

Abstract
Circulating levels of arginine vasopressin (AVP) are elevated during hypovolemia and during cardiac stress. AVP activates arginine vasopressin type 1A (V1A)/Gαq–coupled receptors in the heart and vasculature and V2/Gαs–coupled receptors in the kidney. However, little is known regarding the signaling pathways that influence the effects of V1A receptor (V1AR) activation during cellular injury. Using hypoxia-reoxygenation (H/R) as a cell injury model, we evaluated cell survival and caspase 3/7 activity in H9c2 myoblasts after treatment with AVP. Pretreatment of H9c2 cells with AVP significantly reduced H/R-induced cell death and caspase 3/7 activity, effects that were blocked via both selective V1AR inhibition and mitogen-activated protein kinase (MEK1/2) inhibition. AVP increased extracellular-regulated kinase 1/2 (ERK1/2) phosphorylation in a concentration-dependent manner that was sensitive to MEK1/2 inhibition and V1AR inhibition, but not V1BR or V2R inhibition. Discrete elements of the V1A/Gαq-protein kinase C (PKC) and V1A/G protein–coupled receptor kinase (GRK)/β-arrestin signaling cascades were inhibited to dissect the pathways responsible for the protective effects of V1AR signaling: Gαq (overexpression of Gq-I-ires-green fluorescent protein), PKC (administration of Ro 31-82425; 2-[8-(aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide, HCl, bisindolylmaleimide X, HCl), GRK2 [C-terminal GRK2 peptide overexpression and small interfering RNA (siRNA) knockdown], GRK5 (siRNA knockdown), and β-arrestin1 (siRNA knockdown). These studies demonstrated that both Gαq/PKC- and GRK2/β-arrestin1–dependent V1AR signaling were capable of inducing ERK1/2 phosphorylation in response to AVP stimulation. However, AVP-mediated protection against H/R was elicited only via GRK2- and β-arrestin1–dependent signaling. These results suggest that activation of the V1AR in H9c2 cells mediates protective signaling via a GRK2/β−arrestin1/ERK1/2–dependent mechanism that leads to decreased caspase 3/7 activity and enhanced survival under conditions of ischemic stress.
Introduction
Circulating levels of arginine vasopressin (AVP), which are elevated during hypovolemia and during cardiac stress, mediate important physiologic functions such as osmotic regulation, vasoconstriction, and release of adrenocorticotropic hormone. These physiologic effects are mediated through the interaction of AVP with specific membrane G protein–coupled receptors (GPCRs) in different target tissues: arginine vasopressin type 1A receptor (V1AR; Gαq-coupled; heart and vasculature), V2R (Gαs-coupled; renal collecting tubule), and V1BR (Gαq-coupled; anterior pituitary), also termed V3R (Lolait et al., 1992; Morel et al., 1992; de Keyzer et al., 1994). Although administration of AVP to neonatal mouse cardiomyocytes has been reported to increase cell hypertrophy (Hiroyama et al., 2007), AVP has variable effects on the heart during injury associated with ischemia and reperfusion (Indrambarya et al., 2009; Nazari et al., 2011). Recently, we found that both constitutive and controlled cardiac-specific overexpression of V1AR induced the development of left ventricular hypertrophy, dilatation, diminished contractile performance, and reprogramming of the heart failure (HF) gene program in transgenic mice, effects that were found to be mediated via Gαq protein–dependent signaling (Li et al., 2011).
In the heart, Gαq protein–coupled receptors induce the activation of protein kinase C (PKC), leading to cellular hypertrophy and activation of the HF gene program (Dorn and Force, 2005). Gαq protein–coupled receptors, including V1AR, also undergo agonist-induced phosphorylation by PKC and G protein–coupled receptor kinase (GRK) isoforms (GRK2 and GRK5), leading to internalization and desensitization of the V1AR (Innamorati et al., 1998, 1999; Berrada et al., 2000). The GRKs can also alter cellular physiology independent of PKC through phosphorylation-dependent recruitment of β-arrestins and subsequent activation of downstream pathways, including extracellular-regulated kinase 1/2 (ERK1/2) signaling, which can act to inhibit the ubiquitous responses to cell stress including apoptosis (Dorn, 2009; Huang et al., 2011; Tilley, 2011). Indeed, in the heart, G protein–independent signaling has been shown to promote cardioprotection downstream of angiotensin II- and β-adrenergic receptor activation during conditions of myocardial stress (Noma et al., 2007; Kim et al., 2012). These findings led to the development of biased ligands, GPCR antagonists that activate unique G protein–independent pathways. However, the role of GRK/β-arrestin–mediated V1AR signaling in promoting cell survival is unknown. Thus, in the present study, we sought to determine the effects of V1AR/G protein–independent signaling on cell survival and caspase 3/7 activity in H9c2 cells exposed to the stress of hypoxia and reoxygenation. This has become more than an academic interest because of the recent development of V1A/V2 receptor antagonists for the treatment of patients with hyponatremia secondary to elevated levels of AVP and HF (Rosner and Ronco, 2010; Gheorghiade et al., 2012; Zmily et al., 2013).
Materials and Methods
Penicillin and streptomycin were purchased from Invitrogen (Gaithersburg, MD). AVP (V9879) was from Sigma-Aldrich (St. Louis, MO). 2-[8-(Aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide, HCl, bisindolylmaleimide X, HCl (R0 31-8425) (Ro-31, Cat#557514) and PD 98059 (Cat#513000) were from Calbiochem (San Diego, CA). V1A receptor selective antagonist (2S)-1-[[(2R,3S)-5-chloro-3-(2-chlorophenyl)-1-[(3,4-dimethoxyphenyl)sulfonyl]-2,3-dihydro-3-hydroxy-1H-indol-2-yl]carbonyl]-2-pyrrolidinecarboxamide (SR45059) was from Tocris Bioscience (Cat#2310; Minneapolis, MN). V1B receptor selective antagonist (2S,4R)-1-(5-chloro-1-((2,4-dimethoxyphenyl)sulfonyl)-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl)-4-hydroxy-N,N-dimethyl-2-pyrrolidinecarboxamide (SSR149415) (Cat#1114) and V2 receptor selective antagonist N-[4-(7-chloro-5-hydroxy-2,3,4,5-tetrahydro-benzo[b]azepine-1-carbonyl)-3-methyl-phenyl]-2-methyl-benzamide (OPC41061) (Cat#41061) were purchased from Axon Medchem (Groningen, The Netherlands). A cell viability assay kit and caspase 3/7 assay kit were purchased from Promega (Madison, WI). The hypoxia chamber was purchased from Billups-Rothenberg, Inc. (Del Mar, CA). M199 and Opti-MEM I reduced serum medium were purchased from Invitrogen. Silencer fluorescein-labeled negative control siRNA (AM4620; Ambion/Life Technologies, Carlsbad, CA), GRK2 siRNA (AM16708; Ambion/Life Technologies), GRK5 siRNA (L-080156; Thermo Scientific), and β-arrestin1 siRNA (L-080156; Thermo Scientific, Waltham, MA) were from Ambion/Life Technologies. Adenovirus containing β-galactosidase (β-gal) and green fluorescent protein (GFP) were from Dr. Yibin Wang (University of California, Los Angeles). Gq-I-ires-GFP (GqI) and C-terminal GRK2 peptide (βARKct) were obtained from Dr. Satoru Eguchi and Dr. Walter Koch (Temple University, Philadelphia, PA), respectively. Anti-phosphorylated ERK1/2 (anti–P-ERK1/2), anti–total-ERK1/2, anti–β-arrestin1/2, and anti–glyceraldehyde-3-phosphate dehydrogenase antibodies were purchased from Cell Signaling Technology (Danvers, MA). Anti-GFP, anti-GRK2, and anti-GRK5 were from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Culture and Adenovirus.
H9c2 cells were purchased from American Type Culture Collection (Rockville, MD); maintained in M199 supplemented with 10% fetal calf serum, 100 U/ml penicillin, and 10 μl/ml streptomycin; and grown in an atmosphere of 5% CO/95% humidified air at 37°C on 6-well plates, 100,000 cells seeded per well. Adenoviral infection with βARKct versus β-galactosidase control (Zhu et al., 2001) or Gq-I-ires-GFP versus GFP control was performed at 100 multiplicity of infection when cell confluence reached 60–70%. Twenty-four to thirty-six hours after infection, cells were treated with the reagents under normal conditions or following hypoxia/reoxgenation (H/R).
Transfection of siRNA.
Cells (1 × 105 and 5000 cells per well in 6-well and 96-well plates, respectively) were seeded in plates incubated in M199 with 10% fetal bovine serum. When cell confluent reached 50–60%, the medium was replaced by 1800 µl (6-well plate) or 60 μl (96-well plate) of Opti-MEM I reduced serum medium. The negative control siRNA fluorescein (2.4 μg in 100 μl of Opti-MEM I per well in a 6-well plate and 0.083 μg in 3.2 μl of Opti-MEM I per well in a 96-well plate) and the same amount of scrambled, GRK2, GRK5, and β-arrestin1 siRNA were mixed with the same volume of transfection reagent diluted with Opti-MEM I. The mixture (200 and 20 μl per well in 6-well and 96-well plates, respectively) was added to the cells according to the instructions (at a final concentration of 100 nM). The protein levels of GRK2, GRK5, and β-arrestin1 were determined by Western blot at 48–96 hours after transfection, as indicated.
Hypoxia/Reoxygenation.
A total of 5000 cells per well were seeded in 96-well plates. When the cell confluence reached 80–90%, cells were starved with fetal calf serum–free medium overnight. Prior to H/R, the cells were pretreated with specific reagents, and the culture cell plates were placed in the hypoxia chamber. The chamber was sealed and aerated with 5% CO2 and 95% nitrogen at 3 l/min for 30 minutes. The cells were maintained in the hypoxic environment for 24 hours at 37°C, and then the cells were reoxygenated for 24 hours with 5% CO2/95% humidified air.
Cell Viability and Caspase 3/7 Assay.
Subconfluent cultures grown in 96-well plates with no prior interventions, or infection with adenovirus or transfection with siRNA, as indicated, were starved by withdrawing serum for at least 6 hours. After serum starvation, cells were pretreated with 10 pM to 0.1 μM of vasopressin for 30 minutes in the presence or absence of pharmacological agents, as indicated. Cells were then incubated in the hypoxia chamber for 24 hours followed by reoxygenation for 24 hours. The relative number of live cells and caspase 3/7 activity were measured following the manufacturer’s instructions. Within each assay, triplicates were performed for each condition to attain a single n value.
Immunoblot Analysis of Phosphorylated ERK1/2.
After cells were treated for 2–60 minutes with 1 nM to 10 μM AVP, they were rapidly washed two times with ice-cold phosphate-buffered saline and lysed with 250 μl of ice-cold lysis buffer (50 mM Tris-HCl, 1 mM EGTA, 150 mM NaCl, 1% Triton X-100, 1 mM phenylmethlysulfonyl flouride, 5 mg/ml leupeptin, 20 mg/ml aprotinin, 1 mM NaF, and 1 mM Na3VO4). After centrifugation at 13,000g for 10 minutes, equal amounts of total cell lysate (20 μg of protein) were subjected to 4–12% SDS-PAGE, followed by immunoblotting for phosphorylated ERK1/2 (1:1000), total ERK1/2 (1:1000), or GFP (1:200). Total ERK1/2 was used to normalize P-ERK1/2 responses.
Statistical Analysis.
A commercial software package was used for statistical analysis (GraphPad Software Inc., La Jolla, CA). Comparison of means ± S.E. were compared by unpaired t test, one-, or two-way analysis of variance followed by Tukey’s post-hoc analysis, as indicated in the figure legends. A value of P < 0.05 was considered statistically significant. The number of independent experiments performed in each assay is indicated in the figure legends.
Results
AVP Protects H9c2 Cells against Hypoxia/Reoxygenation-Induced Cell Death via V1AR- and ERK1/2-Dependent Signaling.
To test the ability of AVP to mediate protective signaling, we established an in vitro model of cellular stress wherein H9c2 myoblasts, derived from embryonic rat ventricle, underwent hypoxia for 24 hours followed by reoxygenation for 24 hours (H/R). Under normoxic and serum-free conditions, H9c2 cell survival decreased and caspase 3/7 activity increased, responses that were significantly enhanced when the cells underwent H/R (Fig. 1, A and B). The addition of 10% fetal calf serum was sufficient to prevent these responses under either normoxic or H/R conditions. Since H/R performed in the absence of serum induced the most significant alteration in H9c2 cell survival and caspase 3/7 activity, we used these conditions for all subsequent experiments. As seen in Fig. 1C, pretreatment of the H9c2 cells with AVP significantly attenuated the H/R-induced changes in both cell survival and caspase 3/7 activity in a concentration-dependent manner.
Fig. 1.

AVP protects H9c2 cells against hypoxia/reoxygenation–induced cell death via V1AR- and ERK1/2-dependent signaling. (A) Caspase 3/7 activity in response to serum starvation under conditions of H/R or normoxia. **P < 0.01 versus starvation + normoxia; ††P < 0.01 versus fetal bovine serum (FBS) in same group [one-way analysis of variance (ANOVA)]. (B) Cell survival in response to serum starvation under conditions of H/R or normoxia. *P < 0.05 versus starvation + normoxia; †P < 0.05 versus FBS in same group (one-way ANOVA). (C) AVP concentration dependently increased cell survival and decreased caspase 3/7 activity under conditions of H/R. *P < 0.05 for cell survival; **P < 0.01 for caspase 3/7 activity (repeated two-way ANOVA). Pretreatment of cells with SR49059 (0.1 μM) or PD98059 (10 μM) for 30 minutes blunted effect of AVP on caspase 3/7 activity (D) and cell survival (E). *P < 0.05 versus vehicle (phosphate-buffered saline; PBS)-stimulated control (one-way ANOVA). All data are presented as the mean ± S.E.M. from three independent experiments. RLU, relative luminescence units.
To confirm the role of the cardiac-expressed AVP receptor (V1AR) in mediating AVP-dependent protection against cell death, H9c2 cells underwent H/R in the presence or absence of AVP (10 nM) and the V1AR selective antagonist, SR49059 (0.1 μM). Antagonism of V1AR with SR49059 abolished the ability of AVP to reduce both caspase 3/7 activity and cell death (Fig. 1, D and E). Neither V1BR- nor V2R-selective antagonists (SSR14941 and OPC41061, respectively) had an effect on AVP-dependent changes in caspase 3/7 activity or cell survival during H/R (Supplemental Fig. 1, A and B). Additionally, to assess the potential role of mitogen-activated protein kinase (MEK1/2)-ERK1/2 signaling in mediating the protective effect of AVP, H9c2 cells were treated with the MEK1/2 inhibitor 2′-amino-3′-methoxyflavone (PD98059) (10 μM) prior to H/R induction. Inhibition of MEK1/2 completely blocked the AVP-mediated effects on cell survival and caspase 3/7 activity (Fig. 1, D and E).
AVP-Mediated ERK1/2 Phosphorylation Dynamics.
Since MEK1/2 inhibition abolished the ability of AVP to enhance cell survival during H/R, we sought to define the ERK1/2 signaling response to AVP. AVP stimulation of H9c2 cells for 5 minutes elicited P-ERK1/2 in a concentration-dependent manner with an EC50 of 9.0 ± 2.0 nM (Fig. 2, A and B). This response could only be blocked by V1AR-selective antagonism, but not by V1BR- or V2R-selective antagonism. The IC50 values of SR49059 (V1AR), SSR14941 (V1BR), and OPC41061 (V2R) for inhibition of AVP (10 nM)-induced P-ERK1/2 were 0.38 ± 0.08, 104.4 ±10.2, and 374.9 ± 23.4 nM, respectively (Fig. 2, C and D). ERK1/2 signaling often persists long after receptor stimulation, therefore we assessed the P-ERK1/2 response to AVP at both acute (2–10 minutes) and prolonged (30–60 minutes) time points. AVP-induced P-ERK1/2 peaked at 5 minutes, and was sustained at ∼50% peak activation even to 60 minutes poststimulation (Fig. 2, E and F). As expected, pretreatment of H9c2 cells with the MEK1/2 inhibitor PD98059 (10 μM) significantly attenuated both the acute and prolonged P-ERK1/2 responses to AVP stimulation. Thus far, these results demonstrate that V1AR stimulation enhances H9c2 cell survival via MEK1/2-ERK1/2 signaling.
Fig. 2.
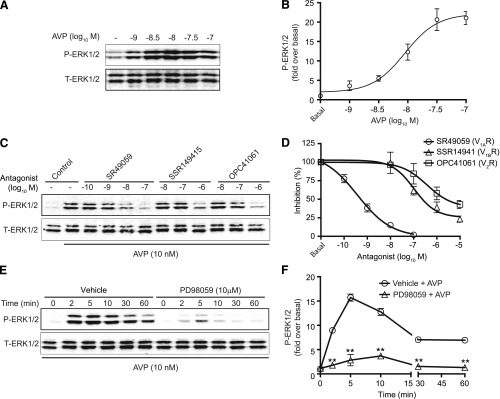
V1AR, not V1BR or V2R, mediates ERK1/2 phosphorylation in H9c2 cells. (A and B) An AVP concentration–P-ERK1/2 response analysis was performed in H9c2 cells, with stimulation for 5 minutes, as determined via immunoblotting. (C and D) Inhibitory concentration responses for V1AR-, V1BR-, or V2R-selective antagonists (30-minute pretreatment at 1 nM to 10 μM) against AVP (10 nM, 5-minute treatment)-induced P-ERK1/2 were generated in H9c2 cells. (E and F) MEK1/2 inhibition with PD98059 (10 μM) blunted the AVP-dependent phosphorylation of ERK1/2 at all time points analyzed. **P < 0.01 versus control at the same time points (unpaired t test). All data are presented as the mean ± S.E.M. from three to four independent experiments. T-ERK1/2, total ERK1/2.
Inhibition of Gαq Protein/PKC Signaling Blunts the AVP-Induced Acute P-ERK1/2 Response but Has No Effect on the AVP-Induced Cell Survival Response.
GPCR stimulation induces the simultaneous activation of both G protein–dependent and –independent signaling pathways that can each act to modulate ERK1/2 activity (Tilley, 2011). Since V1AR/Gαq protein coupling induces the activation of PKC, regulating the hypertrophic gene program (Li et al., 2011), we tested whether Gαq protein/PKC–mediated signaling is involved in AVP-induced P-ERK1/2 and cell survival responses. Overexpression of the peptide inhibitor of Gαq (GqI) inhibited the acute P-ERK1/2 response to AVP (2 and 5 minutes, by 90% ± 3% and 52% ± 6%, respectively, versus GFP control), but had no effect on the AVP-induced prolonged responses at 30 and 60 minutes (Fig. 3, A and C). Further, suppression of Gαq protein by GqI overexpression did not impact AVP-induced effects on caspase 3/7 activity or cell survival (Fig. 3, D and E). Similarly, pretreatment of H9c2 cells with the PKC inhibitor Ro-31 (1 μM) inhibited acute phosphorylation of ERK1/2 (90% ± 6% and 62% ± 3% inhibition at 2 and 5 minutes, respectively), but had no effect on persistent ERK1/2 phosphorylation (30–60 minutes) (Fig. 3, B and C). Pretreatment of H9c2 cells with Ro-31 prior to H/R led to a significant enhancement of H/R-induced cell death and caspase 3/7 activity (Supplemental Fig. 1C). Interestingly, despite the enhanced negative effect on survival mediated by chronic inhibition of PKC by Ro-31, AVP was still capable of inducing a concentration-dependent reduction in cell death and caspase 3/7 activity (Fig. 3F). These results suggest a Gαq protein/PKC-independent component to V1AR-mediated cell survival signaling.
Fig. 3.

Inhibition of Gαq protein/PKC signaling blunts the AVP-induced acute P-ERK1/2 response but has no effect on the AVP-induced cell survival response. (A) Overexpression of GqI (versus GFP alone) with adenovirus or (B) inhibition of PKC with Ro-31 (1 μM, versus 0.1% dimethylsulfoxide vehicle) significantly blunted the acute (2–10 minutes), but not prolonged (30–60 minutes), phosphorylation of ERK1/2 evoked by AVP (10 nM) in H9c2 cells. (C) The averaged data of A and B. *P < 0.05; **P < 0.01 GqI versus GFP at the same time points. †P < 0.05; ††P < 0.01 Ro-31 versus vehicle at the same time points (unpaired t test). Overexpression of GqI failed to block the AVP-mediated decrease in caspase 3/7 activity (D) and increase in cell survival (E). **P < 0.01 versus appropriate vehicle (phosphate-buffered saline; PBS)-stimulated control [one-way analysis of variance (ANOVA)]. (F) Pretreatment of H9c2 cells with Ro-31 (1 μM) did not impact the ability of AVP to protect cells from H/R, as AVP concentration dependently decreased caspase 3/7 activity and increased cell survival. *P < 0.05 for cell survival; **P < 0.01 for caspase 3/7 activity (repeated two-way ANOVA). All data are presented as the mean ± S.E.M. from at least three independent experiments. RLU, relative luminescence units.
Overexpression of βARKct Blunts Persistent AVP-Induced P-ERK1/2 and Abrogates AVP-Mediated H9c2 Cell Survival.
To begin to assess the contribution of G protein–independent signaling to V1AR-mediated P-ERK1/2 and survival responses in H9c2 cells, we overexpressed βARKct, the C-terminal peptide of GRK2 that prevents its recruitment and activation upon GPCR stimulation, or β-gal as a control (Koch et al., 1994; Zhu et al., 2001). Overexpression of βARKct predominantly blunted the persistent P-ERK1/2 responses at 30 and 60 minutes by 45% ± 2% and 39% ± 4%, respectively, versus β-gal control (Fig. 4, A and C). Additionally, AVP-elicited cell survival was completely abolished by overexpression of βARKct (Fig. 4, D and E). Comparison of the effects of the combined inhibition of AVP signaling by simultaneous treatment of H9c2 cells with Ro-31 and βARKct was consistent with these results, as the combination of both Ro-31 and βARKct did not further impact the cell survival responses to AVP (Supplemental Fig. 1, D and E). These results suggest a role for GRK2-dependent V1AR signaling in the promotion of ERK1/2 phosphorylation and cell survival under conditions of cellular stress.
Fig. 4.
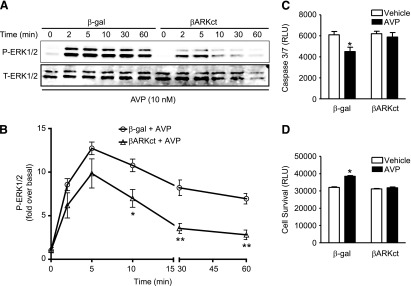
Overexpression of βARKct blunts persistent AVP-induced P-ERK1/2 and abrogates AVP-mediated H9c2 cell survival. (A) Overexpression of βARKct (versus β-gal) significantly blunted the ERK1/2 phosphorylation response (from 10 to 60 minutes) following AVP stimulation. (B) The averaged data of (A). *P < 0.05; **P < 0.01 versus the same time point (unpaired t test). Overexpression of βARKct significantly blocked the AVP-dependent decrease in caspase 3/7 activity (C) and increase in cell survival (D). *P < 0.05 versus vehicle (phosphate-buffered saline; PBS)-stimulated β-gal control (one-way analysis of variance). All data are presented as the mean ± S.E.M. from three independent experiments. RLU, relative luminescence units; T-ERK1/2, total ERK1/2.
siRNA-Mediated Deletion of GRK2 and β-Arrestin1, but Not GRK5, Blunts Persistent AVP-Induced P-ERK1/2 and Cell Survival Responses in H9c2 Cells.
To confirm the role of GRK2 in mediating the effects of AVP on persistent ERK1/2 phosphorylation and survival in H9c2 cells, siRNA-mediated knockdown of GRK2 was performed versus a scrambled siRNA control. After a 48-hour knockdown, GRK2 protein expression was significantly reduced by 68.5% ± 6.5% (Fig. 5, A and D), at which time point the P-ERK1/2 response to AVP was assessed. Consistent with the effects of βARKct on V1AR-mediated signaling, deletion of GRK2 significantly inhibited the persistent P-ERK1/2 response to AVP (Fig. 5, A and E). After 96 hours, including the H/R assay, GRK2 deletion was shown to impair the ability of AVP to promote H9c2 survival during H/R (Fig. 5, F and G). Of note, the H/R procedure did not alter siRNA-mediated silencing of GRK2 in the presence or absence of AVP (Supplemental Fig. 2, A and B). In addition to GRK2, GRK5 is the other predominant GRK cardiac isoform that has been shown to modulate ERK1/2 activity and survival signaling (Huang et al., 2011). To determine if GRK5 contributes to G protein–independent effects of AVP on ERK1/2 phosphorylation and cell survival, we next performed GRK5 siRNA-mediated knockdown experiments. Although knockdown of GRK5 protein expression was attained to a similar extent as that of GRK2 (Fig. 5, B and D), neither the P-ERK1/2 response (Fig. 5, B and E) nor the caspase 3/7 activity and survival responses (Fig. 5, F and G) to AVP were altered in comparison with the scrambled siRNA controls.
Fig. 5.

siRNA-mediated deletion of GRK2 and β-arrestin1, but not GRK5, blunts persistent AVP-induced P-ERK1/2 and cell survival responses in H9c2 cells. Representative immunoblots showing GRK2 (A), GRK5 (B), and β-arrestin1 (βarr1) (C) protein expression after siRNA transfection and the corresponding ERK1/2 phosphorylation responses to AVP stimulation (10 nM, 0–60 minutes) in H9c2 cells. (D) The averaged data of GRK2, GRK5, or β-arrestin1 knockdown, compared with scrambled (Scr) siRNA. **P < 0.01 versus scrambled siRNA control [one-way analysis of variance (ANOVA)]. (E) Summary of the effects of siRNA-mediated deletion of GRK2, GRK5, and βarr1 on the P-ERK1/2 response to AVP (10 nM, 0–60 minutes). **P < 0.01 (GRK2 siRNA); ††P < 0.01 (βarr1 siRNA) versus scrambled siRNA at the same time point (unpaired t test). The effects of siRNA-mediated knockdown of GRK2, GRK5, or βarr1 on AVP-mediated caspase 3/7 activity (F) and cell survival (G) during H/R in H9c2 cells are shown. *P < 0.05 GRK2 siRNA versus Scr siRNA (cell survival, caspase 3/7 activity); ††P < 0.01 βarr1 siRNA versus Scr siRNA (caspase 3/7 activity); and †P < 0.05 βarr1 siRNA versus Scr siRNA (cell survival) (repeated two-way ANOVA). All data are presented as the mean ± S.E.M. of three independent experiments. RLU, relative luminescence units.
β-Arrestins are recruited to GRK–phosphorylated GPCR to induce a number of cellular processes, including persistent ERK1/2 activation, and have been shown to promote cardiac protection under conditions of stress (Luttrell et al., 2001; Noma et al., 2007; Kim et al., 2012). Thus, we sought to determine whether β-arrestins are required for the V1AR/GRK2–dependent effects on ERK1/2 and survival signaling. We began by transfecting the cells with β-arrestin1–selective versus scrambled siRNA. Forty-eight hours later, β-arrestin1 protein expression was significantly reduced by 79.5% ± 3.5% (Fig. 5, C and D). As observed with GRK2 knockdown, deletion of β-arrestin1 inhibited the AVP-induced P-ERK1/2 response, again primarily at the persistent (30–60 minutes) time points (Fig. 5, C and E). Further, β-arrestin1 knockdown significantly impaired the ability of AVP to promote H9c2 cell survival and decrease caspase 3/7 activity during H/R (Fig. 5, F and G).
Via immunoblotting analysis, β-arrestin1 was shown to be the predominant β-arrestin isoform expressed in H9c2 cells (Supplemental Fig. 3A). To determine if there was a low level of β-arrestin2 expression that mediated V1AR effects on ERK1/2 phosphorylation and cell survival, we also transfected H9c2 cells with siRNA directed against β-arrestin2. Perhaps due to the low/nondetectable expression of β-arrestin2 in these cells, β-arrestin2 siRNA induced a substantial, although variable, knockdown of β-arrestin1, which was sufficient to significantly reduce AVP-mediated P-ERK1/2 and survival responses (Supplemental Fig. 3, B–E), essentially recapitulating the data attained using β-arrestin1 siRNA. Altogether, these results reveal that under conditions of hypoxic stress, engagement of a V1AR/GRK2/β-arrestin1/ERK1/2–mediated signaling pathway in H9c2 cells promotes survival.
Discussion
G protein–independent signaling through GRKs and β-arrestins has been shown to mediate a number of beneficial effects on cardiac function and survival during heart failure (Tilley, 2011). GRKs were initially identified as desensitizing regulators of GPCR, terminating their acute signaling responses to agonist stimulation via phosphorylation of the C-terminal tail of the receptor (Benovic et al., 1986; Hausdorff et al., 1990). It has since been clearly demonstrated that beyond desensitization, GRK also initiates G protein–independent signaling events that act to regulate ERK1/2 activity, for instance, via the recruitment of the scaffolding proteins β-arrestins through differential phosphorylation of GPCRs (Heitzler et al., 2012). These events commonly promote more persistent ERK1/2 activity than that mediated by G proteins, and have been demonstrated to impact numerous cellular processes, including regulation of apoptosis and cell survival in numerous cells and, importantly, in the heart (Dorn, 2009; Tilley, 2011; Huang et al., 2011). Although relatively little is known about the pathophysiologic effects of cardiac-expressed V1AR, our recent study utilizing transgenic mice with inducible cardiac-restricted overexpression of V1AR was the first to conclusively demonstrate that enhanced V1AR signaling in the heart itself directly leads to the development of left ventricular hypertrophy, dilatation, diminished contractile performance, and reprogramming of the HF gene profile in a Gαq protein–dependent manner (Li et al., 2011), although a role for Gαq protein–independent signaling in the control of cardiac function and/or survival was not investigated.
In the present study, using H9c2 myoblasts as an in vitro model of H/R, we demonstrate that AVP enhances cell survival during stress in a V1AR/GRK2/β-arrestin1/ERK1/2–dependent manner. H9c2 myoblasts, derived from embryonic rat ventricle, were an excellent model system in which to directly test the effects of V1AR signaling during in vitro stress, as they express cardiac and skeletal isoforms of L-type Ca2+ channels, sarcolemmal ATPase splice variants characteristic of a normal heart and endogenous V1AR, which respond to AVP with typical Gαq protein–coupled receptor responses (Hescheler et al., 1991; Sipido and Marban, 1991; Hammes et al., 1994; Tran et al., 1995; Reilly et al., 1998; Chen and Chen, 1999; Guo et al., 2012). During the development of the H/R model, we initially found that H/R increased caspase 3/7 activity and decreased cell viability with enhanced robustness over normoxic conditions only under serum-free conditions, whereas the presence of fetal bovine serum protected the cells. This would be predicted as the presence of growth factors in the serum could act to constitutively promote cell survival and proliferation through many pathways, including activation of ERK1/2. A high level of ERK1/2 phosphorylation would also mask or blunt AVP-specific effects on ERK1/2 regulation. Since ischemia in vivo promotes a reduction in local oxygen and nutrient delivery, we feel the serum-free condition in our H/R model better replicates this condition. Additionally, it facilitates a controlled environment to specifically add AVP to the media alone to determine its effects on survival, without the confounding presence of other growth- and survival-promoting factors, and the underlying mechanisms, in this case through G protein–independent regulation of ERK1/2.
Our finding that V1AR-mediated cell survival is mediated via G protein–independent regulation of ERK1/2 activity is consistent with other reports describing the molecular mechanisms of GPCR-induced survival signaling, including the cardiac-expressed β-adrenergic receptors and the angiotensin type 1A receptor (AT1R) (Noma et al., 2007; Kim et al., 2012). As with the V1AR, the AT1R is a Gαq-coupled receptor, and studies have shown the survival signaling response to AT1R stimulation depends not on rapid Gαq protein–mediated ERK1/2 signaling, but on prolonged GRK/β-arrestin–mediated ERK1/2 signaling (Ahn et al., 2009; Kim et al., 2012). Our data demonstrate that this pattern of ERK1/2 activation is similar for V1AR in H9c2 myoblasts, wherein Gαq protein/PKC–dependent signaling mediates the acute P-ERK1/2 response, and GRK2-β-arrestin1–dependent signaling primarily regulates the persistent phase of the P-ERK1/2 response. This could account for our previous observation in the V1AR transgenic mice that, although inhibition of Gαq protein blocked the development of the heart failure phenotype, it only partially inhibited activation of ERK1/2 (Li et al., 2011). Thus, although ERK1/2 signaling is required for the AVP-mediated survival response, the G protein–dependent cohort did not contribute to this effect, implying the need for selective subcellular targeting of V1AR/GRK2/β-arrestin1/ERK1/2 signaling in promoting survival.
A distinctive finding in our study is that GRK2, not GRK5, is responsible for driving the β-arrestin1/ERK1/2–signaling response to AVP, whereas previous studies have found that GRK2 mainly induces GPCR desensitization while other GRK isoforms, including GRK5, are responsible for mediating β-arrestin–dependent ERK1/2 signaling responses (Noma et al., 2007; Heitzler et al., 2012). This is an important finding, as inhibition of GRK2 has been shown to be cardioprotective in models of heart failure (Lymperopoulos et al., 2012), but could limit the ability of V1AR to exert survival signaling. In fact, in light of our findings, inhibition of GRK2 could potentially exacerbate negative V1AR-mediated effects by preventing receptor desensitization and augmenting deleterious Gαq protein–dependent signaling during heart failure, in which there are elevated levels of circulating AVP. Additionally, β-arrestin1 and β-arrestin2 are usually ubiquitously expressed and can have redundant or opposing effects on receptor internalization or signaling processes. Thus, our finding that H9c2 cells predominantly express β-arrestin1 with no detectable β-arrestin2 expression is unique, and although in this system β-arrestin1 clearly plays an essential role in mediating the protective effects of V1AR signaling, the contribution of β-arrestin2 to these effects in other cell types, such as fully differentiated adult cardiomyocytes, should be examined on a case-by-case basis.
The importance of understanding the relative contributions of both G protein–dependent and –independent pathways in the regulation of cell survival mechanisms has been recently highlighted in a number of studies investigating biased GPCR ligands, that is, ligands that have agonist-like effects but that are biased toward the activation of one particular pathway over another. A number of AT1R-biased agonists that lack the ability to activate Gαq protein–dependent signaling but do promote GRK/β-arrestin–dependent ERK1/2 signaling have been shown capable of regulating apoptotic pathways in vitro and enhancing cardiac contractility and improved cardiac performance in vivo, one of which is undergoing phase II evaluation for the treatment of heart failure (Violin et al., 2010; Boerrigter et al., 2011). Late-stage heart failure is often associated with high levels of AVP, which has been associated with symptomatic hyponatremia and increased mortality (Francis et al., 1990; Gheorghiade et al., 2012). However, the V2-selective antagonist tolvaptan had no effect on survival and actually increased circulating levels of AVP (Lanfear et al., 2013), whereas the V2-selective antagonist lixivaptan was associated with increased mortality in patients hospitalized with acute heart failure and hyponatremia (Xu, 2012; http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM318867.pdf). The present data suggest that the use of a biased ligand that blocks both V2R- and V1AR-Gαq protein–dependent signaling, while at the same time activating V1AR-mediated GRK2/β-arrestin signaling, would provide a novel new therapeutic for the treatment of patients with chronic heart failure and elevated levels of AVP.
Supplementary Material
Acknowledgments
The authors thank Dr. Satoru Eguchi (Temple University School of Medicine) for kindly providing the Gq-I-ires-GFP adenovirus.
Abbreviations
- AT1R
angiotensin type 1A receptor
- AVP
arginine vasopressin
- βARKct
C-terminal GRK2 peptide
- ERK1/2
extracellular-regulated kinase 1/2
- β-gal
β-galactosidase
- GFP
green fluorescent protein
- GRK
G protein–coupled receptor kinase
- GqI
Gq-I-ires-GFP
- HF
heart failure
- H/R
hypoxia/reoxygenation
- MEK1/2
mitogen-activated protein kinase
- OPC41061
N-[4-(7-chloro-5-hydroxy-2,3,4,5-tetrahydro-benzo[b]azepine-1-carbonyl)-3-methyl-phenyl]-2-methyl-benzamide
- PD 98059
2′-amino-3′-methoxyflavone
- P-ERK1/2
ERK1/2 phosphorylation
- PKC
protein kinase C
- Ro 31-8425/Ro-31
2-[8-(aminomethyl)-6,7,8,9-tetrahydropyrido[1,2-a]indol-3-yl]-3-(1-methyl-1H-indol-3-yl)maleimide, HCl, bisindolylmaleimide X, HCl
- SSR149415
(2S,4R)-1-(5-chloro-1-((2,4-dimethoxyphenyl)sulfonyl)-3-(2-methoxyphenyl)-2-oxo-2,3-dihydro-1H-indol-3-yl)-4-hydroxy-N,N-dimethyl-2-pyrrolidinecarboxamide
- SR45059
(2S)-1-[[(2R,3S)-5-chloro-3-(2-chlorophenyl)-1-[(3,4-dimethoxyphenyl)sulfonyl]-2,3-dihydro-3-hydroxy-1H-indol-2-yl]carbonyl]-2-pyrrolidinecarboxamide
- V1AR
arginine vasopressin type 1A receptor
Authorship Contributions
Participated in research design: Zhu, Tilley, Myers.
Conducted experiments: Zhu, Myers, Coleman.
Performed data analysis: Zhu, Tilley, Myers, Feldman.
Wrote or contributed to the writing of the manuscript: Zhu, Tilley, Feldman.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grants HL105414 and HL091799].
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Ahn S, Kim J, Hara MR, Ren XR, Lefkowitz RJ. (2009) beta-Arrestin-2 Mediates Anti-apoptotic Signaling through Regulation of BAD Phosphorylation. J Biol Chem 284:8855–8865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benovic JL, Strasser RH, Caron MG, Lefkowitz RJ. (1986) Beta-adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc Natl Acad Sci USA 83:2797–2801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berrada K, Plesnicher CL, Luo X, Thibonnier M. (2000) Dynamic interaction of human vasopressin/oxytocin receptor subtypes with G protein-coupled receptor kinases and protein kinase C after agonist stimulation. J Biol Chem 275:27229–27237 [DOI] [PubMed] [Google Scholar]
- Boerrigter G, Lark MW, Whalen EJ, Soergel DG, Violin JD, Burnett JC., Jr (2011) Cardiorenal actions of TRV120027, a novel ß-arrestin-biased ligand at the angiotensin II type I receptor, in healthy and heart failure canines: a novel therapeutic strategy for acute heart failure. Circ Heart Fail 4:770–778 [DOI] [PubMed] [Google Scholar]
- Chen WC, Chen CC. (1999) Signal transduction of arginine vasopressin-induced arachidonic acid release in H9c2 cardiac myoblasts: role of Ca2+ and the protein kinase C-dependent activation of p42 mitogen-activated protein kinase. Endocrinology 140:1639–1648 [DOI] [PubMed] [Google Scholar]
- de Keyzer Y, Auzan C, Lenne F, Beldjord C, Thibonnier M, Bertagna X, Clauser E. (1994) Cloning and characterization of the human V3 pituitary vasopressin receptor. FEBS Lett 356:215–220 [DOI] [PubMed] [Google Scholar]
- Dorn GW., 2nd (2009) GRK mythology: G-protein receptor kinases in cardiovascular disease. J Mol Med (Berl) 87:455–463 [DOI] [PubMed] [Google Scholar]
- Dorn GW, 2nd, Force T. (2005) Protein kinase cascades in the regulation of cardiac hypertrophy. J Clin Invest 115:527–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis GS, Benedict C, Johnstone DE, Kirlin PC, Nicklas J, Liang CS, Kubo SH, Rudin-Toretsky E, Yusuf S. (1990) Comparison of neuroendocrine activation in patients with left ventricular dysfunction with and without congestive heart failure. A substudy of the Studies of Left Ventricular Dysfunction (SOLVD). Circulation 82:1724–1729 [DOI] [PubMed] [Google Scholar]
- Gheorghiade M, Pang PS, Ambrosy AP, Lan G, Schmidt P, Filippatos G, Konstam M, Swedberg K, Cook T, Traver B, et al. (2012) A comprehensive, longitudinal description of the in-hospital and post-discharge clinical, laboratory, and neurohormonal course of patients with heart failure who die or are re-hospitalized within 90 days: analysis from the EVEREST trial. Heart Fail Rev 17:485–509 [DOI] [PubMed] [Google Scholar]
- Guo S, Olm-Shipman A, Walters A, Urciuoli WR, Devito S, Nadtochiy SM, Wojtovich AP, Brookes PS. (2012) A cell-based phenotypic assay to identify cardioprotective agents. Circ Res 110:948–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes A, Oberdorf S, Strehler EE, Stauffer T, Carafoli E, Vetter H, Neyses L. (1994) Differentiation-specific isoform mRNA expression of the calmodulin-dependent plasma membrane Ca(2+)-ATPase. FASEB J 8:428–435 [DOI] [PubMed] [Google Scholar]
- Hausdorff WP, Caron MG, Lefkowitz RJ. (1990) Turning off the signal: desensitization of beta-adrenergic receptor function. FASEB J 4:2881–2889 [PubMed] [Google Scholar]
- Heitzler D, Durand G, Gallay N, Rizk A, Ahn S, Kim J, Violin JD, Dupuy L, Gauthier C, Piketty V, et al. (2012) Competing G protein-coupled receptor kinases balance G protein and β-arrestin signaling (Abstract). Mol Syst Biol 8:590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hescheler J, Meyer R, Plant S, Krautwurst D, Rosenthal W, Schultz G. (1991) Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ Res 69:1476–1486 [DOI] [PubMed] [Google Scholar]
- Hiroyama M, Wang S, Aoyagi T, Oikawa R, Sanbe A, Takeo S, Tanoue A. (2007) Vasopressin promotes cardiomyocyte hypertrophy via the vasopressin V1A receptor in neonatal mice. Eur J Pharmacol 559:89–97 [DOI] [PubMed] [Google Scholar]
- Huang ZM, Gold JI, Koch WJ. (2011) G protein-coupled receptor kinases in normal and failing myocardium. Front Biosci 16:3047–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indrambarya T, Boyd JH, Wang Y, McConechy M, Walley KR. (2009) Low-dose vasopressin infusion results in increased mortality and cardiac dysfunction following ischemia-reperfusion injury in mice (Abstract). Crit Care 13:R98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innamorati G, Sadeghi H, Birnbaumer M. (1998) Transient phosphorylation of the V1a vasopressin receptor. J Biol Chem 273:7155–7161 [DOI] [PubMed] [Google Scholar]
- Innamorati G, Sadeghi H, Birnbaumer M. (1999) Phosphorylation and recycling kinetics of G protein-coupled receptors. J Recept Signal Transduct Res 19:315–326 [DOI] [PubMed] [Google Scholar]
- Kim KS, Abraham D, Williams B, Violin JD, Mao L, Rockman HA. (2012) β-Arrestin-biased AT1R stimulation promotes cell survival during acute cardiac injury. Am J Physiol Heart Circ Physiol 303:H1001–H1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. (1994) Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates G beta gamma-mediated signaling. J Biol Chem 269:6193–6197 [PubMed] [Google Scholar]
- Lanfear DE, Sabbah HN, Goldsmith SR, Greene SJ, Ambrosy AP, Fought AJ, Kwasny MJ, Swedberg K, Yancy CW, Konstam MA, et al. EVEREST trial investigators (2013) Association of arginine vasopressin levels with outcomes and the effect of V2 blockade in patients hospitalized for heart failure with reduced ejection fraction: insights from the EVEREST trial. Circ Heart Fail 6:47–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Chan TO, Myers V, Chowdhury I, Zhang XQ, Song J, Zhang J, Andrel J, Funakoshi H, Robbins J, et al. (2011) Controlled and cardiac-restricted overexpression of the arginine vasopressin V1A receptor causes reversible left ventricular dysfunction through Gαq-mediated cell signaling. Circulation 124:572–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolait SJ, O’Carroll AM, McBride OW, Konig M, Morel A, Brownstein MJ. (1992) Cloning and characterization of a vasopressin V2 receptor and possible link to nephrogenic diabetes insipidus. Nature 357:336–339 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ. (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA 98:2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lymperopoulos A, Rengo G, Koch WJ. (2012) GRK2 inhibition in heart failure: something old, something new. Curr Pharm Des 18:186–191 [DOI] [PubMed] [Google Scholar]
- Morel A, O’Carroll AM, Brownstein MJ, Lolait SJ. (1992) Molecular cloning and expression of a rat V1a arginine vasopressin receptor. Nature 356:523–526 [DOI] [PubMed] [Google Scholar]
- Nazari A, Sadr SS, Faghihi M, Imani A, Moghimian M. (2011) The cardioprotective effect of different doses of vasopressin (AVP) against ischemia-reperfusion injuries in the anesthetized rat heart. Peptides 32:2459–2466 [DOI] [PubMed] [Google Scholar]
- Noma T, Lemaire A, Naga Prasad SV, Barki-Harrington L, Tilley DG, Chen J, Le Corvoisier P, Violin JD, Wei H, Lefkowitz RJ, et al. (2007) Beta-arrestin-mediated beta1-adrenergic receptor transactivation of the EGFR confers cardioprotection. J Clin Invest 117:2445–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reilly BA, Brostrom MA, Brostrom CO. (1998) Regulation of protein synthesis in ventricular myocytes by vasopressin. The role of sarcoplasmic/endoplasmic reticulum Ca2+ stores. J Biol Chem 273:3747–3755 [DOI] [PubMed] [Google Scholar]
- Rosner MH, Ronco C. (2010) Hyponatremia in heart failure: the role of arginine vasopressin and its antagonism. Congest Heart Fail 16 (Suppl 1):S7–S14 [DOI] [PubMed] [Google Scholar]
- Sipido KR, Marban E. (1991) L-type calcium channels, potassium channels, and novel nonspecific cation channels in a clonal muscle cell line derived from embryonic rat ventricle. Circ Res 69:1487–1499 [DOI] [PubMed] [Google Scholar]
- Tilley DG. (2011) G protein-dependent and G protein-independent signaling pathways and their impact on cardiac function. Circ Res 109:217–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran K, Zha X, Chan M, Choy PC. (1995) Enhancement of phospholipid hydrolysis in vasopressin-stimulated BHK-21 and H9c2 cells. Mol Cell Biochem 151:69–76 [DOI] [PubMed] [Google Scholar]
- Violin JD, DeWire SM, Yamashita D, Rominger DH, Nguyen L, Schiller K, Whalen EJ, Gowen M, Lark MW. (2010) Selectively engaging β-arrestins at the angiotensin II type 1 receptor reduces blood pressure and increases cardiac performance. J Pharmacol Exp Ther 335:572–579 [DOI] [PubMed] [Google Scholar]
- Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. (2001) Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci USA 98:1607–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmily HD, Alani A, Ghali JK. (2013) Evaluation of lixivaptan in euvolemic and hypervolemic hyponatremia and heart failure treatment. Expert Opin Drug Metab Toxicol 9:645–655 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.