

Abstract
Surfactants have long been known to have microbicidal action and have been extensively used as antiseptics and disinfectants for a variety of general hygiene and clinical purposes. Among surfactants, quaternary ammonium compounds (QAC) are known to be the most useful antiseptics and disinfectants. However, our previous toxicological studies showed that QAC are also the most toxic surfactants for mammalian cells. An understanding of the mechanisms that underlie QAC toxicity is a crucial first step in their rational use and in the design and development of more effective and safer molecules. We show that QAC-induced toxicity is mediated primarily through mitochondrial dysfunction in mammalian columnar epithelial cell cultures in vitro. Toxic effects begin at sublethal concentrations and are characterized by mitochondrial fragmentation accompanied by decreased cellular energy charge. At very low concentrations, several QAC act on mitochondrial bioenergetics through a common mechanism of action, primarily by inhibiting mitochondrial respiration initiated at complex I and, to a lesser extent, by slowing down coupled ADP phosphorylation. The result is a reduction of cellular energy charge which, when reduced below 50% of its original value, induces apoptosis. The lethal effects are shown to be primarily a result of this process. At higher doses (closer to the critical micellar concentration), QAC induce the complete breakdown of cellular energy charge and necrotic cell death.
INTRODUCTION
The microbicidal activity of surfactants has generally been attributed to their ability to destroy cell membrane structure (1). This destructive action results from preferred partitioning of surfactants from the aqueous phase into cell membranes where, at low concentrations, they may affect some subtle physical properties (lipid bilayer curvature, lateral pressure, surface charge, etc.) which then affect membrane protein function, without a gross destruction of the membrane. At higher concentrations, closer to the surfactant critical micellar concentration (CMC), an equilibrium is established between the cell membrane components associated with the lipid bilayer phase and a coexisting micellar pseudophase in the aqueous medium that results in a dissolution of several components of the lipid bilayer into micelles, destruction of cell membrane integrity, and cell lysis (2, 3).
Our interest in the potential use of surfactants as microbicides in sexually transmitted infections (STI) led us to compare surfactant toxicities to in vitro cultures of mammalian columnar epithelial cells with their ability to kill some bacterial and yeast STI pathogens (4). Interestingly, among all of the surfactant families (nonionic, zwitterionic, anionic, and cationic) examined, cationic surfactants were the only group in which cytotoxicity occurred at concentrations that were too low to provoke cell lysis. Bacterial growth in cultures was also shown to be inhibited at concentrations that were sublethal to epithelial cells. We then systematically evaluated the cytotoxicity of the bromide salts of several monoalkyl quaternary ammonium surfactant compounds (QAC) [alkyl-N,N,N-trimethylammonium (CnTAB), N-alkylpyridinium (CnPB), and alkyl-N-benzyl-N,N-dimethylammonium (CnBZK)] toward several mammalian cell lines (5). That study, performed as a function of exposure time and concentration, compared several CnTAB homologues (with Cn varying between C10 and C16), as well as three different headgroups with the same alkyl chain length, C12. The onset of cytotoxicity was shown to involve neither membrane disintegration, nor perforation, nor cell lysis, suggesting that the susceptible target had to be intracellular. The high values (6 < n < 9) of the Hill coefficients, n, in the logistic fits of our experimental toxicity data (5) suggested that QAC toxicity involved cooperative breakdown of several biochemical processes. The primary site(s) of QAC toxicity would therefore have to be of vital importance to the physiology of the cell. We concentrated our attention upon effects that could hinder mitochondrial energy production (mitochondrial respiration and oxidative phosphorylation) (6) and/or affect DNA structure and thereby compromise vital cellular processes. In agreement with existing literature (7), we found that the equilibrium constants for QAC association with DNA are substantially higher than the lethal QAC concentrations (A. S. Inácio and T. Q. Faria, unpublished data). This line of investigation was therefore not pursued further. Studies on inhibition of mitochondrial respiration and oxidative phosphorylation in isolated mitochondria, however, showed that both processes were inhibited at QAC concentrations that were compatible with the observed cytotoxic concentrations previously measured. We therefore undertook a detailed study of QAC-induced mitochondrial dysfunction and its subsequent consequences for mammalian columnar epithelial cell cultures.
In the present report we show that QAC cytotoxicity to MDCK II cells in vitro is initiated by mitochondrial dysfunction at sublethal concentrations, followed by mitochondrial fragmentation and decreased cellular energy charge at slightly higher concentrations. In isolated mitochondria all of the QAC tested were shown to act via a common mechanism involving inhibition of NADH-ubiquinone oxidoreductase (complex I) and of mitochondrial ADP-phosphorylation. QAC-induced mitochondrial dysfunction results in apoptosis (concentrations ≤ LD90), followed by a shift to necrotic cell death at concentrations above LD90. To our knowledge, this is the first detailed quantitative study concerning the mechanism of QAC toxicity and its dependence on concentration and exposure time.
MATERIALS AND METHODS
Reagents.
Surfactants of the highest commercially available purity were purchased from Sigma-Aldrich (St. Louis, MO) and were used as received. The ApoLive-Glo multiplex assay was from Promega (Madison, WI). The Cytotoxicity Detection KitPLUS and the ATP Bioluminescence Assay Kit CLS II were from Roche Applied Science (Mannheim, Germany). The ADP/ATP ratio assay kit was obtained from Abcam (Cambridge, United Kingdom), and a Pierce BCA protein assay kit was obtained from Thermo Scientific (Waltham, MA). Propidium iodide (PI), rhodamine 123 (Rh123), Hoechst 33342, and hydroethidine (HE) were from Molecular Probes/Invitrogen Corp. (Paisley, Scotland, United Kingdom). Annexin V was from BD Biosciences Pharmingen (San Diego, CA). All other chemicals used were from Sigma-Aldrich.
Cell culture viability and caspase 3/7 activity assay.
MDCK II cells were grown as previously described (5). After achieving complete polarization, the cells were exposed to different concentrations of C10TAB for 3 h. At the end of incubation, Opti-MEM medium containing surfactant was replaced by fresh cell culture medium without phenol red, and the viability and caspase 3/7 activity were assayed using an ApoLive-Glo multiplex assay, according to the manufacturer's instructions. As a positive control for apoptosis, cells were incubated with 1 μM staurosporine for 6 h (8).
Detection of intracellular ROS.
In order to monitor reactive oxygen species (ROS) formation, cells were labeled with HE, a redox-sensitive fluorescent probe that in its reduced form is able to diffuse across the cell membrane. The intracellular oxidation of HE, mainly by superoxide anion, results in the formation of a red fluorescent product that stains the cells and exhibits an increase in fluorescence in the presence of DNA (9). Briefly, cells were incubated for 1 h at 37°C with 10 μM HE. After this, the cell culture medium was removed and replaced with Opti-MEM medium without phenol red containing different concentrations of C10TAB. The rate of intracellular ROS formation was measured by continuously monitoring the changes in the fluorescence signal (518-nm excitation, 605-nm emission) for 3 h at 37°C in a Gemini EM microplate spectrofluorimeter (Molecular Devices). The maximal initial rate of ROS production was calculated for each condition.
LDH cytotoxicity assay.
The cytotoxic effect of C10TAB was evaluated by a lactate dehydrogenase (LDH) leakage assay after 3 h of exposure. At the end of incubation, the activity of the LDH released into the culture medium was measured by using a Cytotoxicity Detection KitPLUS, according to the standard protocol provided by the supplier.
Evaluation of mitochondrial morphology and membrane potential (Δψm) by laser scanning confocal microscopy.
Analysis of mitochondrial morphology and mitochondrial membrane potential was performed by using the mitochondrial membrane potential-sensitive probe Rh123, as described elsewhere (10). At the end of the treatment, cells were labeled with 10 μg of Rh123/ml and 2 μg of Hoechst 33342/ml in Dulbecco modified Eagle medium, without phenol red, buffered with 20 mM HEPES for 30 min at 37°C. Then, 10 μM FCCP was added to Rh123-labeled mock-treated cells in order to completely dissipate the Δψm. Mitochondrial morphometric analysis was carried out in confocal optical sections. Images were acquired using a confocal microscope with a Plan-Apochromat ×63 oil immersion objective (NA = 1.40) using the Carl Zeiss laser scanning system LSM 510 software. The experimental approach used follows the basic outlines described elsewhere (11). Surface rendering was performed in 8- to 15-μm z-stack images composed of 0.43-μm optical slices using Imaris (Bitplane AG, Switzerland) and carried out at the Cell Imaging Unit of Gulbenkian Science Institute (Oeiras, Portugal). From this volumetric reconstruction, the number and volume of distinct mitochondrial particles were calculated.
Evaluation of mitochondrial respiration.
Liver mitochondria were isolated from 8-week-old male Wistar rats according to a well-established protocol (12). Mitochondrial oxygen consumption was measured polarographically with a Clark-type oxygen electrode as previously described (13). The experiments were performed with 1 mg of protein. Different concentrations of the tested cationic surfactants were added to the liver mitochondrial isolates and incubated for 5 min prior to mitochondrial energization with 5 mM glutamate plus 2.5 mM malate or with 5 mM succinate in the presence of 2 μM rotenone. State 3 respiration was elicited by the addition of 125 nmol of ADP/ml. For further details, see Fig. S1 in the supplemental material.
Evaluation of apoptotic and necrotic cell death by annexin V and PI staining.
After 1 or 3 h of incubation with C10TAB, surfactant-containing medium was removed and replaced by fresh medium with 5 μg of PI/ml. The cells were incubated at 37°C and 5% CO2 for 5 min, after which they were washed in phosphate-buffered saline (PBS), followed by 15 min of incubation at room temperature with 2 μg Hoechst 33342/ml and annexin V diluted 1:10 in annexin V binding buffer. Lastly, the cells were washed and kept in annexin V binding buffer for immediate visualization in a confocal microscope.
Quantification of intracellular ATP and ADP levels.
Intracellular ATP levels were determined by the luciferin-luciferase bioluminescent assay. After exposure to QAC, the cells were washed with cold PBS and disrupted with ice-cold lysis buffer (50 mM Tris-HCl, 4 mM EDTA, pH 7.5), for intracellular ATP extraction. An aliquot of the sample was taken out for protein quantification by the bicinchoninic acid method (Pierce BCA protein assay kit). The remaining sample was further treated as described elsewhere (14). The supernatant was then collected, and the ATP content was determined using a CLS II ATP bioluminescence assay kit according to the standard protocol provided by the supplier. The results were normalized to the protein content and were expressed as the percentage of mock-treated control cells. The ATP/ADP ratio of the samples was determined using an ADP/ATP ratio assay kit.
Statistical analysis.
Results are expressed as means ± the standard deviations (SD) unless otherwise stated. The cell dose-response curves were fitted using a four-parameter logistic equation through computer-assisted curve fitting (SigmaPlot 11.0) (5). Statistical analysis was carried out in GraphPad Prism software version 5.0. Comparison between groups was performed using a one-way analysis of variance parametric test. Dunnett's post hoc test was used for multiple comparisons, and statistical significance was assumed when P was <0.05.
RESULTS
Cell viability is reduced prior to QAC-induced membrane damage.
In a detailed study on the toxicity of several QAC toward MDCK II cells (5), we reported that in a homologous series of CnTABs (C10 through C16) irreversible membrane damage resulted from high surfactant concentrations, while reduced cell viability occurred at lower concentrations. Even though this effect describes the toxicity of all CnTAB, it was most prominent for C10TAB. This work, therefore, focused on C10TAB as a representative of the QAC family of surfactants. Figure 1 compares cell viability measured by using a fluorogenic commercial kit (with essentially the same results [5] as the MTT assay), and cell membrane permeabilization was measured by the LDH leakage assay in MDCK cells exposed to C10TAB at different concentrations for a period of 3 h.
Fig 1.
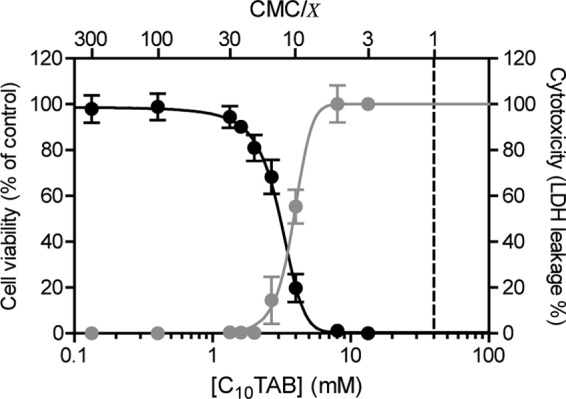
Concentration dependence of the effects of C10TAB on MDCK cell viability (black) and LDH leakage (gray) after 3 h of exposure. The data are presented as means ± the SD of four independent experiments and expressed as a percentage of control (cell viability) or as a percentage of LDH activity from lysed mock-treated control cells (LDH leakage). Surfactant concentrations in the x axis are represented on a logarithmic scale. The CMC of C10TAB is 4.0 × 10−2 M.
Surfactant toxicity is generally understood to be related to their amphiphilicity, reflected in their critical micellar concentration (CMC), which is a measure of the free energy for surfactant partitioning between the aqueous phase and organized apolar phases such as micelles and membranes. Therefore, as previously discussed by us (5), in the present study QAC effects were compared by normalizing their concentrations with respect to their CMCs. For the convenience of the reader the CMC values of the QAC used in the present study are listed in Table 1. MDCK cell viability began to decrease at C10TAB concentrations of 1.3 mM (∼CMC/30) with LD50 at 3.1 mM (∼CMC/13; 95% confidence interval [CI], 2.9 to 3.3 mM). Membrane damage, however, was only detected at 2.7 mM (∼CMC/15) with a 50% lethal dose (LD50) at 3.8 mM (∼CMC/10.5; 95% CI, 3.7 to 4.0 mM). The cells therefore lost viability before their membranes became leaky, and it is possible that the membrane leakage was not a direct action of QAC on the membrane but rather a consequence of prior cell death.
Table 1.
QAC critical micelle concentration
| QAC | CMC (M)a |
|---|---|
| C10TAB | (4.0 ± 0.1) × 10−2 |
| C12TAB | (3.5 ± 0.3) × 10−3 |
| C14TAB | (2.9 ± 0.1) × 10−4 |
| C16TAB | (2.6 ± 0.2) × 10−5 |
| C12PB | (3.9 ± 0.1) × 10−3 |
| C12BZK | (1.7 ± 0.9) × 10−3 |
CMC, critical micelle concentration. Data are from reference 5.
Sublethal concentrations of C10TAB induce changes in mitochondrial network morphology and membrane potential (Δψm).
Mitochondrial morphology (i.e., number, area, and volume of distinct, physically continuous mitochondrial entities) and transmembrane potential, Δψm, were evaluated by confocal microscopic examination of MDCK cells exposed to C10TAB using rhodamine 123 (Rh123). Rh123 easily crosses the cell plasma membrane and distributes electrophoretically into the mitochondrial matrix in response to Δψm (15, 16). It therefore permits visualization of mitochondrial morphology and simultaneously provides a measure of Δψm only if the mitochondrial membrane is intact. Figure 2A to E shows a progressive loss of Rh123 staining of mitochondria, indicating the loss of mitochondrial Δψm and/or of intact mitochondria. In fact, at a C10TAB concentration of 2.7 mM (∼CMC/15) most cells showed a diffuse, low-level Rh123 fluorescence similar to that observed when the mitochondrial Δψm was abolished by addition of FCCP (data not shown). Figure 2A to J show that the mitochondrial morphology changed from a reticulotubular phenotype in untreated cells to a phenotype characterized by numerous, small, rounded mitochondria after C10TAB exposure. These morphological alterations occurred gradually, and their severity was dependent upon surfactant concentration and exposure time. Structural changes were visible after short exposures to very low doses of C10TAB (e.g., exposure to 0.13 mM [∼CMC/300] for 1 h), at which no changes in the intracellular ATP levels and ATP/ADP ratios were detected (Table 2). The exposure to C10TAB concentrations up to 0.4 mM (∼CMC/100) resulted in an apparent increase of the total number of continuous mitochondrial structures, evident for both exposure times. In contrast, concentrations higher than 1.3 mM (∼CMC/30), after 3 h of incubation, caused a progressive decrease in the total number of mitochondria (Fig. 2K). Figure 2L shows that at both exposure times there was also a monotonic C10TAB concentration-dependent decrease in the average mitochondrial volume. Moreover, C10TAB was shown to induce the generation of intracellular ROS, particularly superoxide anion. As can be observed in Fig. 2M, the maximal rate of this oxygen radical production, compared to control (evaluated by the oxidation of the redox-sensitive fluorescent probe HE), was concentration dependent. An increase in the rate of intracellular ROS generation is already detectable at concentrations as low as 0.4 mM (∼CMC/100).
Fig 2.

Morphological changes in the mitochondrial network of MDCK cells after 3 h of exposure to C10TAB. Representative maximal fluorescence intensity projection images obtained by live-cell confocal microscopy, showing the mitochondrial network of MDCK stained with Rh123 (green). Nuclei were visualized with Hoechst 33342 (blue). Zoomed regions of the selected regions in these images are shown in the lower panel. (A) Control cells display reticulotubular mitochondrial morphology, mainly composed of large branched tubules (arrows). Exposure to C10TAB at 0.13 mM (∼CMC/300) (B), 0.4 mM (∼CMC/100) (C), 2.0 mM (∼CMC/20) (D), and 2.7 mM (∼CMC/15) (E) gradually disintegrates the reticulotubular mitochondrial network into a punctiform phenotype, comprising numerous round fragments of various sizes (arrowheads). (E) The loss of Rh123 fluorescence from the mitochondrial regions reflects the dissipation of the Δψm. Scale bars correspond to 10 and 5 μm in the upper and lower (zoomed) images, respectively. (F to J) Mitochondrial distribution and subtyping according to volume in individual representative cells exposed to the same concentrations of C10TAB. Hotter colors indicate larger contiguous mitochondrial volumes. The mitochondrion number (K) and total volume (L) per cell was determined. The data are shown as means ± the SD of 8 to 15 cells per condition of at least three independent experiments. The initial rate of intracellular ROS production measured in cells incubated with increasing concentrations of C10TAB was evaluated using the redox-sensitive fluorescent probe HE. The data are presented as means ± the SD of five independent experiments.
Table 2.
Effects of C10TAB on intracellular ATP levels and ATP/ADP ratios in MDCK cells after 3 h of exposure to C10TAB
| Treatment | Concn (mM) | Mean ± SDa |
|
|---|---|---|---|
| ATP (%)b | ATP/ADP ratio | ||
| Control | 0 | 100.0 ± 10.3 | 2.9 ± 0.5 |
| C10TAB | |||
| CMC/300 | 0.13 | 101.7 ± 11.2 | 2.4 ± 0.3 |
| CMC/100 | 0.40 | 98.0 ± 7.1 | 2.2 ± 0.4 |
| CMC/30 | 1.33 | 84.4 ± 16.1 | 1.4 ± 0.3** |
| CMC/20 | 2.00 | 77.5 ± 13.6* | 1.3 ± 0.4*** |
| CMC/10 | 4.00 | 53.4 ± 12.8*** | 1.1 ± 0.4*** |
| CMC/3 | 13.33 | 0.8 ± 0.5*** | 0.2 ± 0.3*** |
| Staurosporin | 0.001 | 65.4 ± 10.9** | ND |
Data are means of at least three independent experiments. ND, not determined. *, P < 0.05; **, P < 0.01; ***, P < 0.001 (significant differences from the respective control).
Results were normalized to protein content and are expressed as the percentage of mock-treated control cells (24.07 ± 2.48 nmol of ATP/mg of protein).
The observed effects of C10TAB upon the mitochondrial system can thus be summarized as (i) disruption of the reticulotubular phenotype of normal mitochondria to fragmented mitochondria at concentrations up to 0.4 mM (∼CMC/100), (ii) a progressive decrease in mitochondrial Δψm with increasing surfactant concentration, (iii) a progressive C10TAB concentration-dependent decrease in the average volume of the mitochondria at all concentrations examined, and (iv) a linear increase in ROS production rate with increasing C10TAB concentration.
QAC impair respiratory activity in isolated mitochondria.
In order to clarify the mechanisms involved in the toxicity of QAC to mitochondria, basic bioenergetic parameters were evaluated in isolated mitochondria exposed to increasing concentrations of these surfactants (13). This was done using C10TAB and three QAC with the same alkyl chain (C12) but different head groups (TAB, PB, and BZK) to evaluate the general applicability to the QAC group studied.
Mitochondrial production of ATP requires noncompromised mitochondrial respiratory activity, the ability to phosphorylate ADP, and an intact inner mitochondrial membrane that couples the two processes through the formation of a pH gradient across the membrane. Mitochondrial respiration can be evaluated independently of ADP phosphorylation (uncoupled respiration) by making the mitochondrial inner membrane permeable to protons using the protonophore FCCP (see Fig. S1 in the supplemental material). Figure 3A shows the effect of C10TAB upon FCCP-uncoupled respiration rate when mitochondria were energized with glutamate/malate (which generates the complex I substrate, NADH, in the mitochondrial lumen) or with succinate (a complex II substrate, added together with rotenone, a specific inhibitor of complex I). It is evident that while complex I-dependent mitochondrial respiration is strongly inhibited by C10TAB, the complex II-dependent process is not. This argues that C10TAB inhibits complex I and not the respiratory process as a whole. Since complex I activity in intact mitochondria is evaluated by addition of glutamate/malate whose oxidation inside mitochondria produces the complex I substrate, NADH, the question remains open whether the apparent inhibition is a direct effect on complex I or whether it results from an inhibition of glutamate/malate transport or metabolism inside the mitochondria. To resolve this question, we examined oxygen consumption in disrupted mitochondria energized with NADH, the complex I substrate (see Fig. S2 in the supplemental material). The inhibitory effect of QAC was lower in disrupted than in intact mitochondria, suggesting that while part of the inhibitory effect of QAC is directly at the level of complex I, another part is at the level of mitochondrial glutamate/malate transport and/or metabolism. Nevertheless, complex I-dependent respiration is strongly inhibited by C10TAB, whereas complex II-dependent respiration is not, whether in intact or in disrupted mitochondria.
Fig 3.

Effects of QAC on the respiratory activity in isolated mitochondria. The effects of C10TAB (A to C), C12TAB (D to F), C12PB (G to I), and C12BZK (J to L) on FCCP-uncoupled respiration rate (left panels), respiratory control ratio (RCR) (middle panels) and on the ADP/O ratio (right panels) were determined in isolated rat liver mitochondria after 5 min of exposure. The data are presented as means ± the SD of at least three independent experiments. Asterisks (*, P < 0.05; **, P < 0.01; ***, P < 0.001) signify differences from the respective control. The CMC of each QAC is as follows: C10TAB, 4.0 × 10−2 M; C12TAB, 3.5 × 10−3 M; C12PB, 3.9 × 10−3 M; and C12BZK, 1.7 × 10−3 M.
When the rate of ADP-stimulated oxygen consumption in the respiratory chain coupled to ADP-phosphorylation (state 3) is examined, both complex I-initiated and complex II-initiated respiration are seen to be affected (data not shown), the latter being much less affected than the former due to the demonstrated inhibition of complex I by C10TAB. This effect is almost identical to the effect seen upon the respiratory control ratio (RCR; Fig. 3B). On the other hand, the ADP/O ratio (the total amount of oxygen consumed for a given quantity of ADP supplied to the mitochondria) decreases when electrons are supplied at the level of complex I due to specific inhibition of this complex by C10TAB but is unchanged when electrons are supplied at the level of complex II (Fig. 3C). This implies that C10TAB slows down the global process post-complex II without quantitatively affecting its end result. We conclude that QAC have a specific inhibitory effect upon complex I and no effect between complex II and complex IV (see Fig. 3A) but slow down coupled ADP phosphorylation in terms of its rate but not its total outcome. This could be a result of QAC-induced slowing down of ATP-synthetase activity directly at the level of the F1F0-ATPase/ATP synthase or at the level of ADP/ATP and/or phosphate transport across the inner mitochondrial membrane.
Qualitatively, the results obtained with C10TAB and C12TAB with regard to mitochondrial dysfunction were similar, although the CMC-normalized concentrations of C12TAB required to produce the same decreases in the FCCP-uncoupled respiration, RCR and ADP/O ratio were slightly higher (Fig. 3D to F). A further increase in hydrophobic chain length, to 14 or 16 carbons, produced similar effects as those observed for C12TAB, but the CMC-normalized concentrations required to produce the same results were at least 10 times higher (data not shown). The simplest explanation for these quantitative differences is that the concentration of QAC in the membrane at the same CMC-normalized total concentration is lower for amphiphiles with longer hydrophobic chains. The specific effect of the polar group of the QAC analogues was also examined by comparing the homologues C12TAB (Fig. 3D to F), C12PB (Fig. 3G to I), and C12BZK (Fig. 3J to L). The CMC-normalized toxicity ranking was C12PB > C12BZK > C12TAB and is related to their differential partitioning into membranes.
Low doses of C10TAB induce caspase-mediated apoptosis in MDCK cells.
The ability of QAC to promote apoptosis (17, 18) suggested that at least part of the toxic effects described above could result from cell death through apoptosis. The C10TAB-induced activation of the executioner caspases 3 and 7, a relatively early event in the apoptotic process (19), was measured. An increase in caspase 3/7 activity (123.0% ± 18.6% at 1.6 mM; ∼CMC/25) occurred concomitantly with the initiation of C10TAB-induced cell viability loss (Fig. 4A). At higher C10TAB concentrations, 2 mM (∼CMC/20) and 4 mM (∼CMC/10), caspase 3/7 activity increased proportionally to the reduction of cell viability, reaching a maximum of activity close to a C10TAB concentration of 4 mM. The surfactant dose that raised caspase 3/7 activity by 50% compared to control conditions (EC50) was 3.0 mM (∼CMC/13; 95% CI, 2.3 to 3.7), which is the LD50 for C10TAB. At concentrations >6.7 mM (∼CMC/6), the cytotoxicity was ≥90%, but no caspase 3/7 activation was detected. Thus, the decrease in MDCK cell viability, upon exposure to low concentrations of C10TAB, resulted from C10TAB-induced apoptosis, whereas higher concentrations led to necrotic cell death. To further explore this duality of QAC toxicity, treated cells were stained with annexin V (to detect accessible phosphatidylserine [PS] in the plasma membrane) and propidium iodide (PI), which only enters cells with permeabilized membranes. The number of annexin V-positive cells increased proportionally to the surfactant dose and was accompanied by the appearance of pyknotic nuclei (Fig. 4B to D) (20). Moreover, the annexin V staining preceded the emergence of PI-positive cells, which were only visible at higher surfactant concentrations, confirming that cell death through necrosis was restricted to C10TAB concentrations close to CMC. A similar toxicity pattern was also observed for the other QAC tested (see Fig. S3 in the supplemental material). Since the energy state of the cell plays a central role in the mode of cell death (21), the effect of C10TAB on the intracellular ATP levels and, as a measure of the cell energy charge, the ATP/ADP ratio were also determined (22). The results showed that exposure to 1.3 mM (∼CMC/30) C10TAB for 3 h reduced the intracellular ATP concentration to 84.4% ± 16.1% of control and caused a reduction of 50% in the ATP/ADP ratio (2.9 ± 0.5 and 1.4 ± 0.3 for control and [C10TAB] = 1.3 mM [∼CMC/30], respectively; P < 0.01). At this concentration only a slight loss of cell viability (<10%) and a small increase in caspase 3/7 activity (ca. 6%) was detected, suggesting that the cell energy status was affected prior to a commitment to cell death. Furthermore, at concentrations between 2 mM (∼CMC/20) and 4 mM (∼CMC/10) the partial ATP depletion, ranging from 77.5 to 53.4% of control, was accompanied by a simultaneous increase in caspase 3/7 activity. A gradual and significant decrease in the ATP/ADP ratio, from 2.9 ± 0.5 in mock-treated cells to a minimum value of 1.1 ± 0.4 at 4 mM C10TAB (∼CMC/10) was also evident. As a positive control for apoptosis, cells incubated with 1 μM staurosporine for 6 h (23) exhibit a decrease of intracellular ATP (65.4 ± 10.9% of control), reduced cell viability (to 88.9% ± 6.4% of control), and increased caspase 3/7 activity (to 225.4% ± 12.3% of control). This highlights the similarity between the molecular changes elicited by low concentrations of C10TAB and the typical features of apoptosis. In contrast, higher surfactant concentrations induced a distinct pattern of biochemical changes in MDCK cells: exposure to 13.3 mM (∼CMC/3) C10TAB severely depleted ATP (0.8% ± 0.5% of control; P < 0.001) without detectable caspase 3/7 activation. The ATP/ADP ratio also plummeted to 0.2 ± 0.3, reflecting the dissipation of the energy reservoirs that occurs during necrosis.
Fig 4.

C10TAB-induced apoptosis in MDCK cell. Concentration-dependent effects of C10TAB on caspase activation after 3 h of exposure (A). The data are presented as means ± the SD of four independent experiments and are expressed as a percentage of the control. The CMC of C10TAB is represented in the graph by the black dashed line. For comparison purposes cell viability (black) and LDH leakage (gray) dose-response curves (presented in detail in Fig. 1) are also shown. Apoptotic (annexin V+/PI−) and necrotic (annexin V+/PI+) cells were imaged using live-cell confocal fluorescence microscopy after 1 h (B) or 3 h (C) of incubation with C10TAB. Fractional cell death (apoptotic or necrotic) and total cell death (apoptotic and necrotic) data are presented as means ± the SD of at least three independent experiments and are expressed as a percentage of the total cell number. Asterisks (*, P < 0.05; ***, P < 0.001) signify differences from the total percentage of dying cells in control; plus symbols (+++, P < 0.001) signify differences from the percentage of necrotic cells in the control. Representative confocal images showing apoptotic cells stained with annexin V (green) and necrotic cells with PI (red) are shown in panel D. Nuclei were visualized by Hoechst 33342 staining (blue). Scale bar, 10 μm.
DISCUSSION
The use of surfactants as disinfectants and antiseptics has a very long history. Cationic surfactants, in particular, were proposed as antiseptics by Domagk in 1935 (24). They are presently used as preservatives in several topical antiseptic formulations and also as disinfectants. There is also a vast literature on their potential use as disinfectant surface coatings. The potential use of surfactant-containing formulations in prophylaxis of sexually transmitted infections gained much visibility due to at least two unsuccessful large scale preclinical trials (25–27) in the past 20 years. Those trials were driven by the hypothesis that surfactants destroy microbial (encapsulated viral, bacterial, and fungal) membranes but failed to consider that they were equally capable of destroying the membranes of the host's cells exposed to them (28). In fact, this apparently banal fact was only confirmed post factum for one of those trials (26, 27). Another serious flaw in these trials was that there had been no adequate and detailed study of surfactant concentration-dependent and exposure time-dependent toxicity to the exposed cells, and very little, if anything, was known concerning the detailed mechanisms involved, whether toward the potential pathogens or to the human tissue they would eventually be exposed to.
In spite of the negative results obtained in the clinical trials mentioned above, our previous work demonstrated that, contrary to the other surfactant families, QAC may work as bactericides at concentrations that are not toxic to polarized mammalian epithelial cells and suggested their potential utility in the prophylaxis of, at least, bacterial STI (4). However, any such application requires prior detailed knowledge of QAC structure-toxicity relationships, as well as an understanding of the sequence of events underlying surfactant-mediated cell death. Regarding the use of surfactants for topical applications, epithelial cells are the first to come in contact with these molecules, and there is evidence that, at least in vaginal use, columnar epithelia are the most susceptible (26, 27). Thus, in the present study, a columnar epithelial cell line that mimics “tight epithelia” in vitro (MDCK II cells) was used as a model to study the mechanisms of QAC toxicity.
Previous reports have demonstrated that low concentrations of cationic surfactants can induce apoptosis in different mammalian cell types (17, 18), and the involvement of mitochondrial dysfunction in surfactant-induced apoptosis has also been suggested (29, 30), although the underlying mechanisms have not been identified. In the present study, the effects of QAC concentration and exposure time on the morphology, function, and viability of mitochondria in fully polarized columnar epithelial cells were evaluated. Mitochondrial dysfunction was shown to be an early event in QAC-induced toxicity, starting at sublethal concentrations, and characterized by mitochondrial fragmentation accompanied by decreased cellular energy charge in tandem with a dissipation of mitochondrial Δψm, at higher concentrations. QAC also induced loss of mitochondrial connectivity and disintegration of the reticulotubular structures, giving rise to a widespread reorganization into small and round mitochondria. Fluorescence microscopy revealed that mitochondrial fragmentation is one of the earliest manifestations of QAC-induced toxicity and occurred after a short period of exposure to very low doses of QAC. Although mitochondrial fragmentation can occur without activation of apoptosis, many apoptotic stimuli are known to induce mitochondrial fragmentation as an early triggering step in programmed cell death (31, 32). Although the physiological significance of the dynamics of mitochondrial morphology is not completely understood, there is growing evidence to suggest that these dynamics, regulated by fission and fusion events, reflects mitochondrial energetic status (33, 34), and is crucial for key cellular functions (31, 35).
We tested the effects of QAC on standard respiratory parameters of isolated rat liver mitochondria. All of the QAC tested inhibited the NADH-ubiquinone oxidoreductase (complex I). They also lowered the rate of mitochondrial ADP-phosphorylation (RCR) without affecting the overall efficiency (ADP/O ratio) of the process when the respiratory process was initiated at complex II. These results confirm an early study of QAC effects on mitochondrial oxidative phosphorylation (36) that had demonstrated inhibition of the mitochondrial respiration, with a predominant effect on mitochondrial complex I, at low concentrations and uncoupling of respiration and ADP-phosphorylation at high concentrations. The increased production of ROS as a function of increased QAC concentration could be a consequence of slower kinetics of respiration-coupled ADP phosphorylation and/or complex I inhibition. We may therefore conclude that the observed reduction in the intracellular ATP levels of MDCK cells exposed to QAC is a consequence of mitochondrial dysfunction at the level of oxidative phosphorylation, predominantly due to inhibition of respiration initiated at complex I and to a much lesser extent due to a “sluggishness” of the process initiated at complex II.
Above a threshold concentration of QAC, MDCK cell viability decreases, and the cytotoxic effects become progressively more severe with increasing QAC concentration. The initial loss of cell viability is strongly correlated with caspase 3/7 activation, a hallmark of apoptosis. MDCK cells undergoing cell death after exposure to low concentrations of QAC exhibit morphological changes characteristic of apoptosis: cell shrinkage, exposure of PS in the outer leaflet of the plasma membrane, and nuclear DNA condensation and fragmentation. Plasma membrane damage under these conditions is probably a result of secondary necrosis. Exposure of the MDCK cells to QAC concentrations closer to CMC lead to necrotic cell death without caspase activation. Another distinctive physiological feature between apoptosis and necrosis is the intracellular ATP content: whereas programmed cell death requires a minimal level of ATP to be carried out, necrosis is characterized by a severe disruption of bioenergetics and a consequent rapid ATP depletion (37). In the present study, QAC caused a gradual decrease in the intracellular ATP levels starting even before the appearance of any sign of apoptotic signaling, which indicates that the cell energy status can be affected without commitment to cell death. Caspase 3/7 activation was only detectable at QAC concentrations that reduced the ATP/ADP ratio by >50%. Conversely, QAC concentrations close to CMC caused severe ATP depletion and cell necrosis without detectable morphological and biochemical markers of apoptosis.
In conclusion, we have shown that mitochondrial dysfunction is an early event in QAC-induced cytotoxicity, already starting at sublethal concentrations, and characterized by mitochondrial fragmentation accompanied by decreased cellular energy charge at higher concentrations. The results obtained on isolated mitochondria demonstrated that all of the QAC tested acted by a common mechanism that involved the inhibition of the NADH-ubiquinone oxidoreductase (complex I) and slowing down of the mitochondrial phosphorylative system.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by the research grant PTDC/BIA-BCM/112138/2009 from the Foundation for Science and Technology of the Portuguese Ministry of Science and Higher Education (FCT) and Bolsa de Ignição INOV.C (2011) from the University of Coimbra (Coimbra, Portugal), cofunded by COMPETE (Programa Operacional Factores de Competitividade), QREN (Quadro de Referência Estratêgico Nacional), and the European Union (FEDER-Fundo Europeu de Desenvolvimento Regional). A.S.I. is a holder of a doctoral fellowship (SFRH/BD/38397/2007) from the FCT.
We thank Vitor Madeira for his careful reading of the manuscript and very useful comments. We thank Márcio Ribeiro for his help with the protocol for ROS detection.
Footnotes
Published ahead of print 25 March 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02437-12.
REFERENCES
- 1. Hegstad K, Langsrud S, Lunestad BT, Scheie AA, Sunde M, Yazdankhah SP. 2010. Does the wide use of quaternary ammonium compounds enhance the selection and spread of antimicrobial resistance and thus threaten our health? Microb. Drug Resist. 16:91–104 [DOI] [PubMed] [Google Scholar]
- 2. Lichtenberg D, Opatowski E, Kozlov MM. 2000. Phase boundaries in mixtures of membrane-forming amphiphiles and micelle-forming amphiphiles. Biochim. Biophys. Acta 1508:1–19 [DOI] [PubMed] [Google Scholar]
- 3. Prete PS, Domingues CC, Meirelles NC, Malheiros SV, Goni FM, de Paula E, Schreier S. 2011. Multiple stages of detergent-erythrocyte membrane interaction: a spin label study. Biochim. Biophys. Acta 1808:164–170 [DOI] [PubMed] [Google Scholar]
- 4. Vieira OV, Hartmann DO, Cardoso CM, Oberdoerfer D, Baptista M, Santos MA, Almeida L, Ramalho-Santos J, Vaz WLC. 2008. Surfactants as microbicides and contraceptive agents: a systematic in vitro study. PLoS One 3:e2913 doi:10.1371/journal.pone.0002913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Inácio AS, Mesquita KA, Baptista M, Ramalho-Santos J, Vaz WLC, Vieira OV. 2011. In vitro surfactant structure-toxicity relationships: implications for surfactant use in sexually transmitted infection prophylaxis and contraception. PLoS One 6:e19850 doi:10.1371/journal.pone.0019850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brand MD, Nicholls DG. 2011. Assessing mitochondrial dysfunction in cells. Biochem. J. 435:297–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dias RS, Innerlohinger J, Glatter O, Miguel MG, Lindman B. 2005. Coil-globule transition of DNA molecules induced by cationic surfactants: a dynamic light scattering study. J. Phys. Chem. B 109:10458–10463 [DOI] [PubMed] [Google Scholar]
- 8. Bojarski C, Weiske J, Schoneberg T, Schroder W, Mankertz J, Schulzke JD, Florian P, Fromm M, Tauber R, Huber O. 2004. The specific fates of tight junction proteins in apoptotic epithelial cells. J. Cell Sci. 117(Pt 10):2097–2107 [DOI] [PubMed] [Google Scholar]
- 9. Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. 2003. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic. Biol. Med. 34:1359–1368 [DOI] [PubMed] [Google Scholar]
- 10. Koopman WJ, Visch HJ, Smeitink JA, Willems PH. 2006. Simultaneous quantitative measurement and automated analysis of mitochondrial morphology, mass, potential, and motility in living human skin fibroblasts. Cytometry A 69:1–12 [DOI] [PubMed] [Google Scholar]
- 11. Frezza C, Cipolat S, Scorrano L. 2007. Measuring mitochondrial shape changes and their consequences on mitochondrial involvement during apoptosis. Methods Mol. Biol. 372:405–420 [DOI] [PubMed] [Google Scholar]
- 12. Pereira GC, Branco AF, Matos JA, Pereira SL, Parke D, Perkins EL, Serafim TL, Sardao VA, Santos MS, Moreno AJ, Holy J, Oliveira PJ. 2007. Mitochondrially targeted effects of berberine [Natural Yellow 18, 5,6-dihydro-9,10-dimethoxybenzo(g)-1,3-benzodioxolo(5,6-a) quinolizinium] on K1735-M2 mouse melanoma cells: comparison with direct effects on isolated mitochondrial fractions. J. Pharmacol. Exp. Ther. 323:636–649 [DOI] [PubMed] [Google Scholar]
- 13. Silva AM, Oliveira PJ. 2012. Evaluation of respiration with Clark type electrode in isolated mitochondria and permeabilized animal cells. Methods Mol. Biol. 810:7–24 [DOI] [PubMed] [Google Scholar]
- 14. Santos PF, Caramelo OL, Carvalho AP, Duarte CB. 1999. Characterization of ATP release from cultures enriched in cholinergic amacrine-like neurons. J. Neurobiol. 41:340–348 [PubMed] [Google Scholar]
- 15. Chen LB. 1988. Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4:155–181 [DOI] [PubMed] [Google Scholar]
- 16. Rottenberg H. 1984. Membrane potential and surface potential in mitochondria: uptake and binding of lipophilic cations. J. Membr. Biol. 81:127–138 [DOI] [PubMed] [Google Scholar]
- 17. Perani A, Gerardin C, Stacey G, Infante MR, Vinardell P, Rodehuser L, Selve C, Maugras M. 2001. Interactions of surfactants with living cells: induction of apoptosis by detergents containing a beta-lactam moiety. Amino Acids 21:185–194 [DOI] [PubMed] [Google Scholar]
- 18. Debbasch C, Brignole F, Pisella PJ, Warnet JM, Rat P, Baudouin C. 2001. Quaternary ammoniums and other preservatives' contribution in oxidative stress and apoptosis on Chang conjunctival cells. Invest. Ophthalmol. Vis. Sci. 42:642–652 [PubMed] [Google Scholar]
- 19. Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. 1999. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15:269–290 [DOI] [PubMed] [Google Scholar]
- 20. Wyllie AH, Morris RG, Smith AL, Dunlop D. 1984. Chromatin cleavage in apoptosis: association with condensed chromatin morphology and dependence on macromolecular synthesis. J. Pathol. 142:67–77 [DOI] [PubMed] [Google Scholar]
- 21. Eguchi Y, Shimizu S, Tsujimoto Y. 1997. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 57:1835–1840 [PubMed] [Google Scholar]
- 22. Bradbury DA, Simmons TD, Slater KJ, Crouch SP. 2000. Measurement of the ADP:ATP ratio in human leukaemic cell lines can be used as an indicator of cell viability, necrosis and apoptosis. J. Immunol. Methods 240:79–92 [DOI] [PubMed] [Google Scholar]
- 23. Kabir J, Lobo M, Zachary I. 2002. Staurosporine induces endothelial cell apoptosis via focal adhesion kinase dephosphorylation and focal adhesion disassembly independent of focal adhesion kinase proteolysis. Biochem. J. 367(Pt 1):145–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Domagk G. 1935. A new class of disinfectant. Deut Med. Wochenschr. 61:829–832 [Google Scholar]
- 25. Stone A. 2002. Microbicides: a new approach to preventing HIV and other sexually transmitted infections. Nat. Rev. Drug Discov. 1:977–985 [DOI] [PubMed] [Google Scholar]
- 26. Fichorova RN, Tucker LD, Anderson DJ. 2001. The molecular basis of nonoxynol-9-induced vaginal inflammation and its possible relevance to human immunodeficiency virus type 1 transmission. J. Infect. Dis. 184:418–428 [DOI] [PubMed] [Google Scholar]
- 27. Feldblum PJ, Adeiga A, Bakare R, Wevill S, Lendvay A, Obadaki F, Olayemi MO, Wang L, Nanda K, Rountree W. 2008. SAVVY vaginal gel (C31G) for prevention of HIV infection: a randomized controlled trial in Nigeria. PLoS One 3:e1474 doi:10.1371/journal.pone.0001474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aranzazu Partearroyo M, Ostolaza H, Goni FM, Barbera-Guillem E. 1990. Surfactant-induced cell toxicity and cell lysis. A study using B16 melanoma cells. Biochem. Pharmacol. 40:1323–1328 [DOI] [PubMed] [Google Scholar]
- 29. Yip KW, Mao X, Au PY, Hedley DW, Chow S, Dalili S, Mocanu JD, Bastianutto C, Schimmer A, Liu FF. 2006. Benzethonium chloride: a novel anticancer agent identified by using a cell-based small-molecule screen. Clin. Cancer Res. 12:5557–5569 [DOI] [PubMed] [Google Scholar]
- 30. Levine SL, Han Z, Liu J, Farmer DR, Papadopoulos V. 2007. Disrupting mitochondrial function with surfactants inhibits MA-10 Leydig cell steroidogenesis. Cell Biol. Toxicol. 23:385–400 [DOI] [PubMed] [Google Scholar]
- 31. Kiefel BR, Gilson PR, Beech PL. 2006. Cell biology of mitochondrial dynamics. Int. Rev. Cytol. 254:151–213 [DOI] [PubMed] [Google Scholar]
- 32. Karbowski M, Youle RJ. 2003. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 10:870–880 [DOI] [PubMed] [Google Scholar]
- 33. Benard G, Bellance N, James D, Parrone P, Fernandez H, Letellier T, Rossignol R. 2007. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 120(Pt 5):838–848 [DOI] [PubMed] [Google Scholar]
- 34. Osellame LD, Blacker TS, Duchen MR. 2012. Cellular and molecular mechanisms of mitochondrial function. Best Pract Res. Clin. Endocrinol. Metab. 26:711–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen H, Chomyn A, Chan DC. 2005. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J. Biol. Chem. 280:26185–26192 [DOI] [PubMed] [Google Scholar]
- 36. Rogers KS, Higgins ES. 1973. Lipophilic interactions of organic cations with mitochondrial inner membranes during respiratory control. J. Biol. Chem. 248:7142–7148 [PubMed] [Google Scholar]
- 37. Tsujimoto Y. 1997. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 4:429–434 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.