

Abstract
Objectives:
Previous associations between mitochondrial DNA (mtDNA) and idiopathic Parkinson disease (PD) have been inconsistent and contradictory. Our aim was to resolve these inconsistencies and determine whether mtDNA has a significant role in the risk of developing PD.
Methods:
Two-stage genetic association study of 138 common mtDNA variants in 3,074 PD cases and 5,659 ethnically matched controls followed by meta-analysis of 6,140 PD cases and 13,280 controls.
Results:
In the association study, m.2158T>C and m.11251A>G were associated with a reduced risk of PD in both the discovery and replication cohorts. None of the common European mtDNA haplogroups were consistently associated with PD, but pooling of discovery and replication cohorts revealed a protective association with “super-haplogroup” JT. In the meta-analysis, there was a reduced risk of PD with haplogroups J, K, and T and super-haplogroup JT, and an increase in the risk of PD with super-haplogroup H.
Conclusions:
In a 2-stage association study of mtDNA variants and PD, we confirm the reduced risk of PD with super-haplogroup JT and resolve this at the J1b level. Meta-analysis explains the previous inconsistent associations that likely arise through sampling effects. The reduced risk of PD with haplogroups J, K, and T is mirrored by an increased risk of PD in super-haplogroup HV, which increases survival after sepsis. Antagonistic pleiotropy between mtDNA haplogroups may thus be shaping the genetic landscape in humans, leading to an increased risk of PD in later life.
Multiple lines of evidence implicate mitochondria in the pathogenesis of idiopathic Parkinson disease (PD). A defect of respiratory chain complex I has been documented in the substantia nigra of postmortem PD brains,1 and the potent complex I inhibitor MPTP (N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) causes parkinsonism and nigrostriatal dopaminergic degeneration in humans.2 Exposing rats to the complex I inhibitor rotenone causes tremor and rigidity with associated loss of substantia nigra neurons and aggregation of α-synuclein, a major constituent of Lewy bodies, which characterize the neuropathology of PD.3 Furthermore, several monogenic forms of PD are due to mutations in genes coding for mitochondrial proteins or mitochondrial maintenance/autophagy (including PARK2,4 PINK1,5 and DJ5), which compromise oxidative phosphorylation.
Mitochondrial DNA (mtDNA) codes for 13 respiratory chain proteins that are essential for the synthesis of the intracellular energy source adenosine triphosphate. Point mutations of mtDNA compromise oxidative phosphorylation and cause severe multisystem neurologic disorders with extrapyramidal features resembling PD, raising the possibility that more subtle polymorphic variants contribute to genetic susceptibility to PD.
mtDNA is strictly maternally inherited, and mutations acquired throughout human history have subdivided the population into discrete “haplogroups,”6 typically 5,000 to 10,000 years old and each harboring many additional single nucleotide polymorphisms (SNPs).6 Several studies have reported an association of mtDNA haplogroups and specific mtDNA SNPs with PD.7−16 These studies have consistently shown an association between mtDNA and PD, but the precise haplogroup or SNP associations have differed and have often been contradictory (table 1). These inconsistencies mean that the role of inherited mtDNA variants in PD has yet to be fully established.
Table 1.
Previously reported significant associations between mitochondrial DNA and Parkinson diseasea
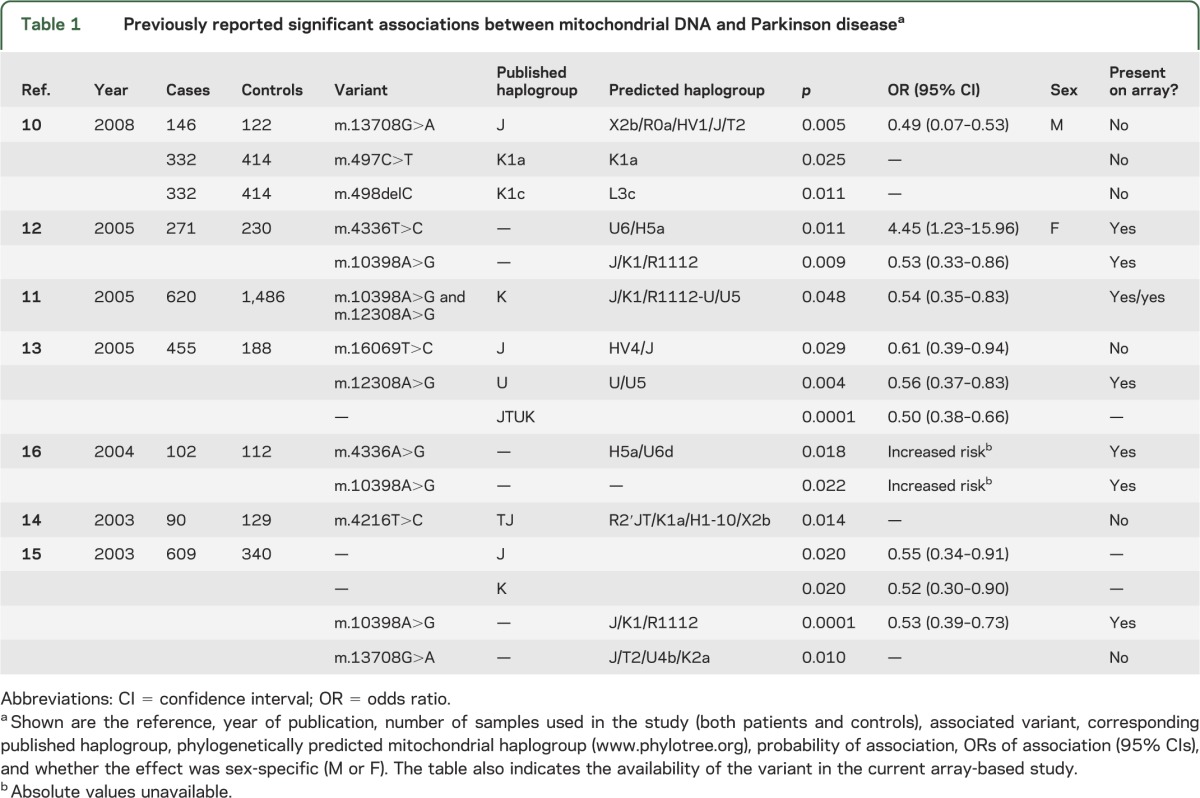
Herein, we report a 2-stage association study of inherited mtDNA variants in PD, which is the largest study to date. Our results confirm earlier haplogroup findings and provide further resolution of the genetic association. In addition, a meta-analysis of the available published data firmly establishes mtDNA as a major determinant of genetic risk in PD, even in the context of reported nuclear genome-wide associations.17
METHODS
We studied 138 mitochondrial variants in 3,074 PD cases and 5,658 ethnically matched controls from the United Kingdom. All samples were Caucasian. We performed a 2-stage genetic association study with discovery and replication phases. In the discovery phase, 2,197 cases (PD1, previously described18) were compared with 2,930 controls from the Wellcome Trust Case Control Consortium–1958 Birth Cohort (WTCCC-58C). In the replication phase, 877 cases (PD2, previously described19) were compared with 2,729 controls from the UK National Blood Service (WTCCC-NBS). Discovery cases (PD1) were genotyped using the Illumina 610K quad array (Illumina, San Diego, CA) and replication cases (PD2) were genotyped using the Affymetrix 550K array (Affymetrix Inc., Santa Clara, CA). All controls were genotyped on the Illumina 1.2M Duo platform.
Stringent quality control (QC) filters were applied to remove poorly performing samples using tools implemented in PLINK v2.05020 as previously described.21 Individuals were excluded from subsequent analysis if data were absent in >10% of SNPs21 (discovery phase, PD1 cases = 472 and WTCCC-58C = 36; replication phase, PD2 = 1 and WTCCC-NBS = 8). After sample QC, 1,725 PD cases and 2,894 controls remained in the discovery phase and 876 PD cases and 2,721 controls remained in the replication phase.
SNPs were excluded if >10% of genotypes were absent21 (discovery = 13 and replication = 6). We also excluded SNPs with a study-wide missing data rate of >5% (if minor allele frequency [MAF] was >5%) or >1% study-wide missing data rate for SNP MAF of <5%21 (discovery = 3 and replication = 4); additionally, SNPs with MAF of <1% were removed (discovery = 43 and replication = 49). Finally, to verify the quality of genotypes, cluster plots of normalized intensity for each SNP were generated using R (http://www.R-project.org) and inspected. To exclude the potential for differential array-based genotyping artifacts, SNP frequencies and subsequent mtDNA haplogroup frequencies were compared between PD1 and PD2 using PLINK v2.050.20 Variants showing significant (p > 0.05) population differences were excluded.
Subsequent analysis was restricted to a concordant dataset of 77 mtDNA variants passing QC in both the discovery and replication phases. Included in the final analysis were m.4667T>C, m.10398A>G, and m.12308A>G, all of which have been previously associated with PD (table 1). Differential missingness tests, which statistically compare the frequency of “missing” genotype data between cases and controls,21 revealed differences in 4 SNPs (p = <10−4), reducing the final number of experimental SNPs to 74. Statistical significance was defined as p < 0.05 in both the discovery and replication phases. Missingness and χ2 test were computed using PLINK v2.050.20
mtDNA haplogroup determination.
The hierarchical relationship among mtDNA variants is represented at www.phylotree.org. Each array variant (n = 74 post-QC) in each subject (n = 8,215 post-QC) was compared with the revised Cambridge Reference Sequence and used to identify haplogroup-specific motifs. Assignment of haplogroup was performed according to published criteria6; for example, haplogroup J is defined by variants m.295 and m.489 (table e-1 on the Neurology® Web site at www.neurology.org). If a no-call was detected in a major haplogroup-defining SNP, then clade-specific subtype variants were used to identify the mtDNA haplogroup. Based on available genotyping data, 99.51% of subjects were assigned to a major European haplogroup. As a final QC metric to remove the effects of phylogenetic heterogeneity, non-European samples (i.e., non-HVJTUKWXI and O haplogroups) were removed from subsequent analysis (discovery: cases = 6 and controls = 5; replication: cases = 25 and controls = 4). Subsequent statistical analysis was based on finalized discovery (PD1 = 1,719 vs WTCCC-58C = 2,889) and replication cohorts (PD2 = 851 vs WTCCC-NBS = 2,717).
Meta-analysis.
A meta-analysis of all available mtDNA haplogroup and PD studies7–15 was conducted using Comprehensive Meta-Analysis (version 2, www.meta-analysis.com).22 In total, 9 published studies (total cases = 3,030 and total controls = 4,862) were combined with our larger dataset (discovery: cases = 1,719 and controls = 2,889; replication: cases = 851 and controls = 2,717).
Standard protocol approvals, registrations, and patient consents.
We received approval from the Northumberland Local Research Ethics Committee (REF 05/Q0902/51) to conduct a genetic association study of PD. Written informed consent was obtained from the patients in all cases.
RESULTS
SNP association.
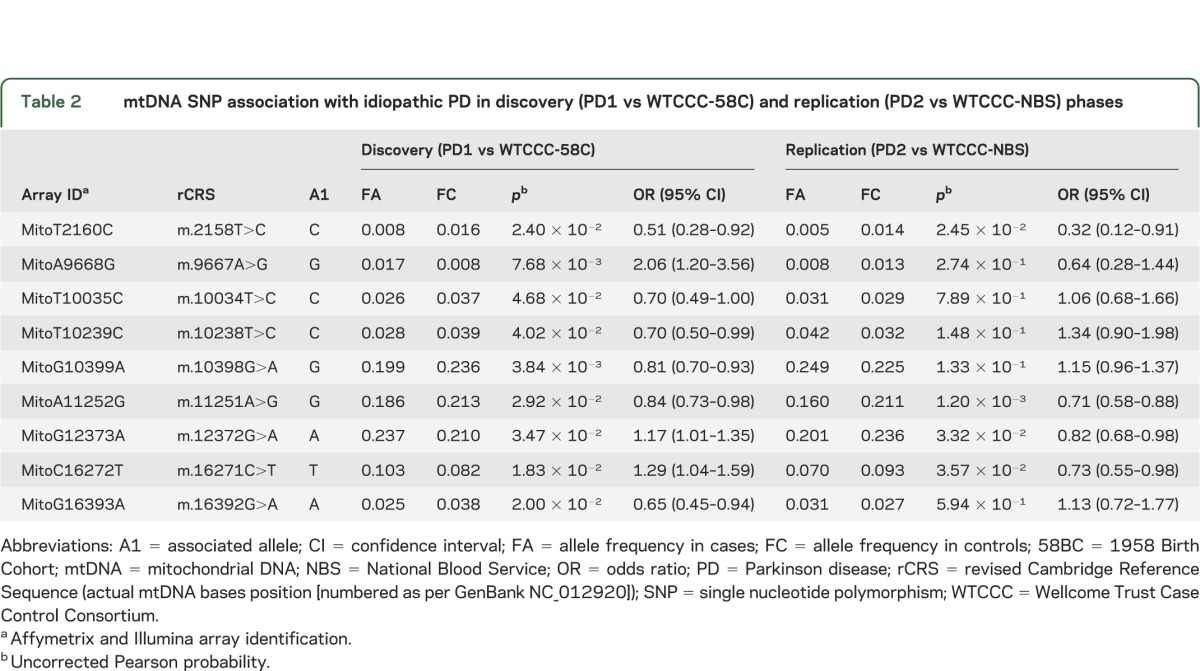
In the discovery cohort, we identified 9 mtDNA variants associated with PD (table 2). Two of these associations were also seen in the replication cohort at the same order of magnitude. Both m.2158T>C (discovery: p = 2.40 × 10−2 and odds ratio [OR] = 0.51; replication: p = 2.45 × 10−2 and OR = 0.32) and m.11251A>G (discovery: p = 2.92 × 10−2 and OR = 0.84; replication: p = 1.20 × 10−3 and OR = 0.71) were associated with a reduced the risk of PD. We found no evidence of a consistent association between PD and m.4667T>C, m.12308A>G, or m.10398A>G, either in the individual cohorts or the combined cohort.
Table 2.
mtDNA SNP association with idiopathic PD in discovery (PD1 vs WTCCC-58C) and replication (PD2 vs WTCCC-NBS) phases
Haplogroup associations.
A comparison of the major European mtDNA haplogroups did not identify a consistent association with PD in the discovery, replication, or combined cohorts (table e-2). However, given previous reports of a reduced risk of PD in subjects belonging to “super-haplogroup JT,” we combined the data from haplogroups J and T. The pooled analysis identified an association with the super-haplogroup JT (p = 3.54 × 10−2, OR = 0.87, 95% CI = 0.77–0.97).
Meta-analysis.
A meta-analysis revealed a significant reduction in PD risk for haplogroups J (p = 1.22 × 10−2, OR = 0.88), T (p = 2.45 × 10−2, OR = 0.87), and K (p = 3.63 × 10−3, OR = 0.84) (figure 1). Combining haplogroups J and T increased the strength of the association but did not change the effect size (p = 5.84 × 10−4, OR = 0.85) (figure 2). When studied in isolation, a meta-analysis of haplogroups H and V failed to show an effect on PD risk (figure e-1). However, a significant increase in the risk of PD was identified when consolidated to the “super-haplogroup” HV (p = 3.64 × 10−3, OR = 1.112) (figure 2). No association was identified between haplogroups U, W, X, and I when analyzed in isolation, or when grouped into phylogenetically related super-haplogroups (figure e-1).
Figure 1. Meta-analysis of previously reported PD and mtDNA haplogroup associations and the data from this study.

Meta-analysis of our data and published European mtDNA haplogroup vs PD associations in isolation. Discovery = Hudson et al., PD1; replication = Hudson et al., PD2. Boxes are proportional to study size, and fixed-model meta-analysis is denoted by a diamond symbol. CI = confidence interval; L95 = lower 95% CI; mtDNA = mitochondrial DNA; OR = odds ratio; PD = Parkinson disease; U95 = upper 95% CI.
Figure 2. Meta-analysis of previously reported PD and mtDNA “super-haplogroup” associations and the data from this study.

Meta-analysis of our data and published European mtDNA haplogroup vs PD associations when combined into super-haplogroups. Discovery = Hudson et al., PD1; replication = Hudson et al., PD2. Boxes are proportional to study size, and fixed-model meta-analysis is denoted by a diamond symbol. CI = confidence interval; L95 = lower 95% CI; mtDNA = mitochondrial DNA; OR = odds ratio; PD = Parkinson disease; U95 = upper 95% CI.
DISCUSSION
Our 2-phased approach identified a consistent association between PD and both m.2158T>C and m.11251A>G. These 2 SNPs are phylogenetically linked, with m.11251A>G defining the mtDNA super-haplogroup JT and m.2158T>C residing on the J-specific subclade (J1b).23 These findings are consistent with the association we observed with super-haplogroup JT, confirm some previous reports, and, more importantly, resolve this association at the subhaplogroup level (J1b). Variant m.11251A>G does not result in an amino acid substitution (MTND4 L164L) and is therefore unlikely to directly contribute to disease pathogenesis. However, m.2158T>C is present in the highly heterogeneous mitochondrial 16s ribosomal RNA gene (MTRNR2). Although it is conceivable that m.2158T>C could alter intramitochondrial synthesis, there are other possibilities. MTRNR2 shows 99% sequence homology to Humanin, a molecule shown to suppress neurotoxicity in Alzheimer disease, leading to the suggestion that Humanin is actually produced from MTRNR2 within the mitochondrion.24,25 It is therefore possible that common mtDNA variation in MTRNR2 has a direct neuroprotective effect.
Despite >99% power to detect the previously reported findings, we were unable to consistently replicate the previous associations to m.4336T>C, m.10398A>G, or m.12308A>G. It is therefore unlikely that these variants are relevant to the etiology of PD. Differences in sample size provide the likely explanation for the discrepancies, with our combined cohorts being >4-fold greater than the largest mtDNA study published to date in PD. We were unable to test the direct association with 6 of the previously associated mtDNA variants because they were not present on the genotyping array; however, the phylogenetic nature of mtDNA allowed us to make direct haplogroup comparisons to these studies using correlated mtDNA variants (table e-1). Combining data from 10 studies (6,140 PD cases and 13,280 controls) confirmed the JT association and also provided sufficient statistical power to show a protective effect from haplogroups J, K, and T. Each one of these haplogroups had been associated with a reduced risk of PD previously but not in the same study. These disparate findings suggested that the associations were spurious false-positive results. However, the results of our meta-analysis indicate that the associations are likely to be robust, with the limited sample sizes in previous studies explaining the inconsistencies.
What are the functional consequences of our findings? mtDNA haplogroups have been shown to modulate the efficiency of adenosine triphosphate synthesis or the production of reactive oxygen species,26 and these effects could directly modulate neuronal protection rather than damage. It also likely that haplogroups J, T, and K are genetic tags of further mtDNA variation not directly genotyped in this study. For example, haplogroup K contains the lowest frequency of nonsynonymous complex I gene variants (compared with the other haplogroups), raising the possibility that natural selection against genetic variation of complex I reduces the risk of PD in haplogroup K individuals.
Conversely, by pooling all of the data, we were able to show that super-haplogroup HV is associated with an increased risk of PD. Whether the increased risk of PD in HV is directly attributable to functional variants within that haplogroup or simply reflects the “mirror image” of the reduced risk of PD in J, K, and T remains to be established. However, it is intriguing that haplogroup H has been associated with an increased propensity to survive severe infection.27 This raises the possibility that natural selection has led to the emergence of genetic variants that predispose toward neurodegenerative disease through antagonistic pleiotropy. Similar to Huntington disease, whereby an increased CAG(n) repeat length in the huntingtin gene causes a neurodegenerative disorder but is associated with a lower incidence of cancer, recent mutations in mtDNA may be tolerated in humans because they increase the chance of surviving early-life insults such as sepsis, but are pathogenic in later life through the increased generation of reactive oxygen species.
Finally, recent data suggest that α-synuclein is localized to mammalian neuronal mitochondria and that its expression is associated with a decrease in complex I activity.28 It is also clear that functional mtDNA is required to modulate α-synuclein toxicity,29 but that overexpression of α-synuclein can result in mitochondrial dysfunction.30 It is therefore likely that α-synuclein accumulation and mtDNA variation interact synergistically to affect neuronal complex I activity. The resultant loss of activity could lead to an increase in reactive oxygen species and a reduction in cellular energy, culminating in the initiation of apoptotic pathways and eventual neuronal cell death.
Supplementary Material
ACKNOWLEDGMENT
This study utilized GWA data generated by the Wellcome Trust Case Control Consortium 2 on UK PD cases and on UK controls from the 1958 Birth Cohort and National Blood Service, and samples and data collected by the International Parkinson’s Disease Genomics Consortium. A full list of consortium members is available online (see Contributors on the Neurology® Web site at www.neurology.org).
GLOSSARY
- 58BC
1958 Birth Cohort
- MAF
minor allele frequency
- mtDNA
mitochondrial DNA
- NBS
National Blood Service
- OR
odds ratio
- PD
Parkinson disease
- QC
quality control
- SNP
single nucleotide polymorphism
- WTCCC
Wellcome Trust Case Control Consortium
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Conceived and designed the experiments: G. Hudson, D. Burn, and P.F. Chinnery. Contributed reagents/materials/analysis tools: G. Hudson, M. Nalls, J.R. Evans, D.P. Breen, S. Winder-Rhodes, K.E. Morrison, H.R. Morris, C.H. Williams-Gray, Wellcome Trust Case Control Consortium 2, R.A. Barker, A.B. Singleton, J. Hardy, N.E. Wood, D.J. Burn, and P.F. Chinnery. Wrote the manuscript: G. Hudson and P.F. Chinnery.
STUDY FUNDING
G.H. is a Parkinson’s UK Senior Fellow. P.F.C. is a Wellcome Trust Senior Fellow in Clinical Science and an NIHR Senior Investigator who also receives funding from the Wellcome Centre for Mitochondrial Research, the Medical Research Council (UK) Translational Muscle Centre, the UK NIHR Biomedical Research Centre for Ageing and Age-Related Disease, and NIHR Dementia Biomedical Research Unit awards to the Newcastle upon Tyne Foundation Hospitals NHS Trust. M.N. and A.S. are supported in part by the Intramural Research Program of the National Institute on Aging, NIH, Department of Health and Human Services, project number Z01 AG000949-06. D.P.B. is supported by Parkinson's UK and has been the recipient of a Raymond and Beverly Sackler studentship.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Schapira AH, Cooper JM, Dexter D, et al. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1989;1:1269. [DOI] [PubMed] [Google Scholar]
- 2.Langston JW, Forno LS, Tetrud J, et al. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann Neurol 1999;46:598–605 [DOI] [PubMed] [Google Scholar]
- 3.Betarbet R, Sherer TB, MacKenzie G, et al. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 2000;3:1301–1306 [DOI] [PubMed] [Google Scholar]
- 4.Matsumine H, Saito M, Shimoda-Matsubayashi S, et al. Localization of a gene for an autosomal recessive form of juvenile parkinsonism to chromosome 6q25.2-27. Am J Hum Genet 1997;60:588–596 [PMC free article] [PubMed] [Google Scholar]
- 5.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004;304:1158–1160 [DOI] [PubMed] [Google Scholar]
- 6.Torroni A, Huoponen K, Francalacci P, et al. Classification of European mtDNAs from an analysis of three European populations. Genetics 1996;144:1835–1850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Simon DK, Mayeux R, Marder K, et al. Mitochondrial DNA mutations in complex I and tRNA genes in Parkinson's disease. Neurology 2000;54:703–709 [DOI] [PubMed] [Google Scholar]
- 8.Mehta P, Mellick GD, Rowe DB, et al. Mitochondrial DNA haplogroups J and K are not protective for Parkinson's disease in the Australian community. Mov Disord 2009;24:290–292 [DOI] [PubMed] [Google Scholar]
- 9.Latsoudis H, Spanaki C, Chlouverakis G, et al. Mitochondrial DNA polymorphisms and haplogroups in Parkinson's disease and control individuals with a similar genetic background. J Hum Genet 2008;53:349–356 [DOI] [PubMed] [Google Scholar]
- 10.Gaweda-Walerych K, Maruszak A, Safranow K, et al. Mitochondrial DNA haplogroups and subhaplogroups are associated with Parkinson's disease risk in a Polish PD cohort. J Neural Transm 2008;115:1521–1526 [DOI] [PubMed] [Google Scholar]
- 11.Ghezzi D, Marelli C, Achilli A, et al. Mitochondrial DNA haplogroup K is associated with a lower risk of Parkinson's disease in Italians. Eur J Hum Genet 2005;13:748–752 [DOI] [PubMed] [Google Scholar]
- 12.Huerta C, Castro MG, Coto E, et al. Mitochondrial DNA polymorphisms and risk of Parkinson's disease in Spanish population. J Neurol Sci 2005;236:49–54 [DOI] [PubMed] [Google Scholar]
- 13.Pyle A, Foltynie T, Tiangyou W, et al. Mitochondrial DNA haplogroup cluster UKJT reduces the risk of PD. Ann Neurol 2005;57:564–567 [DOI] [PubMed] [Google Scholar]
- 14.Ross OA, McCormack R, Maxwell LD, et al. mt4216C variant in linkage with the mtDNA TJ cluster may confer a susceptibility to mitochondrial dysfunction resulting in an increased risk of Parkinson's disease in the Irish. Exp Gerontol 2003;38:397–405 [DOI] [PubMed] [Google Scholar]
- 15.van der Walt JM, Nicodemus KK, Martin ER, et al. Mitochondrial polymorphisms significantly reduce the risk of Parkinson disease. Am J Hum Genet 2003;72:804–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otaegui D, Paisan C, Saenz A, et al. Mitochondrial polymorphisms in Parkinson's disease. Neurosci Lett 2004;370:171–174 [DOI] [PubMed] [Google Scholar]
- 17.Simon-Sanchez J, Schulte C, Bras JM, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet 2009;41:1308–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fung HC, Scholz S, Matarin M, et al. Genome-wide genotyping in Parkinson's disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 2006;5:911–916 [DOI] [PubMed] [Google Scholar]
- 19.Spencer CC, Plagnol V, Strange A, et al. Dissection of the genetics of Parkinson's disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 2011;20:345–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 2007;447:661–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borenstein M, Hedges L, Higgins J, Rothstein H. Comprehensive Meta-Analysis Version 2. Englewood, NJ: Biostat; 2005 [Google Scholar]
- 23.van Oven M, Kayser M. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 2009;30:E386–E394 [DOI] [PubMed] [Google Scholar]
- 24.Maximov V, Martynenko A, Hunsmann G, et al. Mitochondrial 16S rRNA gene encodes a functional peptide, a potential drug for Alzheimer's disease and target for cancer therapy. Med Hypotheses 2002;59:670–673 [DOI] [PubMed] [Google Scholar]
- 25.Tajima H, Niikura T, Hashimoto Y, et al. Evidence for in vivo production of Humanin peptide, a neuroprotective factor against Alzheimer's disease-related insults. Neurosci Lett 2002;324:227–231 [DOI] [PubMed] [Google Scholar]
- 26.Gomez-Duran A, Pacheu-Grau D, Lopez-Gallardo E, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups. Hum Mol Genet 2010;19:3343–3353 [DOI] [PubMed] [Google Scholar]
- 27.Baudouin SV, Saunders D, Tiangyou W, et al. Mitochondrial DNA and survival after sepsis: a prospective study. Lancet 2005;366:2118–2121 [DOI] [PubMed] [Google Scholar]
- 28.Liu G, Zhang C, Yin J, et al. alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett 2009;454:187–192 [DOI] [PubMed] [Google Scholar]
- 29.Buttner S, Bitto A, Ring J, et al. Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J Biol Chem 2008;283:7554–7560 [DOI] [PubMed] [Google Scholar]
- 30.Hsu LJ, Sagara Y, Arroyo A, et al. alpha-Synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol 2000;157:401–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.