

Abstract
Cav1.3 channels mediate Ca2+ influx that triggers exocytosis of glutamate at cochlear inner hair cell (IHC) synapses. Harmonin is a PDZ-domain-containing protein that interacts with the C-terminus of the Cav1.3 α1 subunit (α11.3) and controls cell surface Cav1.3 levels by promoting ubiquitin-dependent proteosomal degradation. However, PDZ-domain-containing proteins have diverse functions and regulate other Cav1.3 properties, which could collectively influence presynaptic transmitter release. Here, we report that harmonin binding to the α11.3 distal C-terminus (dCT) enhances voltage-dependent facilitation (VDF) of Cav1.3 currents both in transfected HEK293T cells and in mouse inner hair cells. In HEK293T cells, this effect of harmonin was greater for Cav1.3 channels containing the auxiliary Cavβ1 than with the β2 auxiliary subunit. Cav1.3 channels lacking the α11.3 dCT were insensitive to harmonin modulation. Moreover, the ‘deaf-circler’dfcr mutant form of harmonin, which does not interact with the α11.3 dCT, did not promote VDF. In mature IHCs from mice expressing the dfcr harmonin mutant, Cav1.3 VDF was less than in control IHCs. This difference was not observed between control and dfcr IHCs prior to hearing onset. Membrane capacitance recordings from dfcr IHCs revealed a role for harmonin in synchronous exocytosis and in increasing the efficiency of Ca2+ influx for triggering exocytosis. Collectively, our results indicate a multifaceted presynaptic role of harmonin in IHCs in regulating Cav1.3 Ca2+ channels and exocytosis.
Key points
Cav1.3 Ca2+ channels mediate sound transmission by triggering presynaptic exocytosis of glutamate from cochlear inner hair cells (IHCs).
Harmonin is a PDZ-domain-containing protein in IHCs that is altered in Usher syndrome, a form of deaf–blindness in humans.
We show that harmonin enhances Cav1.3 voltage-dependent facilitation (VDF) in transfected HEK293T cells in a manner that depends on the identity of the auxiliary Ca2+ channel β subunit.
Cav1.3 VDF is impaired, and synchronous exocytosis and the Ca2+ efficiency of exocytosis are reduced, in IHCs from deaf-circler mice expressing a mutant form of harmonin (dfcr) that cannot interact with Cav1.3.
We conclude that harmonin regulates presynaptic function in mouse IHCs, which adds to our understanding of the factors that may influence hearing impairment in Usher syndrome.
Introduction
Voltage-gated Cav1.3 Ca2+ channels are highly expressed in cochlear inner hair cells (IHCs), where their activity is tightly coupled to the exocytic release from specialized ‘ribbon’ synapses (Platzer et al. 2000; Brandt et al. 2003). Mice lacking Cav1.3 are deaf (Platzer et al. 2000; Dou et al. 2004) as are humans with loss-of-function mutations in the CACNA1D gene encoding the pore-forming Cav1.3 α1 subunit (α11.3) (Baig et al. 2011). Cav1.3 channels are subject to diverse forms of regulation, which can strongly impact neuronal and cardiac signalling (Mangoni et al. 2003; Olson et al. 2005; Hetzenauer et al. 2006; Zhang et al. 2006; Chan et al. 2007; Navedo et al. 2007). Therefore, characterization of the factors that modulate Cav1.3 channels in IHCs is essential for understanding the dynamics of presynaptic Ca2+ signals and sound transmission by IHCs.
Like other Cav1 channels, Cav1.3 can interact directly with various proteins (Calin-Jageman & Lee, 2008). The distal C-terminus (dCT) of the Cav1.3 α11.3 contains a consensus site for binding to PDZ (PSD-95 (postsynaptic density-95)/Discs large/ZO-1 (zona occludens-1)) domains (Songyang et al. 1997). Interactions with PDZ-domain-containing proteins affect the localization and function of Cav1.3 in neurons (Olson et al. 2005; Zhang et al. 2005, 2006). One such protein, erbin, binds to the α11.3 dCT and potentiates Cav1.3 currents in response to depolarizing stimuli through a process known as voltage-dependent facilitation (VDF; Calin-Jageman et al. 2007). Densin-180 also interacts with the α11.3 dCT but does not enhance Cav1.3 VDF. Rather, densin-180 tethers calmodulin-dependent protein kinase II to the Cav1.3 channel complex, which mediates Ca2+-dependent facilitation of Cav1.3 currents in response to high-frequency repetitive stimuli (Jenkins et al. 2010).
Like erbin and densin-180, harmonin is a PDZ-domain-containing protein expressed in the brain, but is additionally localized in IHCs (Verpy et al. 2000; Reiners et al. 2005). The gene encoding harmonin corresponds to the USH1C locus for Usher Type 1 syndrome (Verpy et al. 2000), an autosomal recessive sensory disorder characterized by deafness, vestibular dysfunction, and late-onset retinitis pigmentosa (Kimberling & Moller, 1995). Harmonin is concentrated in the apical hair bundles of cochlear and vestibular hair cells (Adato et al. 2005), where it interacts with multiple proteins and regulates mechanotransduction channels that convert mechanical stimuli into changes in hair cell membrane potential (Grillet et al. 2009; Michalski et al. 2009). In mature IHCs, harmonin is also localized to a subset of ribbon-type active zones. Harmonin binds to the α11.3 dCT, which enhances proteosomal degradation of Cav1.3 and controls Cav1.3 channel density at mouse IHC synapses (Gregory et al. 2011). It is not known whether, like erbin and densin-180, harmonin has other modulatory actions on Cav1.3 that could impact presynaptic function in IHCs. In addition, given the strong localization of harmonin at IHC synapses, harmonin may also play a role in glutamate exocytosis, which has also not been investigated.
To address these open questions, we tested if harmonin influenced additional properties of Cav1.3, and probed the impact of harmonin on exocytosis in IHCs. We found that like erbin, harmonin enhances Cav1.3 VDF, which depends on the interaction of harmonin with the α11.3 dCT and the identity of the Cavβ subunit. Moreover, we established that harmonin regulates Cav1.3 VDF and exocytosis in mouse IHCs. Our results highlight new roles for harmonin as a regulator of presynaptic function in IHCs.
Methods
Ethical approval
All procedures involving mice were approved by the Institutional Animal Care and Use Committee at the University of Iowa in accordance with National Institutes of Health guidelines and animal welfare guidelines at the University of Göttingen and the State of Lower Saxony. After mice were killed by decapitation (for mice less than 10 days old) or isoflurane overdose and/or decapitation (for mice greater than 10 days old), the skull was opened, and the cochlea was removed and opened at the apex so that the apical coil could be harvested for electrophysiological experiments.
Constructs and molecular biology
The following Cav subunit cDNAs were used: α11.3 containing exon 42 (GenBank no. AF370009 and AF370010 for additional sequence encoded by exon 42), β1b (GenBank no. NM017346), α11.2 (GenBank no. M67515), β2A (GenBank no. NM053851), and α2δ-1 (GenBank no. M21948). Expression constructs for FLAG-tagged α11.3, α11.3L-A, α11.3exon42A, and green fluorescent protein (GFP)- and myc-tagged harmonin, dfcr mutant were previously described (Calin-Jageman et al. 2007; Gregory et al. 2011).
Cell culture and transfection
Human embryonic kidney cells transformed with SV40 T-antigen (HEK293T) were maintained in Dulbecco's modified Eagle's medium with 10% fetal bovine serum (Life Technologies, Grand Island, NY, USA) at 37°C in a humidified atmosphere under 5% CO2. Cells were grown to ∼60–90% confluence in 100 mm plates and transfected using GenePorter Reagent (Gene Therapy Systems, San Diego, CA, USA). For immunoprecipitation experiments, HEK293T cells were transfected with cDNAs encoding Cav1.3 (FLAG-α11.3, FLAG-α11.3exon42A, or FLAG-α11.3L-A (6 μg), β1b and α2δ (2 μg each)) and myc-harmonin (4 μg). For electrophysiological experiments, cells were plated on 35 mm culture dishes and transiently transfected using Fugene transfection reagent (Promega, Fitchburg, WI, USA). A total of ∼3 μg total DNA was transfected: α11.3, 1.5 μg; β, 0.8 μg; α2δ, 0.8 μg; ±GFP-tagged harmonin or dfcr mutant, 0.5 μg; or GFP expression plasmid, 0.01 μg.
Coimmunoprecipitation
HEK293T cells were harvested and lysed 48 h after transfection. Lysates were incubated with ANTI-FLAG M2-Agarose Affinity Gel (all reagents from FLAG Immunoprecipitation Kit, Sigma-Aldrich, St Louis, MO, USA) for 2.5 h, rotating at 4°C. After three washes with wash buffer (provided in the FLAG Immunoprecipitation Kit), proteins were eluted with SDS-containing sample buffer and subjected to SDS-PAGE. Coimmunoprecipitated proteins were detected by Western blotting with antibodies against α11.3 (Ab144; Gregory et al. 2011) or myc epitopes (Sigma-Aldrich). Following incubation with appropriate secondary antibodies, WesternC reagent (Bio-rad, Hercules, CA, USA) was utilized for development and the Geldoc Imager for image collection (Bio-rad). Quantification was performed densitometrically with Quantity One software (Bio-rad). To obtain the fraction of harmonin that coimmunoprecipited with α11.3, Western blot signals corresponding to bands for harmonin were divided by those representing the FLAG-immunoprecipitated α11.3. For each experiment, these values for harmonin coimmunoprecipitated with α11.3ex42A or α11.3L-A were normalized to that for α11.3 to determine the percentage change in harmonin that coimmunoprecipitated with α11.3ex42A or α11.3L-A relative to that for α11.3. Data from three independent experiments were averaged (mean ± SEM).
Electrophysiological recordings
Ba2+ currents (IBa) were recorded 48–72 h after transfection at room temperature using the whole-cell patch clamp electrophysiology technique from transiently transfected HEK293T cells. The internal solution contained (in mm): 140 NMDG, 5 EGTA, 10 Hepes, 2 MgCl2, 2 Mg-ATP, pH 7.3 (with methanesulfonate) and adjusted to ∼290 mosmol l−1 with glucose. The external solution contained (in mm): 150 Tris, 2 MgCl2, 10 BaCl2, pH 7.35 (with methanesulfonate) and adjusted to ∼310 mosmol l−1 with glucose. Pipettes of 3–5 MΩ resistance were used. Voltage clamp recordings were performed with an EPC-9 or EPC-10 amplifier under control of PULSE or Patchmaster software (HEKA Elektronik, Lambrecht, Germany). Currents were filtered at 2 kHz and sampled at 10–20 kHz. A P/4 protocol was used to subtract leak currents.
For whole-cell patch clamp recordings of mouse IHCs, cochlear tissue was dissected from mice (postnatal days (p) 6–8 or p16–18) in Minimum Essential Medium (MEM)/Glutamax-1 (Invitrogen, Gaithersburg, MD, USA) supplemented with 10 mm Hepes at room temperature and kept up to 18 h at 37°C prior to recording. IHCs in the apical cochlear turn were visualized on an upright microscope (BX51WI, Olympus) with a ×40 water-immersion objective with DIC optics. The basolateral membrane of IHCs was patch-clamped with electrodes pulled from thick-walled borosilicate glass capillaries (1B150F, Warner Instruments, Camden, CT, USA). The internal solution contained (in mm): 120 caesium gluconate, 80 CsCl, 0.1 CaCl2, 4 MgATP, 5 Hepes and 5 EGTA; pH was adjusted to 7.35 with CsOH; osmolarity ∼305 mosmol l−1. External solution contained (in mm): 105 NaCl, 5.8 KCl, 10 CsCl, 55 TEA-Cl, 10 BaCl2, 1 MgCl, 10 glucose and 10 Hepes supplemented with MEM Vitamins and Amino Acids (Invitrogen, Gaithersburg, MD, USA) at 1X; pH was adjusted to 7.4 with TEA-OH; osmolarity ∼320 mosmol l−1. On the day of recording, 4-aminopyridine (4 mm), apamin (0.3 mm) and TTX (0.5 mm, for p6–8 IHCs) were added to the external solution. Electrode resistances were 3.5–6.2 MΩ in the external solution. Data were acquired with HEKA EPC-9 or EPC-10 amplifiers controlled by Patchmaster software (HEKA Elektronik, Lambrecht, Germany). Leak subtraction was done online with a P/6 protocol. Series resistance was compensated with the patch clamp circuitry (50–70%); average uncompensated series resistance was 12.9 ± 0.6 (n= 89 IHCs). Currents were low-pass filtered at 5 kHz and sampled at 20 kHz except for VDF measurements, where currents were filtered at 2.9 kHz and sampled at 10 kHz. Voltages were not corrected for the liquid junction potential of −7 mV in the external recording solution.
Electrophysiological data were analysed with custom routines in IgorPro software (Wavemetrics, Portland, OR, USA). Average data are expressed as mean ± SEM. Statistical comparisons were done using SigmaPlot software (Systat, Chicago, IL, USA).
Confocal Ca2+ imaging
Confocal Ca2+ imaging was performed in IHCs from dfcr mice and their wild-type littermates as described previously (Frank et al. 2009; Gregory et al. 2011). In brief, synaptic Ca2+ microdomains were identified as hotspots of Fluo-5N fluorescence (low affinity Ca2+ indicator) in XY scans using long (200–254 ms) step depolarizations. We then positioned the laser at the peak pixel of each Ca2+ microdomain as identified in the XY scan (spot detection) and invoked Ca2+ influx by 20 ms step depolarizations to −32 mV. We then applied a 50 ms depolarization to +63 mV preceding the second 20 ms depolarization to −32 mV in order to facilitate the Ca2+ influx. The internal solution contained (in mm): 115 caesium glutamate, 13 TEA-Cl, 1 MgCl2, 1 CaCl2, 10 EGTA, 2 ATP-Mg, 0.3 GTP-Na, 20 Hepes (pH adjusted with CsOH to 7.2, osmolarity ∼295 mosmol l−1) and 0.4 Fluo-5N (penta-potassium salt; Invitrogen). The external solution contained (in mm): 102 NaCl, 35 TEA-Cl, 2.8 KCl, 5 CaCl2, 1 MgCl2, 10 Hepes, 1 CsCl, 11.1 d-glucose (pH adjusted with NaOH to 7.2, osmolarity ∼300 mosmol l−1). Data are presented as mean ± SEM, unless otherwise stated.
Capacitance recordings
For capacitance recordings, IHCs from dfcr mice and their wild-type littermates (p13–19) were subjected to perforated and ruptured patch-clamp recording as described previously (Moser & Beutner, 2000). The internal solution for perforated-patch experiments contained (in mm): 130 caesium gluconate, 10 TEA-Cl, 10 4-aminopyridine, 1 MgCl2, 10 Hepes (pH adjusted with HCl to 7.17, osmolarity ∼290 mosmol l−1) and 300 μg ml−1 amphotericin B. The internal solution for EGTA experiments contained (in mm): 115 caesium glutamate, 13 TEA-Cl, 1 MgCl2, 4 EGTA, 2 CaCl2, 20 Hepes, 2 Mg-ATP, 0.3 Na-GTP (pH adjusted with CsOH to 7.2, osmolarity ∼290 mosmol l−1). The external solution contained (in mm): 100–104 NaCl, 35 TEA-Cl, 2.8 KCl, 10 CaCl2, 1 MgCl2, 10 Hepes, 1 caesium gluconate or CsCl, 5 4-aminopyridine, 11.1 d-glucose (pH adjusted with NaOH to 7.2, osmolarity ∼300 mosmol l−1). For most perforated-patch experiments the external solution also contained apamin (0.1 mm). An EPC-9 amplifier controlled by Pulse software (HEKA Elektronik) was used for measurements. All voltages were corrected for liquid junction potentials. Currents were sampled at 20 kHz and low-pass filtered at 2 kHz. Cells that displayed a holding current exceeding −50 pA were discarded from analysis. Ca2+ currents were further isolated using a P/n protocol. Series resistance (RS) measured at the beginning of the perforated- patch recording was 26.3 ± 1.5 MΩ, n= 15, for control, and 26.9 ± 1.0 MΩ, n= 26, for dfcr. For ruptured-patch recordings, RS was 8.5 ± 0.5 MΩ, n= 10, for control, and 10.4 ± 1.3 MΩ, n= 10, for dfcr. To calculate ΔCm, the initial 30 ms following the voltage step were ignored due to a non-exocytic capacitance artifact (Neef et al. 2007) and the average Cm measured over at least 40 ms. No RS compensation was used.
Immunofluorescence labelling of mouse organ of Corti
Double-labelling for CtBP2/RIBEYE and GluA2/3 was performed as previously described (Khimich et al. 2005). The following antibodies were used: mouse IgG1 anti-CtBP2 (BD Biosciences, 1:200), rabbit anti-GluR2/3 (Chemicon, 1:200) and secondary AlexaFluor488- and AlexaFluor568-labelled antibodies (Molecular Probes, 1:200). Confocal images were acquired using a laser scanning confocal microscope (Leica TCS SP5, Leica MicrosystemsCMS GmbH, Mannheim, Germany) with 488 nm (Ar) and 561 nm (DPSS) lasers for excitation and a ×63 oil immersion objective (NA = 1.4–0.7). For 3-D reconstructions of the specimen, z-axis stacks of 2-D images were taken with a step size of 0.5 μm. Image stacks represent maximum z projections, done in ImageJ. The CtBP-2/RIBEYE and GluA2/3 immunofluorescence spots were counted in the z-stacks and divided by the number of IHCs (number of nuclei in the field of view) in order to yield the number of synapses per IHC. Juxtaposed pre- and postsynaptic spots were considered as mature synapses (Khimich et al. 2005).
Results
Harmonin enhances VDF of Cav1.3 in HEK293T cells
We have previously shown that cotransfection of harmonin with Cav1.3 channels in HEK293T cells increases ubiquitination of α11.3 and decreases Cav1.3 current density through enhanced proteosomal degradation of α11.3 (Gregory et al. 2011). While determining if harmonin affected other parameters of Cav1.3 function, we noted that harmonin had a particularly prominent effect of enhancing VDF in HEK293T cells. We measured VDF with a triple-pulse voltage protocol in which test current amplitudes are compared before (P1) and after (P2) a conditioning prepulse to various voltages (Fig. 1A). Ba2+ was used as the charge carrier to increase resolution of VDF by limiting the competing effects of Ca2+-dependent inactivation (CDI). With this protocol, VDF is evident as an increase in the ratio of the P2:P1 current (Fratio) with depolarized prepulse voltages. Cotransfection of harmonin with Cav1.3 (α11.3, β1b and α2δ subunits) caused a significant increase in maximal VDF seen with a +50 mV prepulse (Fratio,+50= 1.18 ± 0.02 for Cav1.3 alone vs. 1.52 ± 0.08 for Cav1.3 + harmonin; P < 0.01, by Mann–Whitney rank sum test; Fig. 1B). Increased magnitude of IBa with harmonin was not secondary to shifts in the conductance–voltage profile, since Boltzmann fits of tail current activation curves revealed similar parameters (V1/2=−19.6 ± 3.0; k= 7.9 ± 0.8 for Cav1.3 alone vs. V1/2=−18.6 ± 7.5; k= 7.6 ± 1.1 for Cav1.3 + harmonin, P= 0.98 for V1/2 and P= 0.52 for k; Fig. 1C). These results confirm that harmonin increases Cav1.3 VDF, which is consistent with its interaction with Cav1.3 channels in the plasma membrane.
Figure 1. Harmonin enhances VDF of Cav1.3 channels in transfected HEK293T cells.
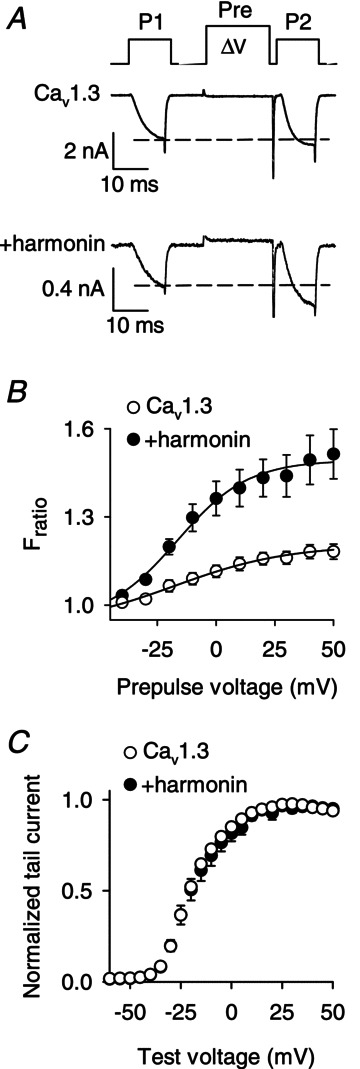
A, voltage protocol for VDF (top) and representative traces showing IBa evoked by 10 ms steps from −90 to −20 mV before (P1) and after (P2) a 20 ms conditioning prepulse (Pre) to +50 mV. B, ratio of P2/P1 current amplitudes (Fratio) is plotted against prepulse voltage for Cav1.3 alone (α11.3, β1b, α2δ; n= 12) or +harmonin (n= 12). C, normalized tail current–voltage relationships obtained for Cav1.3 alone (n= 14) or +harmonin (n= 10). IBa was evoked by test pulses from −90 mV to various voltages. Tail currents measured upon repolarization to −70 mV were normalized to the maximal tail current amplitude and plotted against test voltage.
Cavβ subunits modulate effects of harmonin on Cav1.3 VDF
Although we found that harmonin modulated the VDF of Cav1.3 channels containing the auxiliary Cavβ1b subunit (Cav1.3(β1b), Fig. 1), Cavβ2 is the major Cavβ subunit contributing to Cav1.3 function in mouse IHCs (Neef et al. 2009). Therefore, we characterized the impact of harmonin on VDF in HEK293T cells transfected with Cav1.3 subunits containing Cavβ2A (Cav1.3(β2A)). While Cav1.3(β2A) exhibited marginal VDF (Fratio,+50= 1.17 ± 0.02) that was not significantly different from Cav1.3(β1b) (Fratio,+50= 1.18 ± 0.02; P= 0.22, by t test; Fig. 2A–C), cotransfection with harmonin caused a smaller increase in VDF of Cav1.3(β2A) than Cav1.3(β1b). While Cav1.3(β1b) VDF was increased 28.5 ± 7.2% by harmonin, this increase was only 10.6 ± 1.6% for Cav1.3(β2A) (P < 0.05, by t test; Fig. 2D). These results confirm the importance of Cavβ subunits in modulating responsiveness of Cav1.3 VDF to PDZ-domain-containing proteins (Calin-Jageman et al. 2007), and suggest that native Cav1.3(β2) in IHCs may undergo VDF enhancement by harmonin, although to a lesser extent than Cav1.3(β1b).
Figure 2. The extent of Cav1.3 VDF due to harmonin depends on the identity of the Cavβ subunit.
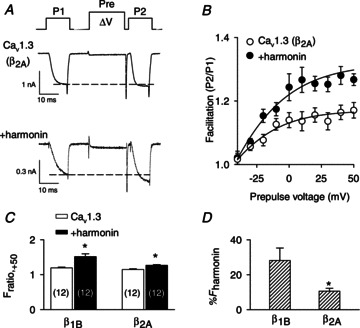
A and B, voltage protocol, representative IBa, and Fratio were as described in Fig. 1A and B except for channels containing the Cavβ2A subunit. n= 12 cells for Cav1.3(β2A) alone, n= 12 cells for +harmonin. C, comparison of facilitation obtained at +50 mV prepulse voltage (Fratio,+50) for β1b- or β2A-containing channels. *P < 0.001 compared to Cav1.3 alone, by t test. Number of cells is indicated in parentheses. D, percentage increase in Fratio,+50 due to harmonin (%Fharmonin) for channels with β1b (n= 12 cells) or β2A (n= 12 cells). %Fharmonin was calculated as (Fratio,+50 for Cav1.3 alone/mean Fratio,+50 for +harmonin) × 100. *P < 0.05 by t test.
Harmonin binding to α11.3 dCT is required for VDF enhancement
To test if increased VDF was due to harmonin binding to the α11.3 dCT, we took advantage of a short splice variant of the rat α11.3 lacking the dCT in which substitution of exon 42A for exon 42 eliminates much of the C-terminal domain including the PDZ-binding sequence in the dCT (Xu & Lipscombe, 2001). We also used α11.3 constructs in which the final leucine residue in the dCT was mutated to alanine (Cav1.3L-A; Fig. 3A). This mutation disrupts the consensus PDZ-binding sequence and prevents binding of harmonin to a C-terminal fragment of α11.3 in vitro (Gregory et al. 2011). To verify that Cav1.342A and Cav1.3L-A have limited interaction with harmonin, we compared their abilities to coimmunoprecipitate with harmonin with that of the wild-type Cav1.3 in transfected HEK293T cells. While harmonin still coimmunoprecipitated with both Cav1.342A and Cav1.3L-A, quantitative analyses revealed a consistent reduction in the amount of harmonin that associated with Cav1.342A and Cav1.3L-A (66.5 ± 8.8% and 82.1 ± 3.9%, respectively) compared to wild-type Cav1.3 (Fig. 3B and C). Evidently, the dCT contributes to, but is not the sole determinant for, harmonin binding to Cav1.3. Yet, electrophysiological recordings revealed that Cav1.342A and Cav1.3L-A underwent VDF that was not changed by cotransfection with harmonin. As we have shown previously, Cav1.342A VDF was greater than for exon 42-containing channels, since the dCT contains a module that normally inhibits VDF (Calin-Jageman et al. 2007). However, there was no difference in maximal VDF in cells transfected with Cav1.342A alone (Fratio,+50= 1.37 ± 0.06) and those cotransfected with harmonin (Fratio,+50= 1.44 ± 0.08; P= 0.48, by t test; Fig. 4A). Similarly, harmonin did not affect VDF of Cav1.3L-A (Fratio,+50= 1.14 ± 0.03 for Cav1.3L-A alone vs. 1.18 ± 0.04 for Cav1.3L-A+ harmonin, P= 0.75, by t test; Fig. 4B). These results demonstrate that, despite other potential harmonin interaction sites within the Cav1.3 channel complex, harmonin binding to the dCT is required for VDF modulation.
Figure 3. Harmonin binding is reduced by disruption of the PDZ-binding site in the α11.3 dCT.
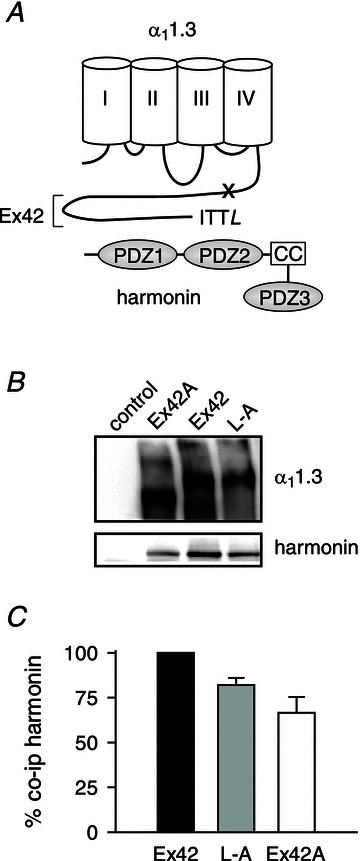
A, schematic diagram of harmonin and α11.3. The long C-terminal domain encoded by exon 42 (Ex42) includes the PDZ-binding motif (ITTL) that interacts with the second of three PDZ domains of harmonin. The coiled-coil domain (CC) that is deleted in the dfcr harmonin mutant is indicated. The ‘x’ marks approximate location of truncation of the dCT due to inclusion of exon 42A. B, coimmunoprecipitation of harmonin with Cav1.3 channels in transfected HEK293T cells. Cells were transfected with harmonin alone (control) or cotransfected with Cav1.3 subunits including α11.3 with full-length (Ex42) or truncated (Ex42A) dCT or α11.3 with terminal leucine substituted with alanine (L-A). Western blotting detected α11.3 (upper panel) and coimmunoprecipitated harmonin (lower panel). C, quantification reflecting percentage change in harmonin coimmunoprecipitated with α11.3Ex42A and α11.3L-A compared to that for α11.3Ex42 (%co-ip harmonin). See Methods for details.
Figure 4. Disruption of the PDZ-binding site in α11.3 prevents modulation of VDF by harmonin.
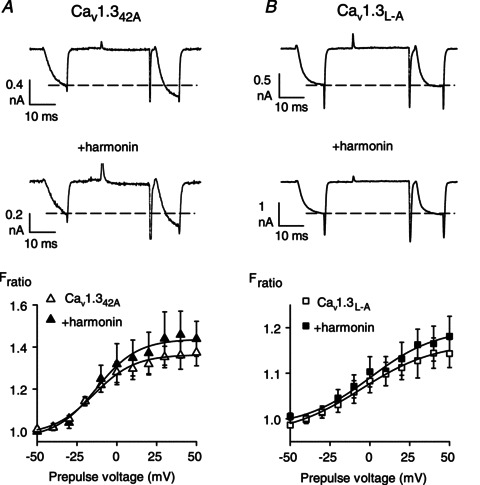
Same as in Fig. 1A and B except for cells transfected with Cav1.342A alone (n= 10) or +harmonin (n= 6) (A), or Cav1.3L-A alone (n= 12) or +harmonin (n= 9) (B).
The dfcr harmonin mutant does not regulate Cav1.3 VDF
‘Deaf-circler’ (dfcr) mice harbour a mutation in the gene encoding harmonin which deletes 132 amino acids between PDZ domains 2 and 3. Similar to humans with Usher syndrome, dfcr mice are deaf and exhibit vestibular defects (Johnson et al. 2003). The dfcr mutant form of harmonin still contains the PDZ domain 2, which interacts with the α11.3 dCT, but the internal deletion removes the coiled-coil domain, which disrupts binding of PDZ ligands, including α11.3 (Gregory et al. 2011). The dfcr mutant does not bind to the α11.3 dCT in vitro and cannot modulate Cav1.3 current density like the wild-type harmonin in transfected cells and in mouse IHCs (Gregory et al. 2011). Since the effects of the dfcr mutant on Cav1.3 VDF have not been characterized, we first compared the effects of co-transfecting wild-type or dfcr harmonin with Cav1.3 in HEK293T cells.
Consistent with the importance of harmonin interactions with the α11.3 dCT for modulation of Cav1.3 VDF (Figs 1–4), dfcr harmonin did not augment VDF of Cav1.3(β2A) (Fratio,+50= 1.15 ± 0.02 for Cav1.3(β2A) alone vs. 1.14 ± 0.02 for Cav1.3(β2A) +dfcr; P= 0.99, by t test; Fig. 5A) or Cav1.3(β1b) (Fratio,+50= 1.21 ± 0.02 for Cav1.3(β1b) alone vs. 1.27 ± 0.04 for Cav1.3(β1b) +dfcr; P= 0.18, by t test; data not shown). To compare a second metric for VDF, we measured the activation kinetics of IBa evoked before and after the conditioning prepulse. Since VDF involves enhanced channel opening in response to depolarization, VDF should manifest as faster activation of the P2 current relative to the P1. To test this, we obtained the time constants (τ) for activation with exponential fits of IBa for P1 and P2, such that enhanced VDF due to harmonin should be measurable as a larger difference in τ for P1 and P2 compared to the dfcr mutant (Δτ, Fig. 5B). While P2 currents activated significantly faster (∼36%) than P1 currents in cells cotransfected with harmonin, no such difference was observed with the dfcr mutant (Fig. 5B). VDF by this metric was significantly greater in cells cotransfected with harmonin than with dfcr mutant (∼70%; Fig. 5B), which further confirms the reduced ability of the dfcr mutant to modulate Cav1.3.
Figure 5. The dfcr mutant of harmonin does not enhance Cav1.3 VDF.
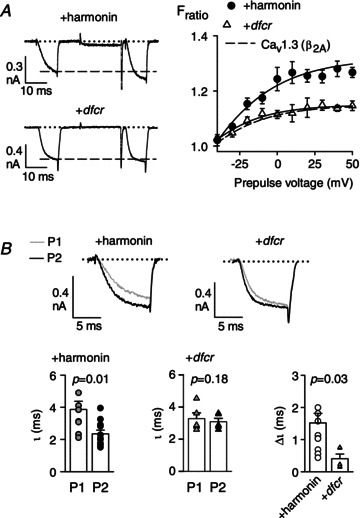
A, same as in Fig. 1A and B except for cells transfected with Cav1.3(β2A) alone (n= 12) or +dfcr (n= 5). Dashed line in graph represents data redrawn from Fig. 2B for Cav1.3(β2A) + harmonin. B: top, representative current traces for P1 and P2 overlaid for comparison; bottom, time constants measured from exponential fits of P1 or P2 current traces for Cav1.3(β2A) + harmonin or Cav1.3(β2A) +dfcr. Δτ represents difference in τ for P1 and P2 currents. P values for τ were determined by t test and for Δτ from paired t test.
Neither wild-type nor dfcr harmonin modulate CDI
To determine if harmonin regulates other Cav1.3 properties in transfected HEK293T cells, we measured the effects of harmonin on inactivation. Like other Cav channels, Cav1.3 undergoes inactivation due to Ca2+- or voltage-dependent mechanisms (CDI or VDI, respectively). CDI is due to calmodulin, which senses local Ca2+ influx due to its direct association with the channel (reviewed in Christel & Lee, 2012). Since Ba2+ ions bind poorly to calmodulin (Wang, 1985), IBa exhibits primarily VDI. Depending on the identity of the auxiliary β subunit, ICa can show both CDI and VDI such that CDI can be isolated as the difference in inactivation of ICa and IBa. With 300 ms step-depolarizations, we measured inactivation as the ratio of the residual current amplitude at the end of the pulse (Ires) and the peak current (Ipk) amplitude (Ires/Ipk), and CDI as the difference in Ires/Ipk for ICa and IBa (Fig. 6A). As expected, ICa inactivated significantly faster than IBa in cells transfected with Cav1.3 alone or cotransfected with harmonin or the dfcr mutant. However, there was no difference in CDI between the three groups (Fig. 6A). Since our metric for CDI may not have detected effects of harmonin or dfcr on the kinetics of CDI, we compared parameters from double exponential fits of the ICa. There was no significant difference in the fractional contribution or time constants for fast or slow inactivation (Fig. 6B). These results indicate that harmonin does not affect CDI in transfected HEK293T cells.
Figure 6. Cav1.3 CDI is not affected by harmonin or the dfcr mutant.
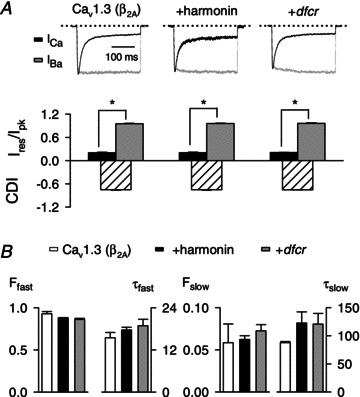
A, top, representative traces for ICa and IBa normalized and overlaid for comparison in cells transfected with Cav1.3(β2A) alone or cotransfected with harmonin or dfcr. Currents were evoked by 300 ms pulses to −20 mV from −90 mV. Bottom, inactivation was calculated as the current amplitude at the end of the pulse normalized to the peak current amplitude (Ires/Ipk) and is shown for ICa and IBa. CDI represents the difference in Ires/Ipk for ICa and the mean Ires/Ipk for IBa. For Cav1.3(β2A) alone, n= 5 for ICa, n= 10 for IBa; for +harmonin, n= 7 for ICa, n= 11 for IBa; for +dfcr, n= 9 for ICa, n= 9 for IBa. *P < 0.001 by t test. B, parameters obtained from double exponential fits of current traces obtained as in A.
Impaired VDF in a mouse model of Usher syndrome
To test the physiological relevance of Cav1.3 VDF modulation by harmonin, we performed whole-cell patch clamp recordings of IHCs from mice expressing the dfcr mutant. We showed previously that Cav1.3 current density is abnormally elevated in IHCs from mature dfcr mice, consistent with decreased proteosomal degradation of Cav1.3 (Gregory et al. 2011). If harmonin also enhances VDF of Cav1.3, we would expect VDF to be reduced in IHCs from dfcr mice. Since the presynaptic localization of harmonin and functional interactions of harmonin with Cav1.3 are characteristic of IHCs from mice at ages (older than p12) after hearing onset (Gregory et al. 2011), modulation of Cav1.3 VDF would be expected in mouse IHCs at p16–18 but not p6–8.
To test these predictions, we compared Cav1.3 VDF in IHCs from heterozygous control and homozygous mutant dfcr mice. Since Cav1.3 accounts for ∼90% of the IHC whole-cell Ca2+ current (Platzer et al. 2000; Brandt et al. 2003), recording solutions were designed to isolate Cav1.3 IBa from other ionic currents without the addition of Cav channel blockers. To determine the optimal conditions for measuring VDF in IHCs, we measured the onset of VDF by comparing test currents evoked before (P1) and after (P2) a conditioning prepulse of varying durations. VDF increased exponentially with prepulse duration with a time constant of ∼11 ms (Fig. 7A). Since maximal VDF was obtained with a 50 ms prepulse duration, we used 50 ms prepulses for the comparisons of VDF in control and dfcr IHCs. Consistent with our findings in HEK293T cells, IBa exhibited modest VDF in mature (p16–18) control IHCs (Fratio,+50= 1.15 ± 0.01) that was significantly weaker in IHCs from dfcr mice (Fratio,+50= 1.07 ± 0.01, P < 001, by t test; Fig. 7B). This difference in VDF was not observed in immature IHCs (p6–8; Fratio,+50= 1.08 ± 0.01 for control vs. 1.07 ± 0.01 for dfcr, P= 0.59, by t test; Fig. 7C). VDF in mature IHCs was associated with significantly faster activation kinetics of IBa evoked after (P2) compared to before (P1) the prepulse (in control IHCs, τP1= 0.60 ± 0.04 ms vs.τP2= 0.37 ± 0.02 ms for +50 mV prepulse; P < 0.001, by paired t test; Fig. 8A and B). By this metric, VDF was still evident in dfcr IHCs (τP1= 0.66 ± 0.05 ms vs.τP2= 0.41 ± 0.03 ms for +50 mV prepulse; P < 0.001, by paired t test; Fig. 8C). However, in dfcr IHCs, the prepulse-induced acceleration of IBa activation was 30–68% weaker than in control IHCs (Δτ= 0.11 ± 0.02 ms for control vs. 0.05 ± 0.01 ms for dfcr, P= 0.02 by Mann–Whitney rank sum test; Fig. 8D). Reduced VDF in mature dfcr IHCs was not secondary to differences in control and dfcr IHCs in terms of CDI or VDI (Supplemental Fig. 1, available online only), or voltage-dependent activation of IBa (Table 1). Therefore, in addition to regulating Cav1.3 channel density in the plasma membrane, harmonin also potentiates Cav1.3 VDF in mature mouse IHCs.
Figure 7. VDF is weaker in dfcr compared to control IHCs after hearing onset in mice.
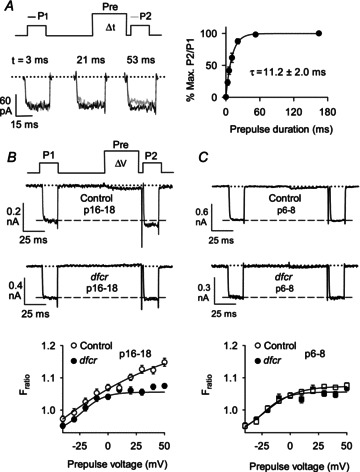
A, measurement of VDF onset. Left, voltage protocol and representative current traces obtained with indicated prepulse durations. P1 and P2 currents are overlaid for comparison. Right, P2/P1 ratios were expressed as percentage of the maximal VDF obtained with a 165 ms prepulse and plotted against prepulse duration. Smooth line represents single-exponential fit. The mean time constant (±SEM) is indicated. B and C, top, voltage protocol for IHC VDF showing 20 ms test pulses (P1, P2) from −75 to −15 mV separated by a 50 ms prepulse to various voltages. Representative traces for IBa obtained with +50 mV prepulse for IHCs from postnatal days (p) 16–18 (B) or p6–8 (C) control (n= 25 for p16–18, n= 18 for p6–8) or dfcr (n= 26 for p16–18, n= 18 for p6–8) mice. Dashed line represents initial amplitude of P1 current. Bottom, Fratio was calculated as P2 divided by P1 current amplitude and plotted against prepulse voltage.
Figure 8. VDF accelerates IBa activation to a greater extent in control than in dfcr IHCs.
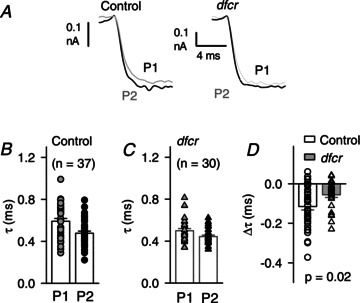
A, voltage protocol and representative traces for P1 and P2 currents overlaid for comparison of activation kinetics. Results are shown from IHCs from p16–18 control or dfcr mice. B and C, time constant (τ) from exponential fit of the rising phase of P1 and P2 IBa traces as shown in A from control (n= 37, B) and dfcr (n= 30, C) IHCs. Symbols represent values from individual cells, bars represent mean ± SEM. D, acceleration in activation due to VDF measured as the difference in τ for P1 and P2 (Δτ) measured as in B for control and dfcr IHCs. P value determined by t test.
Table 1.
I–V parameters for ICa and IBa in control and dfcr IHCs
| IBa (whole-cell) | Control (n= 43) | dfcr (n= 41) | P value |
|---|---|---|---|
| G (nS) | 7.5 ± 0.5 | 7.0 ± 0.5 | 0.49 |
| V1/2 (mV) | −18.2 ± 0.6 | −19.1 ± 0.7 | 0.30 |
| E (mV) | 48.9 ± 1.0 | 49.1 ± 1.0 | 0.91 |
| k | 6.4 ± 0.2 | 6.3 ± 0.2 | 0.57 |
| ICa (whole-cell) | Control (n= 11) | dfcr (n= 13) | P value |
|---|---|---|---|
| G (nS) | 5.41 ± 0.19 | 5.86 ± 0.37 | 0.30 |
| V1/2 (mV) | −21.4 ± 0.8 | −26.1± 1.0 | 0.002 |
| E (mV) | 43.4 ± 0.8 | 43.3 ± 0.6 | 0.94 |
| k | 6.7 ± 0.3 | 7.3 ± 0.3 | 0.26 |
| ICa (perforated- patch) | Control (n= 22) | dfcr (n= 40) | P value |
|---|---|---|---|
| G (nS) | 5.91 ± 0.25 | 5.98 ± 0.28 | 0.87 |
| V1/2 (mV) | −24.3 ± 0.7 | −27.9 ± 0.5 | <0.001 |
| E (mV) | 49.6 ± 1.12 | 47.8 ± 0.8 | 0.20 |
| k | 4.7 ± 0.2 | 4.6 ± 0.2 | 0.71 |
IBa and ICa were recorded in whole-cell (ruptured patch) or perforated-patch configuration and analysed as described in Methods. I-V relationships were fit with the following equation: 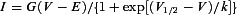 where G is conductance, V is test potential, E is apparent reversal potential, V1/2 is potential of half activation, k is the slope factor. Values represent mean ± SEM. n represents number of IHCs analysed. P values were obtained from unpaired t tests or Wilcoxon rank test.
where G is conductance, V is test potential, E is apparent reversal potential, V1/2 is potential of half activation, k is the slope factor. Values represent mean ± SEM. n represents number of IHCs analysed. P values were obtained from unpaired t tests or Wilcoxon rank test.
We next evaluated modulation of VDF by harmonin using Ca2+ as the charge carrier. We combined whole-cell patch-clamp recordings of total ICa in the plasma membrane and fast confocal Ca2+ imaging of individual IHC active zones (Frank et al. 2009; Gregory et al. 2011). With this approach, we found a small but significant VDF of the P2 relative to the P1 peak ICa and charge integral (Table 2) in control and dfcr IHCs, without any significant difference between the genotypes. As shown previously (Gregory et al. 2011), the amplitude of presynaptic Ca2+ microdomains was significantly elevated in dfcr compared to control IHCs (baseline-normalized fluorescence change ΔF/F0, Table 2). While the ΔF/F0 amplitude did not increase following the conditioning pulse, the Ca2+ microdomains build-up tended to be faster in response to P2 compared to the P1 test pulse indicating the presence of modest VDF. The difference in kinetics of P1 and P2 signals did not reach statistical significance, perhaps related to the slower time course of the active zone Ca2+ signals (τP1∼2 ms) compared to whole-cell measurements of IBa (τP1∼0.6 ms). While multiple factors could hinder detection of differences in VDF of whole-cell ICa and synaptic Ca2+ signals in control and dfcr IHCs (see Discussion), these results demonstrate that VDF is a feature of Cav1.3 channels in IHCs using Ca2+ as the permeant ion.
Table 2.
VDF probed by whole-cell patch clamp recording and Ca2+ imaging at active zones
| Control (n= 41 spots, 14 IHCs) | dfcr (n= 41 spots, 16 IHCs) | P value | |
|---|---|---|---|
| VDF (ICa) | 2.9 ± 1.1% | 3.2 ± 0.7% | 0.79 (ut) |
| ICa (P1) vs. ICa (P2) | P= 0.003 (pt) | P= 0.0004 (pt) | |
| VDF (QCa) | 1.2 ± 0.6% | 2.0 ± 0.6% | 0.16 (w) |
| QCa (P1) vs. QCa (P2) | P= 0.045 (pt) | P= 0.002 (pt) | |
| ΔF/F0 (P1) | 0.64 ± 0.36 | 0.98 ± 0.84 | 0.02 (w) |
| ΔF/F0 (P2) | 0.62 ± 0.34 | 0.95 ± 0.79 | 0.01 (w) |
| τΔF, P1 | 1.6 ± 0.5 ms | 1.8 ± 0.6 ms | 0.40 (w) |
| τΔF, P2 | 1.5 ± 0.4 ms | 1.7 ± 0.5 ms | 0.13 (w) |
Whole-cell Ca2+ currents (ICa) were elicited by 20 ms test pulses (to −32 mV) before (P1) and after (P2) a 50 ms prepulse (to +63 mV) in control and dfcr IHCs. VDF of ICa was calculated as the percentage increase in the peak amplitude (VDF (ICa)) or the current integral (VDF (QCa)) of ICa evoked by the P2 pulse compared to the P1 pulse. Data represent mean ± SEM. Ca2+ influx at individual active zones of these cells was approximated by the change in fluorescence of the low affinity Ca2+ indicator Fluo-5N (background-subtracted and normalized; ΔF/F0). Time constants corresponding to the rise in Ca2+ derived from single exponential fitting to the depolarization-evoked ΔF (τΔF, P1 for P1; τΔF, P2 for P2). Data represent mean ± SD. [Ca2+]i was 5 mm in these experiments to better resolve ΔF/F0. The present analysis was performed on data extracted from the same recordings performed in a previous study in which effects of the dfcr mutation on only the P1 (control) current were reported (Gregory et al. 2011). Data were tested for randomness, normal distribution and equality of variances, then appropriate statistical tests were chosen (pt, paired t test; ut, unpaired t test; w, Wilcoxon rank test).
Impaired exocytosis in dfcr IHCs
Cav1.3 mediates stimulus–secretion coupling in IHCs (Platzer et al. 2000; Brandt et al. 2003), whereby exocytosis of a synaptic vesicle may be controlled by few Cav1.3 channels within nanometer proximity (Brandt et al. 2005). Compared to wild-type harmonin, the dfcr mutant causes abnormally high CaV1.3 current density (Gregory et al. 2011) and reduced VDF (Figs 7 and 8), which could have dual, and potentially complex, effects on IHC exocytosis. To gain insight into the functional consequences of the dfcr mutation on IHC exocytosis, we performed perforated-patch (Fig. 9A–D) and ruptured-patch (Fig. 9E–H) recordings of membrane capacitance changes (ΔCm) evoked by step depolarizations. The test voltage evoking the maximal ICa was determined in current–voltage (I–V) relations (Fig. 9A and E). Interestingly, Boltzmann fits of the I–V curves for ICa revealed a small but significant negative shift in the half-maximal activation voltage for dfcr compared to control IHCs both in perforated-patch and ruptured-patch recordings, which was not observed for IBa (Table 1). The increased Cav1.3 current density in dfcr IHCs was less apparent than reported previously (Gregory et al. 2011), perhaps due to the larger age range of mice in these experiments (p13–20 vs. p16–18). With a 20 ms step to elicit maximal ICa in each cell, ΔCm was significantly reduced in dfcr compared to control IHCs (Fig. 9B).
Figure 9. Presynaptic function is altered in dfcr IHCs.
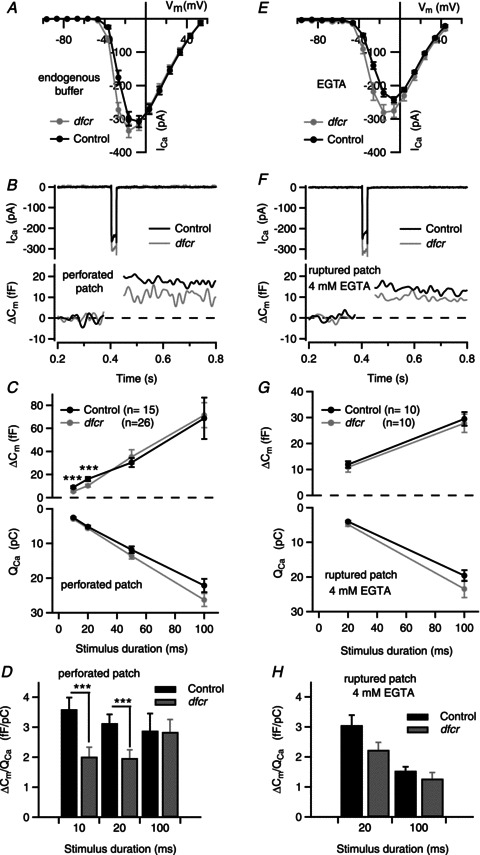
ICa and exocytic membrane capacitance changes (ΔCm) were measured in perforated-patch (A–D) and ruptured-patch configurations (4 mm EGTA + 2 mm Ca2+ in the pipette, E–H). The extracellular Ca2+ was 10 mm to adequately resolve ΔCm. A, I–V relationship for ICa evoked by 10 ms voltage steps from −96.6 mV in perforated-patch recordings (control: n= 23, dfcr: n= 40). B, representative ICa and ΔCm (Cm filtered at 50 Hz) in response to 20 ms depolarization to voltage eliciting peak ICa. C, ΔCm (top) and corresponding ICa integral (QCa, bottom) in response to step pulses for the indicated durations. Depolarizations were from −96.6 mV to the voltage eliciting peak ICa (between −26.6 and −16.6 mV) for each cell. Data represent grand averages (calculated from the means of the individual cells, n= 26 for dfcr and 15 for control IHCs (p13–19) ± SEM. D, reduced Ca2+ efficiency of synchronous exocytosis in dfcr IHCs in response to depolarizations eliciting peak ICa in perforated- patch recordings. ΔCm was normalized to QCa obtained with different stimulus durations in B and C and shown for control and dfcr IHCs (***P < 0.01, by Student's t test). E, I–V relationship for ICa evoked by 10 ms voltage steps from −96.2 mV (control: n= 11, dfcr: n= 13) in ruptured-patch recordings. F and G, same as in B and C, but for data obtained with ruptured-patch configuration with 4 mm EGTA and 2 mm Ca2+ in the intracellular solution. In F, n= 10 for dfcr and 10 for control IHCs. G and H, tendency towards reduced Ca2+ efficiency of synchronous exocytosis was also observed in EGTA-loaded dfcr IHCs. H, same as in D, but for ruptured-patch configuration (n= 10 for control, n= 10 for dfcr).
Previous work has demonstrated the existence of distinct populations of exocytic vesicles that differ in release kinetics and Ca2+ dependence. A readily releasable pool (RRP) of vesicles undergoes fast, synchronous exocytosis and is efficiently recruited by short (10–20 ms) stimuli. A slower phase of exocytosis is observed during sustained depolarizations (Moser & Beutner, 2000; Goutman & Glowatzki, 2007). To further characterize the exocytic defect in dfcr IHCs, we compared ΔCm evoked by varying stimulus durations. With short (10–20 ms) depolarizing pulses, ΔCm was significantly reduced in dfcr compared to control IHCs (∼41%, P < 0.007 for 10 ms pulses, ∼36%, P= 0.011 for 20 ms pulses; Fig. 9C), while there was no difference in ΔCm evoked by longer (50–100 ms) depolarizations (P= 0.85 for 50 ms, P= 0.49 for 100 ms by Wilcoxon rank test; Fig. 9C). These results suggested impairment in synchronous exocytosis of the RRP in dfcr IHCs. Harmonin-dependent VDF may enhance rapid Ca2+ signalling at active zones in control IHCs. If so, then the difference in ΔCm in dfcr and control IHCs should be eliminated upon normalizing ΔCm to the charge integral of ICa (ΔCm/QCa). By this analysis, maximal efficiency of exocytosis was ∼3 fF pC−1 (Fig. 9D), similar to that reported in frog auditory hair cells (Graydon et al. 2011). ΔCm/QCa was still significantly smaller in dfcr compared to control IHCs (Fig. 9D), which suggested a reduced efficiency of Ca2+ influx for driving exocytosis in dfcr compared to control IHCs.
To follow up this possibility, we performed whole-cell patch clamp measurements of ΔCm including EGTA (4 mm+ 2 mm Ca2+, calculated free intracellular [Ca2+]= 106 nm) in the intracellular recording solution. As a slow Ca2+ buffer, EGTA affects sustained exocytosis more strongly than RRP exocytosis, which probably reflects a requirement for long-distance Ca2+ signalling involved in replenishment of depleted vesicles (Moser & Beutner, 2000). If there was looser spatial coupling of Cav1.3 channels and RRP vesicles in dfcr compared to control IHCs, we would expect a stronger effect of EGTA on synchronous exocytosis in dfcr IHCs than in control IHCs. However, with EGTA in ruptured-patch recordings, we found RRP exocytosis of dfcr IHCs to be more similar to control IHCs (Fig. 9G) than in perforated-patch recordings. ΔCm/QCa still tended to be smaller in dfcr than in control IHCs with EGTA (Fig. 9H), although not quite reaching statistical significance (P= 0.053 comparing dfcr vs. control ΔCm/QCa response to 20 ms depolarization in ruptured patch). There was no significant difference in dfcr IHCs in ΔCm/QCa evoked by 20 ms depolarizations in ruptured-patch or perforated-patch recordings (P= 0.12; Fig. 9D and H). These results argue against a looser spatial coupling of the RRP to Cav1.3 channels in dfcr IHCs.
To determine if smaller numbers of IHC synapses could contribute to the exocytic impairment in dfcr IHCs, we quantified afferent IHC synapses in stacks of confocal sections from organs of Corti immunolabelled for presynaptic ribbons (CtBP2/RIBEYE) and postsynaptic AMPA glutamate receptors (GluA2/3; Khimich et al. 2005). However, there was no significant difference between control and dfcr IHCs in the cochlear apex (8.5 ± 0.6 for control, n= 85 cells vs. 9.7 ± 0.4 for dfcr, n= 49 cells; P= 0.2 by t test). These results suggest that the exocytic defect in dfcr IHCs does not involve changes in synapse number, but could depend on presynaptic alterations in RRP dynamics at the subset of synapses characterized by harmonin/Cav1.3 interactions. Our results indicate a multi-faceted role of harmonin in regulating Cav1.3 channel function and RRP exocytosis in mature IHCs, both of which are compromised in the dfcr model of Usher syndrome.
Discussion
Our study provides multiple new insights into the regulation of Cav1.3 channels by harmonin. First, harmonin binding to the distal C-terminus of α11.3 enhances VDF of Cav1.3 current, the extent to which depends on the identity of the Cavβ subunit. Second, in mouse IHCs, VDF characterizes Cav1.3 currents after, but not before, hearing onset and potentiates presynaptic Cav1.3 currents by accelerating Cav1.3 activation kinetics. Third, Cav1.3 VDF and exocytosis are impaired in IHCs from dfcr mice expressing a mutant form of harmonin. We conclude that harmonin is an important determinant of Cav1.3 properties and presynaptic function in mouse IHCs.
Dual regulation of Cav1.3 channels by harmonin
Facilitation of Cav1 channels occurs by multiple mechanisms that have been studied extensively in the context of nerve and muscle (Dolphin, 1996). VDF has been reported for Cav1.3 channels in transformed cell lines (Safa et al. 2001; Calin-Jageman et al. 2007), neonatal mouse outer hair cells (Michna et al. 2003), and mouse sinoatrial nodal cells in the heart (Christel et al. 2012). Our results provide the first evidence that Cav1.3 channels in IHCs undergo VDF and that harmonin contributes to this process. The mechanism involves harmonin binding to the PDZ-binding sequence in the α11.3 dCT, since mutation or deletion of this motif prevents modulation of VDF (Fig. 4). In addition, the dfcr mutant, which cannot bind the α11.3 dCT (Gregory et al. 2011) does not enhance VDF (Fig. 5). Similar to the PDZ protein erbin, binding of harmonin to the α11.3 dCT may relieve an autoinhibitory regulation of VDF imposed by the α11.3 dCT (Calin-Jageman et al. 2007). However, harmonin binding to the α11.3 dCT also promotes ubiquitination of α11.3, which limits Cav1.3 current density by promoting proteosomal degradation of the channel (Gregory et al. 2011). Harmonin may dually regulate Cav1.3 channels by limiting channel trafficking to, or enhancing removal from, the plasma membrane through targeting to proteosomal pathways. Those channels that remain associated with harmonin at the IHC synapse exhibit enhanced VDF due to the α11.3 dCT interaction. Our findings that Cav1.3 current density is increased (Gregory et al. 2011) and VDF is decreased (Figs 7 and 8) in IHCs from dfcr compared to control mice support the bifunctional role of harmonin with respect to Cav1.3 regulation in vivo. Our coimmunoprecipitation experiments suggest that harmonin may interact with sites in the channel complex in addition to the dCT (Fig. 3), raising the possibility that harmonin may regulate yet other aspects of Cav1.3 function.
VDF in HEK293T cells cotransfected with Cav1.3 and harmonin (Figs 1B and 2B) was stronger and occurred at more negative potentials compared to VDF in control IHCs (Fig. 7B). A contributing factor is that VDF is somewhat limited in Cav1.3 channels containing the β2 subunit (Fig. 2C and D), which is the primary Cavβ subunit in mouse IHCs (Neef et al. 2009). In addition, harmonin is found at only ∼50% of mature IHC synapses, whereas Cav1.3 is present at every synapse (Brandt et al. 2005; Gregory et al. 2011). Since our whole-cell recordings summate the activity of all IHC Cav1.3 channels, the impact of harmonin on VDF would be diluted by the nominal VDF exhibited by channels not associated with harmonin. Considering the proportion of harmonin-positive synapses in mature IHCs (∼50%), the ∼13% increase in IBa amplitude due to a +30 mV prepulse in mature IHCs VDF may be twice as large at individual synapses characterized by Cav1.3/harmonin interactions and thus roughly similar to what we find for Cavβ2-containing channels in HEK293T cells (∼25% for a +30 mV prepulse; Fig. 2B).
Considering the above argument, our inability to measure harmonin-dependent VDF using Ca2+ as the charge carrier, both in electrophysiological recordings of ICa and Ca2+ imaging of presynaptic active zones is not unexpected (Table 2). Moreover, VDF of ICa in IHCs is likely to be further diminished by CDI. While not as prominent for Cav1.3 in transfected HEK293T cells as in IHCs (compare Fig. 6 and Supplemental Fig. 1), CDI is still fast enough in IHCs (τfast∼5–7 ms, Supplemental Fig. 1) to partially occlude VDF (τ∼11 ms, Fig. 7A) in whole-cell recordings. Given these caveats, we propose that VDF is physiologically relevant for controlling ICa at the subset of synapses characterized by Cav1.3/harmonin interactions in mature IHCs.
Physiological significance of Cav1.3 VDF in IHCs
VDF due to harmonin is measurable in mature IHCs but not in IHCs prior to hearing onset (Fig. 7B), which coincides with the developmental upregulation of harmonin localization at IHC synapses (Gregory et al. 2011). At very positive voltages (+50 mV), VDF causes ∼20% acceleration in the activation rate of the macroscopic Cav1.3 current (Fig. 8B), an effect that could be twice as large at harmonin-positive synapses. Consistent with this possibility, VDF due to a +50 mV prepulse caused a ∼36% speeding of IBa activation in HEK293T cells cotransfected with Cav1.3 and harmonin (Fig. 5B). Facilitation of ICa in response to prior depolarization has been reported for ICa at rat IHC synapses (Goutman & Glowatzki, 2011; Goutman, 2012) and at retinal bipolar synapses (Cho & von Gersdorff, 2012) and so may be a fundamental form of Cav1 regulation at ribbon synapses.
Exocytosis of the RRP is triggered by the opening of relatively few Cav1.3 channels (Brandt et al. 2005) such that changes in the microscopic properties of Cav1.3 may substantially affect transmitter release. In this respect, modulation of Cav1.3 gating by harmonin is consistent with the impairment of synchronous exocytosis in dfcr IHCs (Fig. 9). By amplifying presynaptic Cav1.3 influx as a function of membrane depolarization, VDF could also contribute to short-term facilitation of IHC transmission. This form of presynaptic plasticity has been documented in afferent recordings at synapses of frog auditory hair cells (Cho et al. 2011) and rat IHCs, where it manifests as a decrease in synaptic failures and delays following pre-depolarizations (Goutman & Glowatzki, 2011). Ca2+ current facilitation in the latter study on immature IHCs was relatively modest and not thought to significantly contribute to the synaptic facilitation, which could be due to the decreased localization of harmonin at immature IHC synapses (Gregory et al., 2011). Thus, at select harmonin-positive IHC synapses, Cav1.3 VDF may increase the gain and enhance the timing of release events, thus improving aspects of intensity and temporal coding of sound, respectively. Maintaining Cav1.3 channel availability during graded changes in the IHC membrane potential is important for the continuous encoding of sound information (Lewis & Hudspeth, 1983; Moser et al. 2006) as well as for ongoing spontaneous afferent firing (Robertson & Paki, 2002; Sueta et al. 2004). Although Cav1.3 channels show less inactivation (both CDI and VDI) in IHCs compared to other cell types (Platzer et al. 2000; Koschak et al. 2001), CDI and VDI are still measurable and can, along with RRP depletion (Moser & Beutner, 2000), lead to significant synaptic depression if unopposed (Cho et al. 2011).
A role for harmonin in synchronous exocytosis
Based on our findings that Ca2+ influx–exocytosis coupling is impaired in dfcr IHCs, we expected that intracellular dialysis with EGTA in whole-cell recordings should more strongly inhibit RRP exocytosis in dfcr IHCs compared to control IHCs, which was not the case (Fig. 9). This result could be explained by a role for harmonin in regulating Ca2+ coupling to only one component of the RRP, which could not be resolved in our measurements of ΔCm. While paired pre- and postsynaptic recordings of synaptic transmitter release indicate two components of the RRP (Wu & Borst, 1999; Sakaba & Neher, 2001; Goutman & Glowatzki, 2007; but see Li et al. 2009), only one RRP was reported with ΔCm measurements of exocytosis (Moser & Beutner, 2000). Thus, our ΔCm recordings may have lacked the sensitivity to detect a stronger effect of EGTA on RRP exocytosis in dfcr IHCs when compared to control, which would further be challenged by an effect of harmonin at only a subset of IHC active zones (Gregory et al. 2011).
Alterations in exocytosis in dfcr IHCs could suggest additional synaptic functions of harmonin that may be independent of Cav1.3 modulation. Analogous to its properties in apical IHC hair bundles (Adato et al. 2005), harmonin could engage in multivalent interactions with Cav1.3 channels and/or other synaptic molecules that could regulate Ca2+ coupling to exocytosis in IHCs. For example, harmonin, either alone or in complex with cadherin-23, binds to phosphatidylinositol 4,5-bisphosphate (PIP2) (Bahloul et al. 2010). PIP2 is a known regulator of exocytosis in other cell types (Koch & Holt, 2012) and reductions in PIP2 synthesis causes defects in Ca2+ signalling and high-frequency hearing-impairment mice (Rodriguez et al. 2012). A detailed understanding of the molecular mechanism by which harmonin regulates the Ca2+ efficiency of exocytosis will require more in-depth analyses, such as paired recordings of IHCs and postsynaptic afferents in genetically modified mice.
A presynaptic role for harmonin in IHCs
In apical hair bundles, the role of harmonin has been elegantly elucidated. The PDZ domains of harmonin bind to multiple proteins (sans, myosin VIIa, cadherin-23) implicated in hair bundle development (Adato et al. 2005). Mutations in harmonin or these interacting proteins cause deafness in humans with Usher syndrome and animal models (Petit, 2001), due to improper formation of hair bundles and subsequent failure of mechanotransduction. In dfcr mice, hair bundles of IHCs are grossly normal (Grillet et al. 2009), but mechanotransduction currents in dfcr outer hair cells show weaker sensitivity and slower kinetics in response to physical displacement of hair bundles, due to the inability of dfcr harmonin to interact with proteins required for gating mechanotransduction channels (Grillet et al. 2009). These results provide an intriguing parallel to our findings that Cav1.3 properties and exocytosis are altered in dfcr IHCs. Other Usher syndrome-associated proteins have also been found to interact with and regulate ion channels. For example, the USH2D protein whirlin associates with Cav1.3 in photoreceptors (Kersten et al. 2010). The Drosophila homologue of whirlin, dysc, interacts with SLO Ca2+-activated K+ channels and enhances the expression of these channels in neurons (Jepson et al. 2012). Understanding how members of the Usher interactome collectively or individually alter membrane excitability and/or synaptic transmission may provide new clues into the cellular pathology leading to deafness and blindness in human patients.
Glossary
- CDI
Ca2+-dependent inactivation
- dCT
distal C-terminus
- IHC
inner hair cell
- p
postnatal day
- RRP
readily releasable pool
- VDF
voltage-dependent facilitation
- VDI
voltage-dependent inactivation
Additional information
Competing interests
None.
Author contributions
The experiments in this study were performed at the University of Iowa, Dominican University and University of Göttingen. Author contributions are as follows: conception and design of experiments, collection analysis, and interpretation of data: F.D.G., I.E.C.-J., T.P., T.M., A.L.; drafting of the article or revising it critically for important intellectual content: F.D.G., T.M., A.L. All authors approved the final version of the manuscript.
Funding
This work was supported by the NIH (DC009433, HL087120 and DC010362 to A.L.; DA015040 and K12/GM00068 to F.D.G.; and DC008417 to I.E.C.-J.), the Carver Research Program of Excellence (to A.L.), a fellowship of the Alexander von Humboldt foundation to T.P., and the German Research Foundation through the Collaborative Research Center 889 (to T.M.).
Supplementary material
Supplemental Fig. 1
References
- Adato A, Michel V, Kikkawa Y, Reiners J, Alagramam KN, Weil D, Yonekawa H, Wolfrum U, El-Amraoui A, Petit C. Interactions in the network of Usher syndrome type 1 proteins. Hum Mol Genet. 2005;14:347–356. doi: 10.1093/hmg/ddi031. [DOI] [PubMed] [Google Scholar]
- Bahloul A, Michel V, Hardelin JP, Nouaille S, Hoos S, Houdusse A, England P, Petit C. Cadherin-23, myosin VIIa and harmonin, encoded by Usher syndrome type I genes, form a ternary complex and interact with membrane phospholipids. Hum Mol Genet. 2010;19:3557–3565. doi: 10.1093/hmg/ddq271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baig SM, Koschak A, Lieb A, Gebhart M, Dafinger C, Nurnberg G, Ali A, Ahmad I, Sinnegger-Brauns MJ, Brandt N, Engel J, Mangoni ME, Farooq M, Khan HU, Nurnberg P, Striessnig J, Bolz HJ. Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- Brandt A, Khimich D, Moser T. Few CaV1.3 channels regulate the exocytosis of a synaptic vesicle at the hair cell ribbon synapse. J Neurosci. 2005;25:11577–11585. doi: 10.1523/JNEUROSCI.3411-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt A, Striessnig J, Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin-Jageman I, Lee A. Cav1 L-type Ca2+ channel signaling complexes in neurons. J Neurochem. 2008;105:573–583. doi: 10.1111/j.1471-4159.2008.05286.x. [DOI] [PubMed] [Google Scholar]
- Calin-Jageman I, Yu K, Hall RA, Mei L, Lee A. Erbin enhances voltage-dependent facilitation of Cav1.3 Ca2+ channels through relief of an autoinhibitory domain in the Cav1.3 α1 subunit. J Neurosci. 2007;27:1374–1385. doi: 10.1523/JNEUROSCI.5191-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. ‘Rejuvenation’ protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Cho S, Li GL, von Gersdorff H. Recovery from short-term depression and facilitation is ultrafast and Ca2+ dependent at auditory hair cell synapses. J Neurosci. 2011;31:5682–5692. doi: 10.1523/JNEUROSCI.5453-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, von Gersdorff H. Ca2+ influx and neurotransmitter release at ribbon synapses. Cell Calcium. 2012;52:208–216. doi: 10.1016/j.ceca.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christel C, Lee A. Ca2+-dependent modulation of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2012;1820:1243–1252. doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christel CJ, Cardona N, Mesirca P, Herrmann S, Hofmann F, Striessnig J, Ludwig A, Mangoni ME, Lee A. Distinct localization and modulation of Cav1.2 and Cav1.3 L-type Ca2+ channels in mouse sinoatrial node. J Physiol. 2012;590:6327–6341. doi: 10.1113/jphysiol.2012.239954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends Neurosci. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- Dou H, Vazquez AE, Namkung Y, Chu H, Cardell EL, Nie L, Parson S, Shin HS, Yamoah EN. Null mutation of α1D Ca2+ channel gene results in deafness but no vestibular defect in mice. J Assoc Res Otolaryngol. 2004;5:215–226. doi: 10.1007/s10162-003-4020-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank T, Khimich D, Neef A, Moser T. Mechanisms contributing to synaptic Ca2+ signals and their heterogeneity in hair cells. Proc Natl Acad Sci U S A. 2009;106:4483–4488. doi: 10.1073/pnas.0813213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD. Transmitter release from cochlear hair cells is phase locked to cyclic stimuli of different intensities and frequencies. J Neurosci. 2012;21:17025–17036. doi: 10.1523/JNEUROSCI.0457-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Glowatzki E. Time course and calcium dependence of transmitter release at a single ribbon synapse. Proc Natl Acad Sci U S A. 2007;104:16341–16346. doi: 10.1073/pnas.0705756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Glowatzki E. Short-term facilitation modulates size and timing of the synaptic response at the inner hair cell ribbon synapse. J Neurosci. 2011;31:7974–7981. doi: 10.1523/JNEUROSCI.0604-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graydon CW, Cho S, Li GL, Kachar B, von Gersdorff H. Sharp Ca2+ nanodomains beneath the ribbon promote highly synchronous multivesicular release at hair cell synapses. J Neurosci. 2011;31:16637–16650. doi: 10.1523/JNEUROSCI.1866-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory FD, Bryan KE, Pangršič T, Calin-Jageman IE, Moser T, Lee A. Harmonin inhibits presynaptic Cav1.3 Ca2+ channels in mouse inner hair cells. Nat Neurosci. 2011;14:1109–1111. doi: 10.1038/nn.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillet N, Xiong W, Reynolds A, Kazmierczak P, Sato T, Lillo C, Dumont RA, Hintermann E, Sczaniecka A, Schwander M, Williams D, Kachar B, Gillespie PG, Müller U. Harmonin mutations cause mechanotransduction defects in cochlear hair cells. Neuron. 2009;62:375–387. doi: 10.1016/j.neuron.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetzenauer A, Sinnegger-Brauns MJ, Striessnig J, Singewald N. Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of CaV1.3 and CaV1.2 isoforms. Neuroscience. 2006;139:1005–1015. doi: 10.1016/j.neuroscience.2006.01.059. [DOI] [PubMed] [Google Scholar]
- Jenkins MA, Christel CJ, Jiao Y, Abiria S, Kim KY, Usachev YM, Obermair GJ, Colbran RJ, Lee A. Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J Neurosci. 2010;30:5125–5135. doi: 10.1523/JNEUROSCI.4367-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JE, Shahidullah M, Lamaze A, Peterson D, Pan H, Koh K. dyschronic, a Drosophila homolog of a deaf-blindness gene, regulates circadian output and Slowpoke channels. PLoS Genet. 2012;8:e1002671. doi: 10.1371/journal.pgen.1002671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KR, Gagnon LH, Webb LS, Peters LL, Hawes NL, Chang B, Zheng QY. Mouse models of USH1C and DFNB18: phenotypic and molecular analyses of two new spontaneous mutations of the Ush1c gene. Hum Mol Genet. 2003;12:3075–3086. doi: 10.1093/hmg/ddg332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten FF, van Wijk E, van Reeuwijk J, van der Zwaag B, Marker T, Peters TA, Katsanis N, Wolfrum U, Keunen JE, Roepman R, Kremer H. Association of whirlin with Cav1.3 (α1D) channels in photoreceptors, defining a novel member of the usher protein network. Invest Ophthalmol Vis Sci. 2010;51:2338–2346. doi: 10.1167/iovs.09-4650. [DOI] [PubMed] [Google Scholar]
- Khimich D, Nouvian R, Pujol R, Tom Dieck S, Egner A, Gundelfinger ED, Moser T. Hair cell synaptic ribbons are essential for synchronous auditory signalling. Nature. 2005;434:889–894. doi: 10.1038/nature03418. [DOI] [PubMed] [Google Scholar]
- Kimberling WJ, Moller C. Clinical and molecular genetics of Usher syndrome. J Am Acad Audiol. 1995;6:63–72. [PubMed] [Google Scholar]
- Koch M, Holt M. Coupling exo- and endocytosis: an essential role for PIP2 at the synapse. Biochim Biophys Acta. 2012;1821:1114–1132. doi: 10.1016/j.bbalip.2012.02.008. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. α1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Lewis RS, Hudspeth AJ. Voltage- and ion-dependent conductances in solitary vertebrate hair cells. Nature. 1983;304:538–541. doi: 10.1038/304538a0. [DOI] [PubMed] [Google Scholar]
- Li GL, Keem E, Andor-Ardo D, Hudspeth AJ, von Gersdorff H. The unitary event underlying multiquantal EPSCs at a hair cell's ribbon synapse. J Neurosci. 2009;29:7558–7568. doi: 10.1523/JNEUROSCI.0514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalski N, Michel V, Caberlotto E, Lefèvre GM, van Aken AF, Tinevez JY, Bizard E, Houbron C, Weil D, Hardelin JP, Richardson GP, Kros CJ, Martin P, Petit C. Harmonin-b, an actin-binding scaffold protein, is involved in the adaptation of mechanoelectrical transduction by sensory hair cells. Pflugers Arch. 2009;459:115–130. doi: 10.1007/s00424-009-0711-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michna M, Knirsch M, Hoda JC, Muenkner S, Langer P, Platzer J, Striessnig J, Engel J. Cav1.3 (α1D) Ca2+ currents in neonatal outer hair cells of mice. J Physiol. 2003;553:747–758. doi: 10.1113/jphysiol.2003.053256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Beutner D. Kinetics of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse of the mouse. Proc Natl Acad Sci U S A. 2000;97:883–888. doi: 10.1073/pnas.97.2.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser T, Brandt A, Lysakowski A. Hair cell ribbon synapses. Cell Tissue Res. 2006;326:347–359. doi: 10.1007/s00441-006-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navedo MF, Amberg GC, Westenbroek RE, Sinnegger-Brauns MJ, Catterall WA, Striessnig J, Santana LF. Cav1.3 channels produce persistent calcium sparklets, but Cav1.2 channels are responsible for sparklets in mouse arterial smooth muscle. Am J Physiol Heart Circ Physiol. 2007;293:H1359–H1370. doi: 10.1152/ajpheart.00450.2007. [DOI] [PubMed] [Google Scholar]
- Neef A, Heinemann C, Moser T. Measurements of membrane patch capacitance using a software-based lock-in system. Pflugers Arch. 2007;454:335–344. doi: 10.1007/s00424-006-0191-1. [DOI] [PubMed] [Google Scholar]
- Neef J, Gehrt A, Bulankina AV, Meyer AC, Riedel D, Gregg RG, Strenzke N, Moser T. The Ca2+ channel subunit β2 regulates Ca2+ channel abundance and function in inner hair cells and is required for hearing. J Neurosci. 2009;29:10730–10740. doi: 10.1523/JNEUROSCI.1577-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a Shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit C. Usher syndrome: from genetics to pathogenesis. Annu Rev Genomics Hum Genet. 2001;2:271–297. doi: 10.1146/annurev.genom.2.1.271. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Reiners J, van Wijk E, Marker T, Zimmermann U, Jurgens K, Te Brinke H, Overlack N, Roepman R, Knipper M, Kremer H, Wolfrum U. The scaffold protein harmonin (USH1C) provides molecular links between Usher syndrome type 1 and type 2. Hum Mol Genet. 2005;14:3933–3943. doi: 10.1093/hmg/ddi417. [DOI] [PubMed] [Google Scholar]
- Robertson D, Paki B. Role of L-type Ca2+ channels in transmitter release from mammalian inner hair cells. II. Single-neuron activity. J Neurophysiol. 2002;87:2734–2740. doi: 10.1152/jn.2002.87.6.2734. [DOI] [PubMed] [Google Scholar]
- Rodriguez L, Simeonato E, Scimemi P, Anselmi F, Cali B, Crispino G, Ciubotaru CD, Bortolozzi M, Ramirez FG, Majumder P, Arslan E, De Camilli P, Pozzan T, Mammano F. Reduced phosphatidylinositol 4,5-bisphosphate synthesis impairs inner ear Ca2+ signaling and high-frequency hearing acquisition. Proc Natl Acad Sci U S A. 2012;109:14013–14018. doi: 10.1073/pnas.1211869109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safa P, Boulter J, Hales TG. Functional properties of CaV1.3 (α1D) L-type Ca2+ channel splice variants expressed by rat brain and neuroendocrine GH3 cells. J Biol Chem. 2001;276:38727–38737. doi: 10.1074/jbc.M103724200. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/s0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Songyang Z, Fanning AS, Fu C, Xu J, Marfatia SM, Chishti AH, Crompton A, Chan AC, Anderson JM, Cantley LC. Recognition of unique carboxyl-terminal motifs by distinct PDZ domains. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- Sueta T, Zhang SY, Sellick PM, Patuzzi R, Robertson D. Effects of a calcium channel blocker on spontaneous neural noise and gross action potential waveforms in the guinea pig cochlea. Hear Res. 2004;188:117–125. doi: 10.1016/S0378-5955(03)00374-5. [DOI] [PubMed] [Google Scholar]
- Verpy E, Leibovici M, Zwaenepoel I, Liu XZ, Gal A, Salem N, Mansour A, Blanchard S, Kobayashi I, Keats BJ, Slim R, Petit C. A defect in harmonin, a PDZ domain-containing protein expressed in the inner ear sensory hair cells, underlies Usher syndrome type 1C. Nat Genet. 2000;26:51–55. doi: 10.1038/79171. [DOI] [PubMed] [Google Scholar]
- Wang CL. A note on Ca2+ binding to calmodulin. Biochem Biophys Res Commun. 1985;130:426–430. doi: 10.1016/0006-291x(85)90434-6. [DOI] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal Cav1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. CaV1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.