

Abstract
Antibiotic discovery has a storied history. From the discovery of penicillin by Sir Alexander Fleming to the relentless quest for antibiotics by Selman Waksman, the stories have become like folklore, used to inspire future generations of scientists. However, recent discovery pipelines have run dry at a time when multidrug resistant pathogens are on the rise. Nature has proven to be a valuable reservoir of antimicrobial agents, which are primarily produced by modularized biochemical pathways. Such modularization is well suited to remodeling by an interdisciplinary approach that spans science and engineering. Herein, we discuss the biological engineering of small molecules, peptides, and non-traditional antimicrobials and provide an overview of the growing applicability of synthetic biology to antimicrobials discovery.
Keywords: synthetic biology, antimicrobial discovery, antibiotics, genetic engineering, protein engineering, biological engineering, bacteriophage
Introduction
The discovery of antibiotics represents one of our greatest scientific achievements, for few other discoveries have had as great an impact on medicine. The rich history of antibiotic discovery can be traced back for over a century, where great intuition was used to recognize that although all microbial pathogens eventually find their way in to the soil, whether through biological decay or animal excrement, the soil is not the source of most infectious diseases.1, 2 This led to an expansive search of soil-dwelling microorganisms for antimicrobial agents culminating in the `golden era' of antibiotic discovery during the mid-twentieth century, where most known classes of antibiotics were discovered.3 Environmental bacteria and fungi have been a great source of bioactive molecules, in particular those with antimicrobial activities. It is estimated that approximately 16,500 antibiotics have been isolated as natural products, the majority of which originate from soil-dwelling Gram-positive bacteria of the order Actinomycetales (Fig. 1a4).5
Fig. 1.
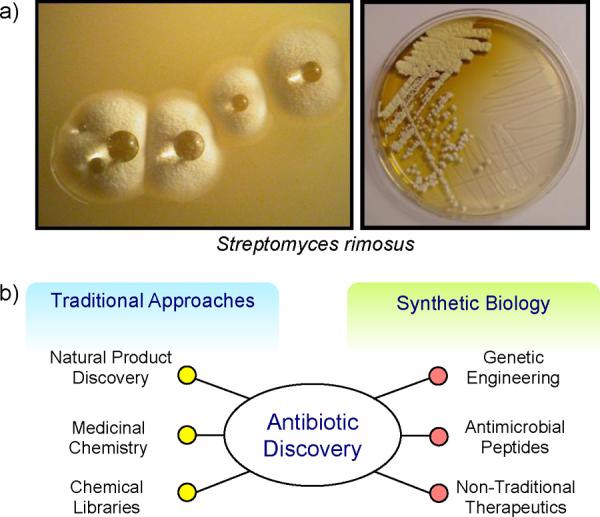
Antibiotic discovery. a) Streptomyces rimosus,4 a soil-dwelling Gram-positive bacteria that produces oxytetracycline, which is visible as a yellow halo around bacterial colonies (right). b) An overview of the approaches utilized for antibiotic discovery.
Although antibiotics are very effective therapeutics that can be used to target diverse species of bacteria, they are highly susceptible to adaptive and environmentally acquired bacterial resistance. This can severely limit their clinical lifetime and render once powerful therapeutics obsolete. Adaptive resistance occurs as a result of changes in gene and protein expression in response to environmental cues and can lead to transient resistance of bacterial populations.6 Alternatively, environmental bacteria harbor reservoirs of resistance genes, where the average bacterium has resistance against 7–8 different antibiotics with varying modes of action.7 As such, there is a large pool of resistance cassettes present in the environment that can be passed on horizontally to infectious bacteria. In fact, such horizontal gene transfer between soil-dwelling bacteria and clinical pathogens was recently demonstrated experimentally, where resistance determinants with significant nucleotide identity were found among diverse species of environmental and pathogenic bacteria.8 Therefore, to combat adaptive and environmentally acquired bacterial resistance, there must be continuous development of novel antibiotics via new discoveries and re-engineering of old therapeutics.
Traditional antibiotic discovery programs have relied on the mining of environmental microbes for natural product biosynthesis via secondary metabolic pathways (Fig. 1b). However, after more than half a century of such investigations, new discoveries are becoming ever more rare. The alleged `low-hanging fruit', or the commonly produced and easily isolated natural product antibiotics, have been picked and it has become exceedingly difficult to find new antimicrobial agents.9, 10 A significant hurdle to overcome with current natural product discovery initiatives is dereplication, the process of rapidly discounting previously identified secondary metabolites.11 Considering that approximately 1% of Actinomycetes produce the antibiotic streptomycin,10 it can be easily understood why an efficient and high-throughput approach is required to prevent the re-discovery of old antibiotics. Alternative approaches to antibiotic discovery have included the use of medicinal chemistry to expand the chemical diversity of commonly utilized antimicrobials.3 Although the total synthesis of natural products is not feasible due to the complexity of the molecules,12, 13 the chemical modification of antibiotic scaffolds to evade resistance has been quite successful, as seen for example with the second- and third-generation tetracyclines.14 Another widely used drug discovery approach has been the screening of large chemical libraries for antimicrobial activity. However, this approach has achieved little success as synthetic chemical libraries cannot mimic the structural complexity inherent to bioactive molecules.15
Although the discovery of new antibiotics continues to become ever more difficult owing to an exhausted scientific approach and a drop in industry participation, the need for new antimicrobials continues to grow in the clinic due to the emergence of multidrug resistant pathogens, as exemplified by methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE). To tackle these new challenges, multi-disciplinary approaches are required that can take advantage of the efficiency and versatility of biological platforms within a user defined context. The nexus between such a biological and engineering approach is synthetic biology.
By modularizing biological elements using genetic and protein engineering, synthetic biologists are able to incorporate new functionality within pre-existing biological platforms to sample new chemical space. Herein, synthetic biology approaches to antibiotic development will be discussed in the context of genetic engineering for small-molecule development, peptide antimicrobials, and non-traditional therapeutics.
Small-Molecule Antibiotics
Small-molecule antibiotics represent the largest class of antimicrobial agents and include both natural products and synthetic molecules that encompass a diverse array of molecular architectures. Most small molecule antibiotics are synthesized as natural products by environmental microbes using simple building blocks, which are assembled into elaborate structures via secondary metabolic pathways. These large biochemical pathways possess inherent modularity that make them attractive platforms for synthetic biology.16
Secondary Metabolic Pathways
Synthetic biology strides to implement a modular design for the engineering of biological molecules.17–21 Such approaches can be applied to engineer the secondary metabolic biosynthetic pathways of Actinomycetes. These soil-dwelling microbes can be thought of as chemical factories that produce secondary metabolites in a `conveyor-belt'-like fashion.14 Such versatility is afforded by the use of large enzymatic complexes that allow for the coordinated action of many different enzymes to build complex small molecules from basic building blocks.22
The polyketide class of secondary metabolites, which include the clinically relevant macrolide and tetracycline antibiotics, are synthesized by large enzymatic complexes called polyketide synthases (PKS). A detailed overview of the biochemistry of PKS pathways is beyond the scope of this review and the interested reader is referred to other reviews.22–26 Although there are different types of PKS enzymes with varying mechanistic complexities, the basic process of polyketide assembly follows a similar path. The carbon backbone of a polyketide molecule is assembled by a designated PKS complex through sequential or iterative condensation of acyl-CoA building blocks.22, 27 Subsequently, the polyketide backbone is decorated with a variety of different functional groups such as sugars, alcohols, aromatic rings, methyl groups, and amino groups via the action of specific tailoring enzymes.25, 26 These functional groups are responsible for mediating interactions that are key to the biological activity of polyketides.28 For example, even though there are three generations of tetracyclines with diverse chemical structures, the ribosome inhibitory action of tetracyclines are imparted by only the keto-enol functionalities at the base of the molecules.29, 30 Therefore, at the latter stages of tetracycline biosynthesis, synthetic biology could be utilized to increase the chemical diversity of natural products by modulating tailoring reactions. By taking advantage of the modular nature of PKS and tailoring enzymes, one can mix-and-match them to develop new chemical entities with engineered biological platforms.
Type I PKS Assembly: Erythromycin
The biosynthetic assembly of the macrolide antibiotic erythromycin produced by Saccharopolyspora erythraea represents the best studied PKS pathway. A type I PKS produces erythromycin, where three mega-enzymes (DEBS-1, DEBS-2, and DEBS-3) constituting 7 modules and 28 enzymatic domains catalyze the production of 6-deoxyerthronolide B (6-DEB), the aglycone scaffold of erythromycin (Fig. 2a).23 6-DEB production proceeds in an assembly-line fashion where at each stage one acyl-CoA intermediate is incorporated (1 propionyl-CoA and 6 methylmalonyl-CoA units).31 Following polyketide assembly, 6-DEB is decorated by regiospecific hydroxylation and glycosylation reactions via dedicated tailoring enzymes.31, 32
Fig. 2.

PKS assembly of natural products. a) Type I PKS assembly. Erythromycin is produced 3 mega-enzyme complexes (DEBS-1, DEBS-2, and DEBS-3) that constitute 7 individual domains (blue brackets). Subsequent to the assembly of the aglycone 6-DEB, tailoring enzymes incorporate hydroxyl and sugar moieties to produce erythromycin.31, 88 b) Type II PKS assembly. The oxytetracycline biosynthetic cluster is composed of 21 genes.47 The minimal PKS assembles the polyketide scaffold, which is then highly modified by tailoring enzymes to produce oxytetracycline. The `variable' side of tetracyclines represents regions in the molecule where modifications can be made to increase chemical diversity, whereas the `non-variable' side constitutes functional moieties required for biological activity and hence modifications are not tolerated.
The modular nature of the erythromycin biosynthetic machinery was apparent when the biosynthetic gene cluster was initially sequenced in 1990.33, 34 The repetitive utilization of standardized catalytic units provides opportunities to mix-and-match the enzymatic domains to sample new chemical space and create `unnatural' natural products. Engineering of unnatural erythromycin derivatives can be achieved at 2 distinct steps: during PKS assembly of the polyketide scaffold or during the tailoring reactions.
The first stage of erythromycin assembly involves loading the PKS with a starter unit. The loading module in DEBS-1 (N-terminal acyltransferase and acyl carrier protein) naturally loads a propionate unit onto the PKS. However, loading modules from the macrolide antibiotics oleandomycin or tylosin can be incorporated into DEBS-1 to provide control over starter unit specificity, namely a propionate or acetate starter unit.35 Alternatively, diversity can be introduced by swapping the methylmalonyl specific acyltransferase domains of DEBS with domains that have specificity for methoxymalonyl, ethylmalonyl, or malonyl extender units.36–39 Changing the number of modules present in the DEBS can also change the scaffold of erythromycin. For example, a three-module DEBS derivative results in the production of an 8-membered ring lactone.40
The final stage of erythromycin assembly involves the decoration of 6-DEB with hydroxyl and sugar group, which are required for biological activity.28, 32 At this stage, modularity can be observed in the substrate flexibility of the glycosyltransferase enzymes. For instance, EryBV catalyzes the incorporation of TDP-L-mycarose on to 6-DEB (Fig. 2a). However, it has been shown that EryBV is not only able to catalyze the reverse reaction as well, but it can also incorporate other TDP-L-sugar analogues on to the aglycon macrolide.41 Similarly, in addition to its native substrate TDP-D-desosamine, EryCIII is also able to incorporate other sugars on to the erythromycin scaffold including TDP-D-mycaminose and TDP-D-angolosamine.42, 43 Therefore, the substrate promiscuity of tailoring enzymes could provide a useful avenue for the development of unnatural polyketides.
Such modularity and substrate promiscuity at almost every stage of type I PKS polyketide assembly affords many exciting opportunities for a synthetic biological approach to drug development. In an innovative experiment to develop `unnatural' natural products, Menzella et al. validated this approach by dismantling type I PKS genes in to synthetic building blocks that could be reassembled in a combinatorial fashion.44 By using a standardized loading module, intra- and inter-peptide linkers, and a polyketide cleavage module, the authors used 8 different PKS clusters to synthesize 14 individual modules each encoded in an expression vector either as a donor or acceptor module. Subsequently, donor and acceptor modules were co-transformed into an Escherichia coli strain engineered to synthesize polyketide compounds resulting in 154 bimodular combinations. Intriguingly, all of the different synthetic components showed great compatibility, where 47% of the bimodular PKSs were active and produced the expected triketide lactone products. The bimodular PKSs were further engineered to accept alternate starter units to further expand product diversity.45 The same group further advanced this technology to develop combinatorial trimodule PKSs that were able to assemble tetraketide molecules.46 The trimodular PKSs achieved remarkable efficiency, where 96% of the combinations tested were able to produce the expected tetraketide molecules.
Thus, type I PKS enzymes have proven to be versatile platforms that can be manipulated in innovative ways in the quest for chemical diversity. This notion can be further extended to type II PKS enzymes, which are more disseminated and complex chemical assembly lines.
Type II PKS Assembly: Oxytetracycline
Unlike the type I PKS enzymes, where each module catalyzes the addition of one acyl-CoA unit to produce the polyketide backbone, type II PKS assembly involves a `minimal' PKS complex (acyl-carrier protein, chain length factor, and ketosynthase units) that iteratively catalyze the condensation of malonyl-CoA extender units.27, 47 Subsequently, the polyketides are modified by a host of tailoring enzymes including: aromatases, cyclases, oxygenases, methyltransferases, ketoreductases, acyltransferases, and aminotransferases.27, 47 Accordingly, a intriguing path to broaden the chemical diversity of these natural products would be via the tailoring reactions.
The tetracycline class of antibiotics represents one of the most medically significant natural products synthesized by type II PKS complexes.27 Tetracycline biosynthesis has been investigated for almost half a century starting with early studies involving blocked mutants.48–52 However, it is only recently that we are beginning to understand the detailed biochemistry of the biosynthetic pathway as a result of advances in genetic engineering and the availability of heterologous expression systems.47, 53
Streptomyces rimosus, first discovered at Pfizer in 1950, produces the tetracycline antibiotic oxytetracycline originally marketed as Terramycin® (Fig. 1a).4, 14, 54 Oxytetracycline is produced by a type II PKS encoded within a 24 kb biosynthetic gene cluster that contains 21 genes flanked by 2 resistance genes (Fig. 2b).47, 55. Oxytetracycline biosynthesis has recently been reviewed and as such will not be discussed here.56.
A lack of appropriate genetic engineering tools for S. rimosus stemming from genomic instability and inefficient DNA transformation procedures,55 has led to the use of heterologous hosts for the study of oxytetracycline biosynthesis. Although heterologous expression of PKS genes in E. coli leads to the formation of inclusion bodies,57Streptomyces coelicolor CH999 and Myxococcus xanthus have been successfully used for the heterologous expression of pathway genes.47, 58 In a series of studies over the past few years, Tang and co-workers have demonstrated the modular nature of the oxytetracycline pathway enzymes by reconstituting the oxytetracycline minimal PKS in combination with varying tailoring enzymes in S. coelicolor CH999. This has led to the production of a variety of tetracycline analogues and pathway intermediates.47, 53, 59, 60
A rational synthetic biology approach to tetracycline production would be fundamentally different than the combinatorial methods used with type I PKS pathways. Through the studies of tetracycline analogues and tetracycline-ribosomal structural data, it has been well established that the keto-enol functionalities on the `non-variable' side of tetracyclines make key contacts to the ribosome and are essential for biological activity, whereas the `variable' side of the molecule affords opportunities for great chemical diversity (Fig. 2b).14, 29, 30, 61, 62 Accordingly, different combinations of the oxytetracycline biosynthetic enzymes (or engineered versions with enhanced substrate promiscuity) can be used to develop analogues that contain chemically modifiable functional groups on the `variable' side. This could provide opportunities to introduce new chemical moieties that do not interfere with ribosomal binding, but do disrupt molecular interactions between tetracyclines and efflux proteins (the main mode of clinical resistance), because of an altered molecular shape.14, 63
Synthetic Biology: Tools of the Trade
As with any ambitious engineering project, we are often at the mercy of the diversity and quality of tools at our disposal or a lack thereof. However, the great advances made in synthetic biology over the past decade have provided us with powerful tools to rewire natural networks. The development of recombineering technology based on the λ-Red system over a decade ago laid the foundation for the simple and efficient swapping of genetic material in a seamless fashion.64, 65 By simply expressing exo, bet, and gam genes from the bacteriophage λ-Red recombination system, one can efficiently induce the recombination of homologous DNA elements between chromosomal/plasmid DNA and transformed linear DNA containing as little as 30–50 bp of homology.64, 66 Furthermore, recombineering can also be performed with single stranded DNA oligonucleotides as short as 30 bp to provide a highly efficient method by which one can mutagenize genomic DNA.67 Recombineering has revolutionized genetic engineering and is commonly used to modify natural product biosynthetic pathways.
Additional advances in DNA synthesis technologies68 and DNA assembly techniques (Gibson,69 Biobricks,70 BglBricks,71 Golden Gate shuffling,72 Circular Polymerase Extension Cloning73) have provided facile solutions for the genetic manipulation of large secondary metabolic gene clusters. Natural product biosynthesis requires the coordinated action of many enzymes and subsequent to DNA assembly, the expression of individual genes must be tuned to achieve maximum efficiency. The genomic evolution MAGE (Multiplex Automated Genome Engineering) technology is a powerful method to achieve this. By using 90 bp oligonucleotides, one can simultaneously and rapidly target multiple short DNA elements for directed evolution.74 In a demonstration of the efficiency of MAGE for biosynthetic pathway tuning, Wang et al. optimized lycopene production in E. coli by evolving 24 genes, where 20 ribosome binding sites were tuned and 4 genes were inactivated. This resulted in a variant that produced 5-fold more lycopene than the parental strain.74 Recently, Carr et al. have improved on this platform by developing co-selection MAGE, where a selectable marker (turned off) is placed within 500 kb of sites targeted for evolution.75 By turning on the selectable marker while simultaneously evolving multiple genomic targets, sequence diversity can introduced more efficiently.75 This approach is highly relevant to the tuning of PKS gene clusters, since individual operons are located in close proximity and antibiotic resistance genes (selectable marker) are generally present within each respective biosynthetic cluster.
Additionally, protein engineering and directed evolution for tuning of polypeptide sequences are well-established arts that have been around for decades. Techniques such as rational protein modifications based on structure-activity relationships can be used to engineer enzymes with altered substrate specificities. Notably, by modifying two active site residues of the serine protease subtilisin, the Wells groups were able to produce the derivative subtiligase that catalyzes peptide bond formation rather than hydrolysis.76, 77 For more combinatorial sequence optimization approaches, display technologies such as phage,78 bacterial,79 yeast,80 ribosome,81 or DNA82 display can be used, among which phage display is most widely utilized. Recently, Liu and co-workers further improved the efficiency of phage display by developing phage-assisted continuous evolution (PACE), where directed evolution can be accomplished in a continuous and automated process with minimum researcher intervention.83 Furthermore, at the post-translational level proteins can also be ligated together much like DNA, to develop the `conveyor belt' structures of PKS enzymes or to co-localize the many different tailoring enzymes, especially within heterologous hosts. Intein trans-splicing can be used to seamlessly link two polypeptide strands, although the reactions are dependent on the redox status of the environment and can produce undesired side products.84, 85 Recently, Zakeri et al. developed short peptide tags (isopeptag and SpyTag) that can be used to covalently and irreversibly link polypeptide strands via spontaneous isopeptide bond formation to highly specific protein partners.86, 87 Importantly, the peptide-protein pairs are genetically encodable and can therefore be used to rapidly and spontaneously link proteins both in vitro and in vivo.86, 87 This technology creates `protein building blocks' to forge peptide linkages in both linear and branched configurations to develop novel protein architectures. Consequently, the synthetic biological tools at both the DNA and protein levels are available to not only modify natural secondary metabolic pathways, but also to engineer completely synthetic ones as well.
Antimicrobial peptides
Peptides represent the other major class of biopolymers that serve as scaffolds for antimicrobials. Peptide antimicrobials can be separated into two major classes: nonribosomal peptides (NRPs) and antimicrobial peptides (AMPs). Similar to polyketides, NRPs are small molecule antibiotics that are produced by environmental microbes using both proteinogenic and nonproteinogenic amino acids.88 Alternatively, AMPs are produced by multi-cellular organisms as part of their immune systems using the standard 20 amino acids.89 Both classes of peptide antibiotics are assembled through pathways that are suitable for engineering.
Nonribosomal Peptides
Nonribosomal peptides comprise some of the most clinically important drugs including β-lactams, glycopeptides, and lipopeptides.90 These natural products are most commonly produced by nonribosomal peptide synthetases (NRPSs), which are the molecular assembly lines that fabricate peptide antibiotics using both natural and non-natural amino acid building blocks.88 The biochemistry of NRPS pathways have been studied in great detail and are reviewed elsewhere.91–93 Briefly, the general mechanism of NRPS assembly closely resembles that of the type I PKS machinery, where large multi-domain complexes catalyze the sequential addition of amino acids in to the growing peptide chain.94 For example, the heptapeptide core of the glycopeptide antibiotic chloroeremomycin is produced by three large enzymatic complexes containing a total of 7 modules, each responsible for the incorporation of 1 amino acid. Subsequently, tailoring enzymes modify the peptide scaffold via methylation, oxidative cross-linking, and glycosylation reactions.91, 95 Since the mechanism of NRPS and type I PKS assembly is similar, the same general synthetic biology principles can be applied to modify NRPS complexes to expand chemical diversity. Accordingly, the modularization of NRPS pathways will only be briefly discussed.
Interestingly, there exist natural PKS-NRPS hybrid biosynthetic pathways that demonstrate the compatibility of the two natural product assembly machineries. To illustrate, the antibiotic bacillaene is produced by a 2.5 MDa PKS-NRPS hybrid complex (13 PKS modules and 3 NRPS modules) in Bacillus subtilis that is encoded on an ±80 kb biosynthetic cluster.96 Consequently, both polyketides and amino acids can be used as building blocks by the same protein machinery thereby allowing for a new category of chemicals that can be engineered for antibiotic development. Intriguingly, the PKS-NRPS hybrid complexes in the bacterium further associate and co-localize adjacent to the cell membrane to produce `biosynthetic factories' that are 10–100 MDa in size.97 This unusual arrangement of biosynthetic enzymes can allow for the engineering of cells that can redirect energy resources for antibiotic production by rewiring natural pathways, thereby significantly reducing the cost of drug development.
Since NRPS systems use natural and unnatural amino acids as building blocks that are then linked by stable peptide bonds, one can sample a more diverse chemical space than PKS systems that use a few simple acyl-CoA subunits that result in unstable intermediates during assembly (Fig. 3). Therefore, it is possible to rationally engineer NRPS complexes by substituting modules that have specificity for different amino acids to impart desirable features in to peptide antibiotics. When structure-activity relationships are available, a rational engineering approach can often be more informative and fruitful in the development of bioactive compounds than a randomized combinatorial library, which can often be limited by the quality of the utilized high-throughput biological screens.
Fig. 3.

Structural diversity of peptide antibiotics. The large number of natural and unnatural amino acids provides great opportunities to develop antimicrobial agents with diverse structural architectures. Indolicidin (PDB 1G89), β-defensin-1 (PDB 1IJV), and plectasin (PDB 1ZFU) where visualized with PyMOL.
To illustrate, Baltz and co-workers were able to expand the antibacterial spectrum of a lipopeptide by producing rationally engineered hybrid NRPSs.98 The cyclic lipopeptide antibiotic daptomycin has bactericidal activity against Gram-positive bacteria, and is used to treat complicated skin infections and infections caused by multidrug resistant organisms.99 Although daptomycin is active against Streptococcus pneumonia with a minimal inhibitory concentration (MIC) of 0.06 μg/mL, it cannot be used to treat pneumonia infections because it is sequestered by pulmonary surfactant.98, 100 Based on previous knowledge of amino acid substitutions that overcome surfactant sequestration within the similarly structured lipopeptide antibiotic A54145, Baltz and co-workers developed hybrid A54145 NRPSs by substituting modules from the daptomycin NRPS.98 In doing so, the authors were able to produce 16 A54145 derivatives that had up to 6 amino acid modifications on the tridecapeptide backbone. One of these derivatives had a 32-fold lower MIC than daptomycin in the presence of 1% surfactant, and showed improved antibacterial activity relative to daptomycin in a S. pneumoniae lung infection mouse model.
Further modularization of NRP assembly can also be performed at the stage of tailoring enzymes that decorate the peptide scaffolds with various functional groups. For example, the methyltransferase MtfA from the chloroeremomycin biosynthetic cluster, a glycopeptide antibiotic with a vancomycin peptide scaffold produced by Amycolatopsis orientalis, can be used to methylate desulfo-A47934, a glycopeptide antibiotic with a teicoplanin peptide scaffold produced by Streptomyces toyocaensis.101 Methylation of the glycopeptide by MtfA could be performed both in vitro and in vivo. Such modifications not only increase the chemical diversity of glycopeptides that may be used to combat resistance mechanisms, but they can also allow for the masking or unmasking of functional groups that could be used by medicinal chemists to explore novel chemical architectures.
Antimicrobial Peptides
Multicellular organisms from the plant and animal kingdoms also possess small peptide-based antimicrobial defense mechanisms.102 These defense peptides, aptly named antimicrobial peptides, are far less recognized than the traditional small molecule antibiotics that are routinely used in the clinic. Nevertheless, their utility as natural antimicrobials amongst such diverse forms of life and their amenability for synthetic biology opens up new avenues for antibiotic discovery.
The biological activity of AMPs can derive from direct killing of pathogenic bacteria or indirect induction of killing by modulating the innate immune system.103 Here, we shall focus on the direct interactions between AMPs and their bacterial targets as a more conventional and clinically relevant form of therapy. The structure and physical properties of AMPs are markedly different than the aforementioned NRP antibiotics (Fig. 3). For example, whereas the well-studied glycopeptide vancomycin is composed of a rigid 7-member peptide scaffold that inhibits cell wall biosynthesis by specifically binding to a peptidoglycan intermediate,91 AMPs have a more generalized mode of action where they disrupt membrane integrity without targeting any specific cellular substructures.103 Although some AMPs are able to cross the cellular membrane and interact with cellular components,104 cell membrane disruption appears to be the main antimicrobial mode of action.
AMPs possess several structural and biophysical properties that allow for the specific targeting of bacterial as opposed to mammalian membranes. Generally, AMPs are short peptides containing 10–50 amino acids and are composed primarily of cationic and hydrophobic residues that provide amphiphilic characteristics.102, 105 Although there is little sequence conservation amongst different peptides, they do adopt secondary α-helical and β-sheet structures that fall in to several well recognized motifs.89, 105 While mammalian membranes are primary composed of neutral lipids and contain cholesterol, bacterial membranes on the other hand are mainly composed of anionic lipids and do not contain cholesterol.89, 102 Therefore, AMPs are electrostatically attracted to bacterial membranes, where they integrate in to the lipid bilayer due to their amphiphilic properties and disrupt membrane integrity by forming various porous structures, or at high concentrations they can act like detergents to disintegrate membranes leading to cell death.106 Accordingly, large concentrations of AMPs are required to achieve antibacterial activity with MICs in the micromolar range.106 This is not unexpected when one considers the typical biophysical characteristics of peptides. Due to their small size and limited surface area, peptides lack well-defined structures and have few molecular `handles' to mediate strong non-covalent binding interactions, whether electrostatic or hydrophobic in nature.107 Thus, they can be thought of as flexible and `slippery' molecules when one considers global biochemical interactions in general terms. However, it is indeed these common structural features and versatile physiochemical characteristics of AMPs that afford opportunities to re-engineer the peptides to impart more potent biological activity.
A synthetic biology approach to engineering novel AMPs is quite different than the aforementioned engineering of natural secondary metabolic pathways. Due to the small size of AMPs, common physiochemical properties, and an ambiguous membrane-disrupting mechanism, AMP re-engineering efforts are well-suited for computational modeling and peptide design.108 For instance, Tsai et al. used molecular dynamics simulations to rationally design improved analogs of the 13-residue AMP indolicidin.109 Indolicidin has broad-spectrum antimicrobial activities and although the detailed molecular mechanism of the peptide is not well understood, it is clear that interaction with and disruption of the cell membrane is key to its mode of action.109, 110 However, the clinical application of indolicidin is inhibited in part due to cytotoxicity, where the peptide causes the lysis of erythrocytes, but synthetic analogs have been made with reduced cytotoxic effects.111 Using molecular dynamics simulations, Tsai et al. sought to better understand the molecular interactions between indolicidin and hypothetical erythrocyte/bacterial membranes.109 The simulations revealed that indolicidin perturbs both membranes, but by different mechanisms. Disruption of the hypothetical erythrocyte membrane was largely due to insertion of the peptide in to the lipid bilayer and the formation of key hydrophobic contacts. Alternatively, disruption of the hypothetical bacterial membrane was primarily the result of adsorption of the peptide on to the surface of the lipid bilayer and the formation of important electrostatic interactions at the water-lipid interface. After identifying key residues for mediating insertion and adsorption interactions, the authors designed synthetic indolicidin analogs to enhance electrostatic interactions for improved antimicrobial activity, while weakening hydrophobic interactions to reduce hemolytic activity. This resulted in the development of an indolicidin analog (IL-K7F89) with 3 altered residues, which had a 2-fold lower MIC and a 10-fold decrease in hemolysis relative to indolicidin. This study demonstrates that although detailed mechanistic information may not be available for most AMPs, their small size and general membrane disruptive properties make them highly amenable for computation simulation to both study their molecular mechanisms and design synthetic variants with improved properties.
Further applications of computational methodology include the virtual screening of synthetic peptides for antimicrobial activity. Such an approach can significantly reduce the cost and time associated with large-scale peptide synthesis and screening efforts. Recently, artificial intelligence algorithms were used to virtually screen a 100,000 9-mer peptide library in the search for potent AMPs.112, 113 Quantitative structure-activity relationships were used to analyze the antibiotic properties of peptides based on atomic scale molecular data. Subsequently, artificial neural networks were used to design models that were able to predict the antibacterial potential of the peptides. The designed algorithm was able to predict highly active AMPs with a 94% rate of accuracy.113 When the two most active AMPs were tested in an in vivo model of Staphylococcus aureus infection, they demonstrated superior efficacy compared to the most clinically advanced AMP.112
AMPs do have some limitations that need to be addressed, namely protease degradation and acquired resistance.103 Protease degradation could be overcome by changing the chemical properties of the peptides, such as chemically modifying peptides to prevent protease recognition or incorporating unnatural or D-amino acids, although such modifications may affect peptide activity, stability, or toxicity.103 Contrary to previous beliefs,102 it has been experimentally demonstrated that bacteria are able to acquire heritable resistance to AMPs.114 However, even though resistance may inhibit the bacterial killing activity of AMPs, it does not affect their immunomodulatory activities and there is limited cross-resistance between different AMPs.103, 115
Due to the availability of a larger subset of building blocks including both natural and unnatural amino acids, peptides provide more versatile scaffolds to engineer antibiotics when compared to polyketides. This is apparent in the structural diversity observed among the vastly different NRPs and AMPs (Fig. 3). Structural diversity can be further increased due to facile chemical synthesis techniques. For example, solid-phase peptide synthesis was recently used to construct branched chimeric AMPs, where two recognition regions were linked to a killing moiety to allow for species-specific bacterial targeting within mixed bacterial populations.116 Additionally, the physiochemical properties of peptides can be tuned by simple amino acid substitutions, whereas polyketide antibiotics require complex genetic engineering efforts. Therefore, synthetic biology could provide many exciting opportunities in the near future for the development of novel peptide antibiotics.
Non-Traditional Therapeutics
Current clinical antimicrobials are small molecules that target a handful of key cellular targets that are unique to bacteria. The revolutionary discovery of penicillin by Sir Alexander Fleming in 1929117 and its rapid production and clinical application came to define what we generally regard as an antibiotic. However, before antibiotics, bacteriophages were explored as an antimicrobial therapy and even after antibiotics were widely adopted in the Western world, bacteriophage therapy continued to be used in Eastern Europe and the Soviet Union.118, 119 Due to the alarming emergence of multidrug resistant bacterial pathogens over recent decades along with a diminishing antibiotic discovery pipeline,120 there has been a resurgence of interest in non-traditional antimicrobials, especially ones that are amenable to engineering with synthetic biology. Bacteriophage-based therapeutics face a host of challenges in being adopted for clinical therapy118 but there are academic and industrial groups pursuing engineering strategies to overcome these hurdles.121
Phage Engineering
In addition to adaptive and acquired antibiotic resistance mechanisms, bacterial tolerance to certain antibiotics can also be increased as a result of extracellular barriers, such as those erected by biofilms, produced by some bacterial populations. Biofilms are microbial communities that are formed when bacteria transition from a planktonic to a surface bounds state.122 As certain bacteria adhere to and colonize various solid surfaces, they secrete extracellular polymeric substances to produce three-dimensional structures containing fluid channels.122–124 It is well established that bacteria present in biofilms are far more tolerant of antibiotics compared to their planktonic counterparts, although the exact mechanisms of resistance are diverse.124, 125
To aid in biofilm formation, bacteria use outer-membrane appendages to promote surface attachment, colonization, and biofilm formation.123, 126, 127 Recent studies on the outer-membrane proteins of Gram-positive bacterial pathogens have further shed light on the biochemistry of Gram-positive bacterium-host interactions. Remarkably, in the human pathogen Streptococcus pyogenes, two autocatalyzed isopeptide bonds stabilize the major pilin subunit that is polymerized to form the mature pilus,128 an important outer-membrane appendage for bacterial surface attachment.123 This provides the pilin subunit with dramatically enhanced proteolytic and thermodynamic stability, as well as tolerance to extreme amounts of shear force.129, 130 Each subunit is then covalently attached to the next via sortase catalyzed intermolecular isopeptide bonds, and similarly adhesin proteins are also attached to the pilus to mediate host cell recognition and binding.126, 127, 131, 132 Intriguingly, the S. pyogenes adhesin contains an internal thioester bond that is proposed to mediate covalent attachment to host cells.133 Therefore, during S. pyogenes infection there is a linear path of covalent connectivity between the bacterium and host. Coupled to biofilm formation that can provide a physical drug barrier, it can be envisioned that to combat such infections a dual strategy would be best suited, where one can first breakdown biofilms before applying antibiotics to kill the pathogens.
Over the past decade, advances in synthetic biology methodologies and genetic engineering technologies have provided exciting opportunities to engineer biological platforms. As an extension of phage therapy, where phage are used to specifically target and kill bacteria, Lu and Collins devised a two-pronged strategy to combat biofilm-producing bacteria by engineering phage to be able to enzymatically degrade biofilms (Fig. 4).134 The cell-bound polymeric polysaccharide β-1,6-N-acetyl-d-glucosamine is an important component of the extracellular polymeric substances within E. coli and Staphylococcus biofilms.135 The β-hexosaminidase DspB from Actinobacillus actinomycetemcomitans has been shown to hydrolyze β-1,6-N-acetyl-d-glucosamine polymers and disperse biofilms.135 Accordingly, Lu and Collins integrated the dspB gene in to the genome of T7 phage along with the T3 phage gene 1.2 to extend the T7 phage host range to include F-plasmid containing E. coli.134 The rationale was that the newly engineered T7 phage (T7DspB) would be able to infect E. coli within biofilms and then the phage would use the bacterial machinery to express DspB, which would get released in to the biofilm after cell lysis. This would aid in dispersal of the biofilm and improve the efficiency with which phage can spread and infect surrounding bacteria in the biofilm. As hypothesized, the engineered T7DspB phage was approximately 100-fold more efficient at reducing bacterial biofilm cell counts than a control lacking dspB, and resulted in ~99.997% bacterial removal.134 Biofilm degradation could be visually observed using scanning electron microscopy, and phage replication within E. coli was not affected by the introduction of the dspB gene. Therefore, to combat persistent bacterial infections engineered phage can be used to breakdown protective barriers constructed by bacteria, thereby making them more vulnerable to subsequent treatments.
Fig. 4.

Non-traditional phage antimicrobials. Phage can be engineered to express enzymes that disperse biofilms (blue) or as adjuvants that disrupt bacterial cellular regulatory networks and sensitize cells for further antibiotic treatment. Alternatively, phage lysin can be applied extracellularly to degrade cell walls. PlyC (PBD 4F88) is used to depict phage lysins, visualized with PyMOL.
In addition to targeting biofilms, phage can also be engineered to target key bacterial processes aside from those traditionally targeted by small molecule antibiotics. However, a major issue with phage therapy is the emergence of bacterial resistance, which can arise through a variety of mechanisms including blocking of phage DNA entry, restriction-modification of phage DNA, interference with phage adsorption onto bacterial cell surfaces, or abortive infection systems.118, 136 To address these issues, Lu and Collins sought to engineer phage that targeted nonessential bacterial gene networks (Fig. 4).137 Such phage would serve dual roles – since nonessential genes are targeted, reduced evolutionary pressure should be imposed on the bacteria leading to reduced emergence of phage resistance, and targeting nonessential gene networks should weaken bacteria and make them more susceptible to subsequent antibiotic therapy. To develop phage as adjuvants for antibiotic therapy, the authors integrated an inducible lexA3 gene in to the genome of the filamentous M13mp18 phage to create φlexA3.137 It has been shown that bactericidal antibiotics induce the production of reactive hydroxyl radicals, leading to DNA damage and subsequent activation of the DNA repair SOS response system, which is in turn inhibited by LexA3.138, 139 Therefore, expression of LexA3 in bacteria would disrupt the DNA repair mechanism and enhance the effects of bactericidal antibiotics. Indeed, when φlexA3 was used in combination with quinolone, aminoglycoside, or β-lactam antibiotics it enhanced cell killing by several orders of magnitude compared to the antibiotic alone or the antibiotic with an unmodified phage. Furthermore, φlexA3 also enhanced the efficacy of a quinolone antibiotic against antibiotic resistant bacteria within a mouse model of E. coli infection, thereby demonstrating its potential for clinical applications.137 To illustrate the generality of developing phage adjuvants for antibiotic therapy, the authors also successfully targeting gene networks involved in oxidative stress response, biofilm formation, and outer membrane permeability where in each case enhanced efficacy was observed with combination therapy.
Phage can also be used to reverse bacterial resistance. Streptomycin resistance in bacteria can arise as a result of mutations in the rpsL gene, which encodes a protein in the 30S ribosomal subunit.140 However, wild-type rpsL has a dominant sensitive phenotype and when introduced in to resistance bacteria, it can restore the antibiotic sensitive phenotype.140 Edgar et al. have engineered a lysogenic phage λ to contain 1–2 copies of a sequence optimized rpsL gene that can reverse bacterial resistance similar to the wild-type rpsL gene, while minimizing recombination between the phage and bacterial copies of the rpsL gene due to different codon usage.140 Also, rather than using another antibiotic selection marker for phage maintenance, the authors used resistance to the toxic compound tellurite to maintain phage in bacteria. Interestingly, the engineered phage were able to dramatically increase the sensitivity of bacteria to streptomycin, reducing the MIC of a resistant strain from 200 μg/mL to 1.56 μg/mL. The authors further demonstrated this approach by engineering phage that can reverse resistance to a quinolone antibiotic.
The ability of engineered phage to disrupt bacterial biofilms and gene networks can be used to increase the susceptibility of bacteria to antibiotics. In order to combat the emergence of widespread antibiotic resistance, we need to not only discover new antibiotics but also to find ways to revitalize old antibiotics that have been rendered ineffective due to bacterial resistance. Similar to how clavulanic acid and penicillins have enjoyed great clinical success as combination therapeutics,141 phage may one day be a useful adjuvant to traditional antibiotics.
Other Non-Traditional Therapeutics
Phage have been evolutionarily optimized to efficiently kill bacteria. As such, they can serve as a source for novel antimicrobial products. In addition to engineering phage as adjuvants to antibacterial therapy, we can also explore how phage kill their bacterial hosts and exploit this processes to develop new therapeutics. During a typical bacterial phage infection, bacteria are only killed when they are lysed by phage enzymes. Thus, the enzymes responsible for mediating this process, namely phage lysins, can serve as new sources of antimicrobial agents (Fig. 4).142, 143 This expands the molecular toolbox of antimicrobial agents to include enzymes as well as polyketides, peptides, and phage.
The modular nature of phage lysins makes them amenable for synthetic biology applications. Lysins are composed of two domains, a catalytic N-terminal domain that is responsible for degrading cell wall components, and a cell-binding C-terminal domain that mediates species-specific cell wall binding.142 When different phage lysins are compared, high sequence homology is observed in the catalytic domain and little sequence conservation is seen in the cell-binding domain, thereby allowing for specificity of host bacterial cell walls.142 This provides opportunities to mix-and-match domains and engineer lysins with enhanced functionalities. For example, the Fischetti group developed a chimeric lysin by fusing the catalytic and cell-binding domains of two different staphylococcal lysins.144 The rationale was to utilize a cell-binding domain that did not have homology to SH3b domains that bind peptide cross-bridges for which resistance is easily acquired. Also, a catalytic domain was sought that was both soluble and highly active. The authors engineered over 15 chimeric lysins and were able to isolate ClyS, a lysin that was both specific for Staphylococci and could be purified as a soluble protein.144 Not only was ClyS active against both antibiotic sensitive and resistant Streptococci, but it also acted synergistically with other small molecule antibiotics.144 Moreover, ClyS was able to protect mice in a systemic staphylococcal infection model and could be formulated within an ointment for topical skin application.144, 145 Further engineering of lysin proteins may enhance the limited host range of the antimicrobial enzymes and increase the repertoire of available antimicrobials.
Pyocins are another class of bacterial antimicrobial agents that are produced by Pseudomonas aeruginosa, although they structurally resemble phage tails.146 R-type pyocins are composed of a hollow sheath core and a tail fiber region that specifically bind to the surfaces of target cells.147 Subsequently, the hollow sheath core inserts in to the bacterial inner and outer membranes through the cell wall, resulting in cell death via cytoplasmic membrane depolarization due to leakage of intracellular ions.147 Although pyocins have a limited antimicrobial spectrum, they can be engineered to extend their target bacterial range.147 To illustrate, Scholl et al. engineered a R-type pyocin from P. aeruginosa to contain tail fiber fusion proteins that incorporated tail spikes from the E. coli O157 specific phage ϕV10.148 The ϕV10 tail spike proteins mediate recognition of target E. coli O157 strains and degradation of the surface polysaccharide structures.148 Interestingly, the hybrid pyocin was able kill E. coli O157 strains and do so by degrading the surface polysaccharide structures. As a demonstration of the utility of this technology for food preservation, the hybrid pyocin was also able to significantly reduce the colonization of beef surfaces by E. coli O157.
Alternatively, bacteria can be engineered to detect and kill cells using pyocins. For instance, E. coli can be engineered to contain a quorum-sensing module that is able to detect a chemical signal from P. aeruginosa, and in response express a pyocin and a lytic protein to allow for pyocin release.149 When co-cultured with P. aeruginosa, the engineered E. coli is able to reduce the viability of the pathogen by over 99%.149
Another interesting class of non-traditional antimicrobials is bacteriocins, which are short ribosomally synthesized peptides that are produced by bacteria.150 Similar to AMPs, bacteriocins function by permeabilizing membranes via pore formation leading to efflux of intracellular ions and small molecules.150 Bacteriocins are encoded within gene clusters that also encode other proteins responsible for post-translational modifications, self-immunity, and protein transport.151 Diverse peptide structures are observed within the various classes of bacteriocins, including both linear and cyclical peptides that adopt stable secondary structures.150, 151 These peptides have been extensively used as food preservatives due to their potent antimicrobial activity as well as resistance to proteases, heat, and acidic environments.150, 151 Based on the similarity of bacteriocins to the other peptide antimicrobials discussed earlier, many of the same synthetic biology principles can be used to engineer new functionalities in to these molecules.
Conclusions
Over the past century, antibiotics have revolutionized science and medicine. Antibiotics have become essential components in our daily lives whether in the fight against infectious disease or use in basic scientific research, agriculture, and industry applications. Their elegance is in their simplicity – when they work, they are highly effective and curative. Unfortunately, their Achilles heel – resistance – has led to the dramatic emergence of bacteria that cannot be treated effectively with current therapies.
Antibiotics and resistance go hand-in-hand in Nature – the organisms that produce antibiotics must also produce self-defense mechanisms against their activity. In fact, a recent study of ancient resistance determinants taken from ice cores from the Canadian arctic demonstrates the presence of environmental antibiotic resistance genes that date back 30,000 years.152 Therefore, antibiotic resistance is certainly not a new concept but is one that has risen to the forefront of medicine due to extensive use in the environment and in the clinic. Throughout evolutionary history, bacteria have existed within environments containing hotbeds of antimicrobial agents and resistance genes that come not only from other antibiotic-producing bacteria but from other fungi and plants as well.153 Accordingly, resistance has evolved and been passed on to other organisms through simple genetic exchange. Therefore, in the quest for antibiotic discovery, one must be wary of various antimicrobial resistance mechanisms and find new ways to overcome them. That being said, we must also accept that most therapeutics, whether for infectious disease or other ailments, have a limited window of opportunity for effectiveness and that we must always remain a step ahead of resistance with new technologies.
Biology is based on the assemblage of a small number of simple building blocks that together form highly complex and sophisticated structures. Scientific advances over the past several decades have afforded us a unique opportunity to not only sit on the sidelines and observe how biology functions, but to step in and modify natural systems and instill new functionality. At the heart of this scientific-engineering interdisciplinary approach lies synthetic biology. By breaking down biology in to small interchangeable biological components, synthetic biology can reassemble them in imaginative ways to sample new chemical space that cannot be reached by current biological systems or chemical approaches.57, 154–156 Here, we provided examples where synthetic biology can be used to construct new antimicrobial agents by building upon natural systems. By swapping biological assembly modules in secondary metabolic pathways, we can create entirely new chimeric chemical entities. Alternatively, we can use computational approaches to create small focused `smart' libraries of antimicrobial peptides and significantly reduce the costs associated with drug discovery. We can re-engineer phage therapeutics and leverage phage-derived products to create programmable therapeutics that extend our notions of traditional antibiotics. Antibiotic discovery has been defined by periods of great ingenuity that have culminated in the golden era of antibiotic discovery and medicinal chemistry, where most of our current arsenal of antimicrobials originated. A new interdisciplinary approach is required to tackle the challenges that we face today in regards to antimicrobial discovery. By exploiting our enhanced ability to program biology and remodel natural systems, we are poised to potentially usher in a new era of antimicrobial discovery in the early 21st century.
Acknowledgements
We appreciate comments on this manuscript from Sara Cleto. T.K.L. acknowledges support from the Office of Naval Research, Army Research Office, Defense Advanced Research Projects Agency, National Science Foundation, National Institutes of Health (DP2 OD008435), Ellison Medical Foundation, and Institute for Soldier Nanotechnologies. B.Z. is supported by the Defense Advanced Research Projects Agency.
References
- (1).Pasteur L, Joubert J. Charbon et septicemie. Comptes Rendus Chimie. 1877;85:101–105. [Google Scholar]
- (2).Waksman SA, Woodruff HB. The Soil as a Source of Microorganisms Antagonistic to Disease-Producing Bacteria. J. Bacteriol. 1940;40:581–600. doi: 10.1128/jb.40.4.581-600.1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wright GD. The antibiotic resistome: the nexus of chemical and genetic diversity. Nat. Rev. Microbiol. 2007;5:175–186. doi: 10.1038/nrmicro1614. [DOI] [PubMed] [Google Scholar]
- (4).Zakeri B. Methyltransferases as agents of chemical diversity in natural products. McMaster University: 2008. [Google Scholar]
- (5).Berdy J. Bioactive microbial metabolites. J. Antibiot. (Tokyo) 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- (6).Fernandez L, Hancock RE. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012;25:661–681. doi: 10.1128/CMR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).D'Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- (8).Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337:1107–1111. doi: 10.1126/science.1220761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Baltz RH. Antibiotic discovery from actinomycetes: Will a renaissance follow th decline and fall? SIM News. 2005;55:186–196. [Google Scholar]
- (10).Baltz RH. Renaissance in antibacterial discovery from actinomycetes. Curr. Opin. Pharmacol. 2008;8:557–563. doi: 10.1016/j.coph.2008.04.008. [DOI] [PubMed] [Google Scholar]
- (11).Qiu F, Imai A, McAlpine JB, Lankin DC, Burton I, Karakach T, Farnsworth NR, Chen SN, Pauli GF. Dereplication, residual complexity, and rational naming: the case of the Actaea triterpenes. J. Nat. Prod. 2012;75:432–443. doi: 10.1021/np200878s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Charest MG, Siegel DR, Myers AG. Synthesis of (−)-Tetracycline. J. Am. Chem. Soc. 2005;127:8292–8293. doi: 10.1021/ja052151d. [DOI] [PubMed] [Google Scholar]
- (13).Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. A Convergent Enantioselective Route to Structurally Diverse 6-Deoxytetracycline Antibiotics. Science. 2005;308:395–398. doi: 10.1126/science.1109755. [DOI] [PubMed] [Google Scholar]
- (14).Zakeri B, Wright GD. Chemical biology of tetracycline antibiotics. Biochem. Cell Biol. 2008;86:124–136. doi: 10.1139/O08-002. [DOI] [PubMed] [Google Scholar]
- (15).Fernandes P. Antibacterial discovery and development--the failure of success? Nat. Biotechnol. 2006;24:1497–1503. doi: 10.1038/nbt1206-1497. [DOI] [PubMed] [Google Scholar]
- (16).Khosla C, Kapur S, Cane DE. Revisiting the modularity of modular polyketide synthases. Curr. Opin. Chem. Biol. 2009;13:135–143. doi: 10.1016/j.cbpa.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lam CM, Suarez Diez M, Godinho M, Martins dos Santos VA. Programmable bacterial catalysis - designing cells for biosynthesis of value-added compounds. FEBS Lett. 2012;586:2184–2190. doi: 10.1016/j.febslet.2012.02.030. [DOI] [PubMed] [Google Scholar]
- (18).Yadav VG, De Mey M, Lim CG, Ajikumar PK, Stephanopoulos G. The future of metabolic engineering and synthetic biology: towards a systematic practice. Metab. Eng. 2012;14:233–241. doi: 10.1016/j.ymben.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lee H, DeLoache WC, Dueber JE. Spatial organization of enzymes for metabolic engineering. Metab. Eng. 2012;14:242–251. doi: 10.1016/j.ymben.2011.09.003. [DOI] [PubMed] [Google Scholar]
- (20).Keasling JD. Synthetic biology and the development of tools for metabolic engineering. Metab. Eng. 2012;14:189–195. doi: 10.1016/j.ymben.2012.01.004. [DOI] [PubMed] [Google Scholar]
- (21).Felnagle EA, Chaubey A, Noey EL, Houk KN, Liao JC. Engineering synthetic recursive pathways to generate non-natural small molecules. Nat. Chem. Biol. 2012;8:518–526. doi: 10.1038/nchembio.959. [DOI] [PubMed] [Google Scholar]
- (22).Lai JR, Koglin A, Walsh CT. Carrier protein structure and recognition in polyketide and nonribosomal peptide biosynthesis. Biochemistry. 2006;45:14869–14879. doi: 10.1021/bi061979p. [DOI] [PubMed] [Google Scholar]
- (23).Walsh CT, Fischbach MA. Natural products version 2.0: connecting genes to molecules. J. Am. Chem. Soc. 2010;132:2469–2493. doi: 10.1021/ja909118a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Meier JL, Burkart MD. The chemical biology of modular biosynthetic enzymes. Chem. Soc. Rev. 2009;38:2012–2045. doi: 10.1039/b805115c. [DOI] [PubMed] [Google Scholar]
- (25).Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- (26).Gulder TA, Freeman MF, Piel J. The Catalytic Diversity of Multimodular Polyketide Synthases: Natural Product Biosynthesis Beyond Textbook Assembly Rules. Top. Curr. Chem. 2011 doi: 10.1007/128_2010_113. [DOI] [PubMed] [Google Scholar]
- (27).Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Type II polyketide synthases: gaining a deeper insight into enzymatic teamwork. Nat. Prod. Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]
- (28).Weymouth-Wilson AC. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 1997;14:99–110. doi: 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]
- (29).Brodersen DE, Clemons WM, Jr., Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell. 2000;103:1143–1154. doi: 10.1016/s0092-8674(00)00216-6. [DOI] [PubMed] [Google Scholar]
- (30).Pioletti M, Schlunzen F, Harms J, Zarivach R, Gluhmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, Hartsch T, Yonath A, Franceschi F. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. EMBO J. 2001;20:1829–1839. doi: 10.1093/emboj/20.8.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Walsh CT. Combinatorial biosynthesis of antibiotics: challenges and opportunities. Chembiochem. 2002;3:125–134. doi: 10.1002/1439-7633(20020301)3:2/3<124::AID-CBIC124>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- (32).Park SR, Han AR, Ban YH, Yoo YJ, Kim EJ, Yoon YJ. Genetic engineering of macrolide biosynthesis: past advances, current state, and future prospects. Appl. Microbiol. Biotechnol. 2010;85:1227–1239. doi: 10.1007/s00253-009-2326-8. [DOI] [PubMed] [Google Scholar]
- (33).Cortes J, Haydock SF, Roberts GA, Bevitt DJ, Leadlay PF. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature. 1990;348:176–178. doi: 10.1038/348176a0. [DOI] [PubMed] [Google Scholar]
- (34).Donadio S, Staver MJ, McAlpine JB, Swanson SJ, Katz L. Modular organization of genes required for complex polyketide biosynthesis. Science. 1991;252:675–679. doi: 10.1126/science.2024119. [DOI] [PubMed] [Google Scholar]
- (35).Long PF, Wilkinson CJ, Bisang CP, Cortes J, Dunster N, Oliynyk M, McCormick E, McArthur H, Mendez C, Salas JA, Staunton J, Leadlay PF. Engineering specificity of starter unit selection by the erythromycin-producing polyketide synthase. Mol. Microbiol. 2002;43:1215–1225. doi: 10.1046/j.1365-2958.2002.02815.x. [DOI] [PubMed] [Google Scholar]
- (36).Stassi DL, Kakavas SJ, Reynolds KA, Gunawardana G, Swanson S, Zeidner D, Jackson M, Liu H, Buko A, Katz L. Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering. Proc. Natl. Acad. Sci. U. S. A. 1998;95:7305–7309. doi: 10.1073/pnas.95.13.7305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2006;128:3067–3074. doi: 10.1021/ja058093d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kato Y, Bai L, Xue Q, Revill WP, Yu TW, Floss HG. Functional expression of genes involved in the biosynthesis of the novel polyketide chain extension unit, methoxymalonyl-acyl carrier protein, and engineered biosynthesis of 2-desmethyl-2-methoxy-6-deoxyerythronolide B. J. Am. Chem. Soc. 2002;124:5268–5269. doi: 10.1021/ja0127483. [DOI] [PubMed] [Google Scholar]
- (39).Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J. Am. Chem. Soc. 2003;125:5366–5374. doi: 10.1021/ja029539i. [DOI] [PubMed] [Google Scholar]
- (40).Kao CM, McPherson M, McDaniel RN, Fu H, Cane DE, Khosla C. Gain of Function Mutagenesis of the Erythromycin Polyketide Synthase. 2. Engineered Biosynthesis of an Eight-Membered Ring Tetraketide Lactone. J. Am. Chem. Soc. 1997;119:11339–11340. [Google Scholar]
- (41).Zhang C, Fu Q, Albermann C, Li L, Thorson JS. The in vitro characterization of the erythronolide mycarosyltransferase EryBV and its utility in macrolide diversification. Chembiochem. 2007;8:385–390. doi: 10.1002/cbic.200600509. [DOI] [PubMed] [Google Scholar]
- (42).Yuan Y, Chung HS, Leimkuhler C, Walsh CT, Kahne D, Walker S. In vitro reconstitution of EryCIII activity for the preparation of unnatural macrolides. J. Am. Chem. Soc. 2005;127:14128–14129. doi: 10.1021/ja053704n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Schell U, Haydock SF, Kaja AL, Carletti I, Lill RE, Read E, Sheehan LS, Low L, Fernandez MJ, Grolle F, McArthur HA, Sheridan RM, Leadlay PF, Wilkinson B, Gaisser S. Engineered biosynthesis of hybrid macrolide polyketides containing D-angolosamine and D-mycaminose moieties. Org. Biomol. Chem. 2008;6:3315–3327. doi: 10.1039/b807914e. [DOI] [PubMed] [Google Scholar]
- (44).Menzella HG, Reid R, Carney JR, Chandran SS, Reisinger SJ, Patel KG, Hopwood DA, Santi DV. Combinatorial polyketide biosynthesis by de novo design and rearrangement of modular polyketide synthase genes. Nat Biotech. 2005;23:1171–1176. doi: 10.1038/nbt1128. [DOI] [PubMed] [Google Scholar]
- (45).Menzella HG, Carney JR, Li Y, Santi DV. Using chemobiosynthesis and synthetic mini-polyketide synthases to produce pharmaceutical intermediates in Escherichia coli. Appl. Environ. Microbiol. 2010;76:5221–5227. doi: 10.1128/AEM.02961-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Menzella HG, Carney JR, Santi DV. Rational design and assembly of synthetic trimodular polyketide synthases. Chem. Biol. 2007;14:143–151. doi: 10.1016/j.chembiol.2006.12.002. [DOI] [PubMed] [Google Scholar]
- (47).Zhang W, Ames BD, Tsai SC, Tang Y. Engineered biosynthesis of a novel amidated polyketide, using the malonamyl-specific initiation module from the oxytetracycline polyketide synthase. Appl. Environ. Microbiol. 2006;72:2573–2580. doi: 10.1128/AEM.72.4.2573-2580.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McCormick JR, Joachim UH, Jensen ER, Johnson S, Sjolander NO. Biosynthesis of The Tetracyclines. VII. 4-Hydroxy-6-methylpretetramid, an Intermediate Accumulated by a Blocked Mutant of Streptomyces aureofaciens. J. Am. Chem. Soc. 1965;87:1793–1794. doi: 10.1021/ja01086a033. [DOI] [PubMed] [Google Scholar]
- (49).McCormick JR, Jensen ER, Johnson S, Sjolander NO. Biosynthesis of the tetracyclines. IX. 4-Aminodedimethylaminoanhydrodemethylchlortetracycline from a mutant of Streptomyces aureofaciens. J. Am. Chem. Soc. 1968;90:2201–2202. doi: 10.1021/ja01010a063. [DOI] [PubMed] [Google Scholar]
- (50).McCormick JR, Jensen ER. Biosynthesis of the tetracyclines. XII. Anhydrodemethylchlortetracycline from a mutant of Streptomyces aureofaciens. J. Am. Chem. Soc. 1969;91:206. doi: 10.1021/ja01029a046. [DOI] [PubMed] [Google Scholar]
- (51).McCormick JR, Jensen ER. Biosynthesis of The Tetracyclines. VIII. Characterization of 4-Hydroxy-6-methylpretetramid. J. Am. Chem. Soc. 1965;87:1794–1795. doi: 10.1021/ja01086a034. [DOI] [PubMed] [Google Scholar]
- (52).Barbatschi F, Dann M, Martin JH, Miller P, Mitscher LA, Bohonos N. 4-Dedimethylamino-4-epiamino-5a,6-anhydrotetracycline. Experientia. 1965;21:162–163. doi: 10.1007/BF02141995. [DOI] [PubMed] [Google Scholar]
- (53).Zhang W, Watanabe K, Wang CC, Tang Y. Investigation of early tailoring reactions in the oxytetracycline biosynthetic pathway. J. Biol. Chem. 2007;282:25717–25725. doi: 10.1074/jbc.M703437200. [DOI] [PubMed] [Google Scholar]
- (54).Finlay AC, Hobby GL, Pan YS, Regna PP, Routien JB, Seeley DB, Shull GM, Sobin BA, Solomons IA, Vinson JW, Kane JH. Terramycin, a new antibiotic. Science. 1950;111:85. doi: 10.1126/science.111.2874.85. [DOI] [PubMed] [Google Scholar]
- (55).Petkovic H, Cullum J, Hranueli D, Hunter IS, Peric-Concha N, Pigac J, Thamchaipenet A, Vujaklija D, Long PF. Genetics of Streptomyces rimosus, the oxytetracycline producer. Microbiol. Mol. Biol. Rev. 2006;70:704–728. doi: 10.1128/MMBR.00004-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Pickens LB, Tang Y. Oxytetracycline biosynthesis. J. Biol. Chem. 2010;285:27509–27515. doi: 10.1074/jbc.R110.130419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Winter JM, Tang Y. Synthetic biological approaches to natural product biosynthesis. Curr. Opin. Biotechnol. 2012;23:736–743. doi: 10.1016/j.copbio.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Stevens DC, Henry MR, Murphy KA, Boddy CN. Heterologous expression of the oxytetracycline biosynthetic pathway in Myxococcus xanthus. Appl. Environ. Microbiol. 2010;76:2681–2683. doi: 10.1128/AEM.02841-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wang P, Zhang W, Zhan J, Tang Y. Identification of OxyE as an ancillary oxygenase during tetracycline biosynthesis. Chembiochem. 2009;10:1544–1550. doi: 10.1002/cbic.200900122. [DOI] [PubMed] [Google Scholar]
- (60).Zhang W, Watanabe K, Cai X, Jung ME, Tang Y, Zhan J. Identifying the minimal enzymes required for anhydrotetracycline biosynthesis. J. Am. Chem. Soc. 2008;130:6068–6069. doi: 10.1021/ja800951e. [DOI] [PubMed] [Google Scholar]
- (61).Nelson ML, Park BH, Levy SB. Molecular Requirements for the Inhibition of the Tetracycline Antiport Protein and the Effect of Potent Inhibitors on the Growth of Tetracycline-Resistant Bacteria. J. Med. Chem. 1994;37:1355–1361. doi: 10.1021/jm00035a016. [DOI] [PubMed] [Google Scholar]
- (62).Nelson ML. Chapter 11. The chemistry and biology of the tetracyclines; Annual Reports in Medicinal Chemistry. Vol. olume 37. Academic Press; 2002. pp. 105–114. [Google Scholar]
- (63).Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001;65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Sharan SK, Thomason LC, Kuznetsov SG, Court D. Recombineering: a homologous recombination-based method of genetic engineering. Nat. Protocols. 2009;4:206–223. doi: 10.1038/nprot.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Court DL, Sawitzke JA, Thomason LC. Genetic engineering using homologous recombination. Annu. Rev. Genet. 2002;36:361–388. doi: 10.1146/annurev.genet.36.061102.093104. [DOI] [PubMed] [Google Scholar]
- (67).Ellis HM, Yu D, DiTizio T, Court DL. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc. Natl. Acad. Sci. U. S. A. 2001;98:6742–6746. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Carr PA. DNA construction: homemade or ordered out? Nat. Methods. 2010;7:887–889. doi: 10.1038/nmeth1110-887. [DOI] [PubMed] [Google Scholar]
- (69).Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, III, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- (70).Knight T. Idempotent vector design for standard assembly of Biobricks. DSpace. 2003 [Google Scholar]
- (71).Anderson JC, Dueber JE, Leguia M, Wu GC, Goler JA, Arkin AP, Keasling JD. BglBricks: A flexible standard for biological part assembly. J. Biol. Eng. 2010;4:1. doi: 10.1186/1754-1611-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Engler C, Gruetzner R, Kandzia R, Marillonnet S. Golden gate shuffling: a one-pot DNA shuffling method based on type IIs restriction enzymes. PLoS One. 2009;4:e5553. doi: 10.1371/journal.pone.0005553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Quan J, Tian J. Circular polymerase extension cloning of complex gene libraries and pathways. PLoS One. 2009;4:e6441. doi: 10.1371/journal.pone.0006441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Carr PA, Wang HH, Sterling B, Isaacs FJ, Lajoie MJ, Xu G, Church GM, Jacobson JM. Enhanced multiplex genome engineering through co-operative oligonucleotide co-selection. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Abrahmsen L, Tom J, Burnier J, Butcher KA, Kossiakoff A, Wells JA. Engineering subtilisin and its substrates for efficient ligation of peptide bonds in aqueous solution. Biochemistry (N. Y. ) 1991;30:4151–4159. doi: 10.1021/bi00231a007. [DOI] [PubMed] [Google Scholar]
- (77).Chang TK, Jackson DY, Burnier JP, Wells JA. Subtiligase: a tool for semisynthesis of proteins. Proc. Natl. Acad. Sci. U. S. A. 1994;91:12544–12548. doi: 10.1073/pnas.91.26.12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Paschke M. Phage display systems and their applications. Appl. Microbiol. Biotechnol. 2006;70:2–11. doi: 10.1007/s00253-005-0270-9. [DOI] [PubMed] [Google Scholar]
- (79).Lofblom J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011;6:1115–1129. doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- (80).Gai SA, Wittrup KD. Yeast surface display for protein engineering and characterization. Curr. Opin. Struct. Biol. 2007;17:467–473. doi: 10.1016/j.sbi.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Pluckthun A. Ribosome display: a perspective. Methods Mol. Biol. 2012;805:3–28. doi: 10.1007/978-1-61779-379-0_1. [DOI] [PubMed] [Google Scholar]
- (82).Bertschinger J, Grabulovski D, Neri D. Selection of single domain binding proteins by covalent DNA display. Protein Eng Des Sel. 2007;20:57–68. doi: 10.1093/protein/gzl055. [DOI] [PubMed] [Google Scholar]
- (83).Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Lockless SW, Muir TW. Traceless protein splicing utilizing evolved split inteins. Proc. Natl. Acad. Sci. U. S. A. 2009;106:10999–11004. doi: 10.1073/pnas.0902964106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Saleh L, Perler FB. Protein splicing in cis and in trans. Chem. Rec. 2006;6:183–193. doi: 10.1002/tcr.20082. [DOI] [PubMed] [Google Scholar]
- (86).Zakeri B, Howarth M. Spontaneous intermolecular amide bond formation between side chains for irreversible peptide targeting. J. Am. Chem. Soc. 2010;132:4526–4527. doi: 10.1021/ja910795a. [DOI] [PubMed] [Google Scholar]
- (87).Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. U. S. A. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 2006;106:3468–3496. doi: 10.1021/cr0503097. [DOI] [PubMed] [Google Scholar]
- (89).Fjell CD, Hiss JA, Hancock RE, Schneider G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 2011;11:37–51. doi: 10.1038/nrd3591. [DOI] [PubMed] [Google Scholar]
- (90).Baltz RH. Molecular engineering approaches to peptide, polyketide and other antibiotics. Nat. Biotechnol. 2006;24:1533–1540. doi: 10.1038/nbt1265. [DOI] [PubMed] [Google Scholar]
- (91).Hubbard BK, Walsh CT. Vancomycin assembly: nature's way. Angew. Chem. Int. Ed Engl. 2003;42:730–765. doi: 10.1002/anie.200390202. [DOI] [PubMed] [Google Scholar]
- (92).Koglin A, Walsh CT. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat. Prod. Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Strieker M, Tanovic A, Marahiel MA. Nonribosomal peptide synthetases: structures and dynamics. Curr. Opin. Struct. Biol. 2010;20:234–240. doi: 10.1016/j.sbi.2010.01.009. [DOI] [PubMed] [Google Scholar]
- (94).Koglin A, Walsh CT. Structural insights into nonribosomal peptide enzymatic assembly lines. Nat. Prod. Rep. 2009;26:987–1000. doi: 10.1039/b904543k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 2005;105:425–448. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- (96).Butcher RA, Schroeder FC, Fischbach MA, Straight PD, Kolter R, Walsh CT, Clardy J. The identification of bacillaene, the product of the PksX megacomplex in Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:1506–1509. doi: 10.1073/pnas.0610503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Straight PD, Fischbach MA, Walsh CT, Rudner DZ, Kolter R. A singular enzymatic megacomplex from Bacillus subtilis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:305–310. doi: 10.1073/pnas.0609073103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Nguyen KT, He X, Alexander DC, Li C, Gu JQ, Mascio C, Van Praagh A, Mortin L, Chu M, Silverman JA, Brian P, Baltz RH. Genetically engineered lipopeptide antibiotics related to A54145 and daptomycin with improved properties. Antimicrob. Agents Chemother. 2010;54:1404–1413. doi: 10.1128/AAC.01307-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Arbeit RD, Maki D, Tally FP, Campanaro E, Eisenstein BI, Daptomycin 98–01 and 99–01 Investigators The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin. Infect. Dis. 2004;38:1673–1681. doi: 10.1086/420818. [DOI] [PubMed] [Google Scholar]
- (100).Silverman JA, Mortin LI, Vanpraagh AD, Li T, Alder J. Inhibition of daptomycin by pulmonary surfactant: in vitro modeling and clinical impact. J. Infect. Dis. 2005;191:2149–2152. doi: 10.1086/430352. [DOI] [PubMed] [Google Scholar]
- (101).Shi R, Lamb SS, Zakeri B, Proteau A, Cui Q, Sulea T, Matte A, Wright GD, Cygler M. Structure and function of the glycopeptide N-methyltransferase MtfA, a tool for the biosynthesis of modified glycopeptide antibiotics. Chem. Biol. 2009;16:401–410. doi: 10.1016/j.chembiol.2009.02.007. [DOI] [PubMed] [Google Scholar]
- (102).Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- (103).Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- (104).Henriques ST, Melo MN, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem. J. 2006;399:1–7. doi: 10.1042/BJ20061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- (106).Melo MN, Ferre R, Castanho MA. Antimicrobial peptides: linking partition, activity and high membrane-bound concentrations. Nat. Rev. Microbiol. 2009;7:245–250. doi: 10.1038/nrmicro2095. [DOI] [PubMed] [Google Scholar]
- (107).Houk KN, Leach AG, Kim SP, Zhang X. Binding affinities of host-guest, protein-ligand, and protein-transition-state complexes. Angew. Chem. Int. Ed Engl. 2003;42:4872–4897. doi: 10.1002/anie.200200565. [DOI] [PubMed] [Google Scholar]
- (108).Tyo KE, Alper HS, Stephanopoulos GN. Expanding the metabolic engineering toolbox: more options to engineer cells. Trends Biotechnol. 2007;25:132–137. doi: 10.1016/j.tibtech.2007.01.003. [DOI] [PubMed] [Google Scholar]
- (109).Tsai CW, Hsu NY, Wang CH, Lu CY, Chang Y, Tsai HH, Ruaan RC. Coupling molecular dynamics simulations with experiments for the rational design of indolicidin-analogous antimicrobial peptides. J. Mol. Biol. 2009;392:837–854. doi: 10.1016/j.jmb.2009.06.071. [DOI] [PubMed] [Google Scholar]
- (110).Chan DI, Prenner EJ, Vogel HJ. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim. Biophys. Acta. 2006;1758:1184–1202. doi: 10.1016/j.bbamem.2006.04.006. [DOI] [PubMed] [Google Scholar]
- (111).Falla TJ, Hancock RE. Improved activity of a synthetic indolicidin analog. Antimicrob. Agents Chemother. 1997;41:771–775. doi: 10.1128/aac.41.4.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Cherkasov A, Hilpert K, Jenssen H, Fjell CD, Waldbrook M, Mullaly SC, Volkmer R, Hancock RE. Use of artificial intelligence in the design of small peptide antibiotics effective against a broad spectrum of highly antibiotic-resistant superbugs. ACS Chem. Biol. 2009;4:65–74. doi: 10.1021/cb800240j. [DOI] [PubMed] [Google Scholar]
- (113).Fjell CD, Jenssen H, Hilpert K, Cheung WA, Pante N, Hancock RE, Cherkasov A. Identification of novel antibacterial peptides by chemoinformatics and machine learning. J. Med. Chem. 2009;52:2006–2015. doi: 10.1021/jm8015365. [DOI] [PubMed] [Google Scholar]
- (114).Perron GG, Zasloff M, Bell G. Experimental evolution of resistance to an antimicrobial peptide. Proc. Biol. Sci. 2006;273:251–256. doi: 10.1098/rspb.2005.3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Samuelsen O, Haukland HH, Jenssen H, Kramer M, Sandvik K, Ulvatne H, Vorland LH. Induced resistance to the antimicrobial peptide lactoferricin B in Staphylococcus aureus. FEBS Lett. 2005;579:3421–3426. doi: 10.1016/j.febslet.2005.05.017. [DOI] [PubMed] [Google Scholar]
- (116).He J, Anderson MH, Shi W, Eckert R. Design and activity of a 'dual-targeted' antimicrobial peptide. Int. J. Antimicrob. Agents. 2009;33:532–537. doi: 10.1016/j.ijantimicag.2008.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Fleming A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. British Journal of Experimental Pathology. 1929;10:226–236. [Google Scholar]
- (118).Lu TK, Koeris MS. The next generation of bacteriophage therapy. Curr. Opin. Microbiol. 2011;14:524–531. doi: 10.1016/j.mib.2011.07.028. [DOI] [PubMed] [Google Scholar]
- (119).Sulakvelidze A, Alavidze Z, Morris JG., Jr Bacteriophage therapy. Antimicrob. Agents Chemother. 2001;45:649–659. doi: 10.1128/AAC.45.3.649-659.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Jabes D. The antibiotic R&D pipeline: an update. Curr. Opin. Microbiol. 2011;14:564–569. doi: 10.1016/j.mib.2011.08.002. [DOI] [PubMed] [Google Scholar]
- (121).Gravitz L. Turning a new phage. Nat. Med. 2012;18:1318–1320. doi: 10.1038/nm0912-1318. [DOI] [PubMed] [Google Scholar]
- (122).Petrova OE, Sauer K. Sticky situations: key components that control bacterial surface attachment. J. Bacteriol. 2012;194:2413–2425. doi: 10.1128/JB.00003-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Telford JL, Barocchi MA, Margarit I, Rappuoli R, Grandi G. Pili in gram-positive pathogens. Nat. Rev. Microbiol. 2006;4:509–519. doi: 10.1038/nrmicro1443. [DOI] [PubMed] [Google Scholar]
- (124).Seneviratne CJ, Wang Y, Jin L, Wong SS, Herath TD, Samaranayake LP. Unraveling the resistance of microbial biofilms: has proteomics been helpful? Proteomics. 2012;12:651–665. doi: 10.1002/pmic.201100356. [DOI] [PubMed] [Google Scholar]
- (125).Smith AW. Biofilms and antibiotic therapy: is there a role for combating bacterial resistance by the use of novel drug delivery systems? Adv. Drug Deliv. Rev. 2005;57:1539–1550. doi: 10.1016/j.addr.2005.04.007. [DOI] [PubMed] [Google Scholar]
- (126).Kline KA, Dodson KW, Caparon MG, Hultgren SJ. A tale of two pili: assembly and function of pili in bacteria. Trends Microbiol. 2010;18:224–232. doi: 10.1016/j.tim.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Scott JR, Zahner D. Pili with strong attachments: Gram-positive bacteria do it differently. Mol. Microbiol. 2006;62:320–330. doi: 10.1111/j.1365-2958.2006.05279.x. [DOI] [PubMed] [Google Scholar]
- (128).Kang HJ, Coulibaly F, Clow F, Proft T, Baker EN. Stabilizing isopeptide bonds revealed in gram-positive bacterial pilus structure. Science. 2007;318:1625–1628. doi: 10.1126/science.1145806. [DOI] [PubMed] [Google Scholar]
- (129).Kang HJ, Baker EN. Intramolecular isopeptide bonds give thermodynamic and proteolytic stability to the major pilin protein of Streptococcus pyogenes. J. Biol. Chem. 2009;284:20729–20737. doi: 10.1074/jbc.M109.014514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Alegre-Cebollada J, Badilla CL, Fernandez JM. Isopeptide bonds block the mechanical extension of pili in pathogenic Streptococcus pyogenes. J. Biol. Chem. 2010;285:11235–11242. doi: 10.1074/jbc.M110.102962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (131).Linke C, Young PG, Kang HJ, Bunker RD, Middleditch MJ, Caradoc-Davies TT, Proft T, Baker EN. Crystal structure of the minor pilin FctB reveals determinants of Group A streptococcal pilus anchoring. J. Biol. Chem. 2010;285:20381–20389. doi: 10.1074/jbc.M109.089680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Kang HJ, Baker EN. Intramolecular isopeptide bonds: protein crosslinks built for stress? Trends Biochem. Sci. 2011;36:229–237. doi: 10.1016/j.tibs.2010.09.007. [DOI] [PubMed] [Google Scholar]
- (133).Pointon JA, Smith WD, Saalbach G, Crow A, Kehoe MA, Banfield MJ. A highly unusual thioester bond in a pilus adhesin is required for efficient host cell interaction. J. Biol. Chem. 2010 doi: 10.1074/jbc.M110.149385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Lu TK, Collins JJ. Dispersing biofilms with engineered enzymatic bacteriophage. Proc. Natl. Acad. Sci. U. S. A. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Itoh Y, Wang X, Hinnebusch BJ, Preston JF, 3rd, Romeo T. Depolymerization of beta-1,6-N-acetyl-D-glucosamine disrupts the integrity of diverse bacterial biofilms. J. Bacteriol. 2005;187:382–387. doi: 10.1128/JB.187.1.382-387.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010;8:317–327. doi: 10.1038/nrmicro2315. [DOI] [PubMed] [Google Scholar]
- (137).Lu TK, Collins JJ. Engineered bacteriophage targeting gene networks as adjuvants for antibiotic therapy. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4629–4634. doi: 10.1073/pnas.0800442106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- (139).Cirz RT, Chin JK, Andes DR, de Crecy-Lagard V, Craig WA, Romesberg FE. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005;3:e176. doi: 10.1371/journal.pbio.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Edgar R, Friedman N, Molshanski-Mor S, Qimron U. Reversing bacterial resistance to antibiotics by phage-mediated delivery of dominant sensitive genes. Appl. Environ. Microbiol. 2012;78:744–751. doi: 10.1128/AEM.05741-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Song JY, Jensen SE, Lee KJ. Clavulanic acid biosynthesis and genetic manipulation for its overproduction. Appl. Microbiol. Biotechnol. 2010;88:659–669. doi: 10.1007/s00253-010-2801-2. [DOI] [PubMed] [Google Scholar]
- (142).Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr. Opin. Microbiol. 2008;11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Abaev I, Foster-Frey J, Korobova O, Shishkova N, Kiseleva N, Kopylov P, Pryamchuk S, Schmelcher M, Becker SC, Donovan DM. Staphylococcal Phage 2638A endolysin is lytic for Staphylococcus aureus and harbors an inter-lytic-domain secondary translational start site. Appl. Microbiol. Biotechnol. 2012 doi: 10.1007/s00253-012-4252-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Daniel A, Euler C, Collin M, Chahales P, Gorelick KJ, Fischetti VA. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2010;54:1603–1612. doi: 10.1128/AAC.01625-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Pastagia M, Euler C, Chahales P, Fuentes-Duculan J, Krueger JG, Fischetti VA. A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive Staphylococcus aureus strains. Antimicrob. Agents Chemother. 2011;55:738–744. doi: 10.1128/AAC.00890-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Michel-Briand Y, Baysse C. The pyocins of Pseudomonas aeruginosa. Biochimie. 2002;84:499–510. doi: 10.1016/s0300-9084(02)01422-0. [DOI] [PubMed] [Google Scholar]
- (147).Williams SR, Gebhart D, Martin DW, Scholl D. Retargeting R-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 2008;74:3868–3876. doi: 10.1128/AEM.00141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Scholl D, Cooley M, Williams SR, Gebhart D, Martin D, Bates A, Mandrell R. An engineered R-type pyocin is a highly specific and sensitive bactericidal agent for the food-borne pathogen Escherichia coli O157:H7. Antimicrob. Agents Chemother. 2009;53:3074–3080. doi: 10.1128/AAC.01660-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Saeidi N, Wong CK, Lo TM, Nguyen HX, Ling H, Leong SS, Poh CL, Chang MW. Engineering microbes to sense and eradicate Pseudomonas aeruginosa, a human pathogen. Mol. Syst. Biol. 2011;7:521. doi: 10.1038/msb.2011.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Zendo T, Yoneyama F, Sonomoto K. Lactococcal membrane-permeabilizing antimicrobial peptides. Appl. Microbiol. Biotechnol. 2010;88:1–9. doi: 10.1007/s00253-010-2764-3. [DOI] [PubMed] [Google Scholar]
- (151).van Belkum MJ, Martin-Visscher LA, Vederas JC. Structure and genetics of circular bacteriocins. Trends Microbiol. 2011;19:411–418. doi: 10.1016/j.tim.2011.04.004. [DOI] [PubMed] [Google Scholar]
- (152).D'Costa VM, King CE, Kalan L, Morar M, Sung WW, Schwarz C, Froese D, Zazula G, Calmels F, Debruyne R, Golding GB, Poinar HN, Wright GD. Antibiotic resistance is ancient. Nature. 2011;477:457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- (153).Wright GD, Poinar H. Antibiotic resistance is ancient: implications for drug discovery. Trends Microbiol. 2012;20:157–159. doi: 10.1016/j.tim.2012.01.002. [DOI] [PubMed] [Google Scholar]
- (154).Medema MH, Alam MT, Breitling R, Takano E. The future of industrial antibiotic production: from random mutagenesis to synthetic biology. Bioeng. Bugs. 2011;2:230–233. doi: 10.1111/j.1751-7915.2010.00226.x. [DOI] [PubMed] [Google Scholar]
- (155).Ruder WC, Lu T, Collins JJ. Synthetic biology moving into the clinic. Science. 2011;333:1248–1252. doi: 10.1126/science.1206843. [DOI] [PubMed] [Google Scholar]
- (156).Cheng AA, Lu TK. Synthetic biology: an emerging engineering discipline. Annu. Rev. Biomed. Eng. 2012;14:155–178. doi: 10.1146/annurev-bioeng-071811-150118. [DOI] [PubMed] [Google Scholar]