyl, and product complexes of cephalothin with AmpC beta-lactamase. Structure 10:413–424 [DOI] [PubMed] [Google Scholar]
- 38. Meroueh SO, Minasov G, Lee W, Shoichet BK, Mobashery S. 2003. Structural aspects for evolution of beta-lactamases from penicillin-binding proteins. J. Am. Chem. Soc. 125:9612–9618 [DOI] [PubMed] [Google Scholar]
- 39. Jacoby GA. 2009. AmpC beta-lactamases. Clin. Microbiol. Rev. 22:161–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Beadle BM, Shoichet BK. 2002. Structural basis for imipenem inhibition of class C beta-lactamases. Antimicrob. Agents Chemother. 46:3978–3980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xu H, Hazra S, Blanchard JS. 2012. NXL104 irreversibly inhibits the beta-lactamase from Mycobacterium tuberculosis. Biochemistry 51:4551–4557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen Y, Shoichet B, Bonnet R. 2005. Structure, function, and inhibition along the reaction coordinate of CTX-M beta-lactamases. J. Am. Chem. Soc. 127:5423–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Johnson F. 1968. Allylic strain in six-membered rings. Chem. Rev. 68:375 [Google Scholar]
- 44. Bürgi HB, Dunitz JD, Shefter E. 1973. Geometrical reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc. 95:5065–5067 [Google Scholar]
- 45. Wilmouth RC, Kassamally S, Westwood NJ, Sheppard RJ, Claridge TD, Aplin RT, Wright PA, Pritchard GJ, Schofield CJ. 1999. Mechanistic insights into the inhibition of serine proteases by monocyclic lactams. Biochemistry 38:7989–7998 [DOI] [PubMed] [Google Scholar]
- 46. Kaur K, Lan MJ, Pratt RF. 2001. Mechanism of inhibition of the class C beta-lactamase of Enterobacter cloacae P99 by cyclic acyl phosph(on)ates: rescue by return. J. Am. Chem. Soc. 123:10436–10443 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.