

Abstract
Islet transplantation is an effective method to obtain long-term glycemic control for patients with type 1 diabetes, yet its widespread use is limited by an inadequate supply of donor islets. The hormone leptin has profound glucose-lowering and insulin-sensitizing action in type 1 diabetic rodent models. We hypothesized that leptin administration could reduce the dose of transplanted islets required to achieve metabolic control in a mouse model of type 1 diabetes. We first performed a leptin dose-response study in C57Bl/6 mice with streptozotocin (STZ)-induced diabetes to determine a leptin dose insufficient to reverse hyperglycemia. Subsequently, we compared the ability of suboptimal islet transplants of 50 or 125 syngeneic islets to achieve glycemic control in STZ-induced diabetic C57Bl/6 mice treated with or without this dose of leptin. The dose-response study revealed that leptin reverses STZ-induced diabetes in a dose-dependent manner. Supraphysiological leptin levels were necessary to restore euglycemia but simultaneously increased risk of hypoglycemia, and also lost efficacy after 12 days of administration. In contrast, 1 µg/day leptin only modestly reduced blood glucose but maintained efficacy throughout the study duration. We then administered 1 µg/day leptin to diabetic mice that underwent transplantation of 50 or 125 islets. Although these islet doses were insufficient to ameliorate hyperglycemia alone, coadministration of leptin with islet transplantation robustly improved control of glucose and lipid metabolism, without increasing circulating insulin levels. This study reveals that low-dose leptin administration can reduce the number of transplanted islets required to achieve metabolic control in STZ-induced diabetic mice.
The current state-of-the-art for achieving long-term glycemic control in type 1 diabetic patients is transplantation of cadaveric donor islets. Whereas frequent episodes of hyperglycemia and hypoglycemia occur with insulin therapy, islet transplantation can effectively eliminate these excursions and maintain glycemia within a target range of 3.3 to 7.8 mmol/L (1). Unfortunately, islet transplantation is not widely available because of limited donor islet supply. Most transplant recipients require islets from at least two cadaveric donors to achieve target glycemia (1,2), and the decline of graft function within 5 years of transplantation necessitates that most patients resume insulin therapy (2). Thus, a strategy to reduce the number of islets needed to achieve insulin independence is essential for widespread application of islet transplantation from cadaveric donor islets.
The hormone leptin has a well-recognized role in glucose homeostasis (3). Recent studies have demonstrated that high-dose leptin administration reverses hyperglycemia and dyslipidemia in type 1 diabetic rodent models (4–8). However, leptin is unlikely to replace insulin as a therapy for type 1 diabetes because it offers little, if any, advantage over insulin injections with regard to metabolic control and quality of life. Alternatively, glycemic control and insulin requirements for type 1 diabetic patients may be improved by leptin and insulin cotherapy. In diabetic mice, leptin administration reduced the insulin dose needed to ameliorate hyperglycemia (9), and combined leptin and insulin administration achieved better glycemic control than insulin alone (6).
Because islet transplantation provides superior metabolic control over insulin injections, we investigated whether leptin as an adjunct to islet transplantation could provide tighter glycemic control with fewer transplanted islets. Such an effect could increase the availability and efficacy of islet transplantation as a treatment. To test this, we examined whether leptin administration could reduce the number of transplanted islets needed to reverse streptozotocin (STZ)-induced diabetes in mice (STZ-diabetic mice). Because high-dose leptin alone can restore normoglycemia in STZ-diabetic rodents (4–8), which thereby can enhance islet graft function (10,11), we first performed a dose-response study in STZ-diabetic mice to identify a leptin dose that was insufficient to reverse hyperglycemia. Subsequently, we administered this dose of leptin to diabetic mice transplanted with 50 or 125 syngeneic islets (17 and 42% of an optimal dose of 300 islets, respectively) to determine whether leptin cotherapy could enhance the ability of these suboptimal islet doses to achieve metabolic control.
RESEARCH DESIGN AND METHODS
Animals.
C57Bl/6 male mice (Jackson Laboratories, Bar Harbor, ME) were housed with a 12-/12-h light/dark cycle with ad libitum access to chow diet (2918; Harlan Laboratories, Madison, WI) and water. All procedures with animals were approved by the University of British Columbia Animal Care Committee and performed in accordance with the Canadian Council on Animal Care guidelines.
STZ administration.
STZ (Sigma-Aldrich, St. Louis, MO) prepared in acetate buffer, pH 4.5, was administered to 10-week-old C57Bl/6 mice at 180 mg/kg via intraperitoneal injection 6 days prior to surgery. Nondiabetic control mice received an intraperitoneal injection of acetate buffer alone. Diabetes was defined as fasting blood glucose >16 mmol/L on 2 consecutive days.
Leptin administration via mini-osmotic pump.
Recombinant murine leptin (PeproTech, Rocky Hill, NJ) was reconstituted in water according to the manufacturer’s instructions and loaded in osmotic pumps (DURECT Corporation, Cupertino, CA) designed for either 4-week or 6-week infusion. Pumps were implanted subcutaneously on day 0. Diabetic vehicle-treated mice received osmotic pumps loaded with water only. Nondiabetic controls received sham surgery without pump implantation. For the leptin dosing study, the concentration of leptin loaded into the pumps was adjusted to deliver doses of 1, 3, 5, and 10 µg/day per mouse, whereas for the islet transplant study a single dose of 1 µg/day per mouse was used.
Islet isolation and transplantation.
Islets were isolated from 12-week-old male C57Bl/6 mice (Centre for Disease Modeling, Vancouver, Canada) as previously described (12) and cultured in Ham F-10 (Sigma-Aldrich) supplemented with 0.5% BSA, 100 units/mL penicillin, and 100 µg/mL streptomycin until transplantation. Immediately prior to transplantation, islets were hand-picked into aliquots of 50, 125, or 300 islets and transplanted under the left kidney capsule of recipient mice on day 0. Control mice received sham surgery.
Metabolic measurements.
Body weight, blood glucose, plasma leptin, insulin, IGF binding protein 2 (IGFBP2), triglycerides, β-hydroxybutyrate, and free fatty acids (FFA) were measured after a 4-h fast from samples collected through saphenous vein as previously described (8,13). Fasting plasma leptin and insulin levels in the islet transplant study were measured from samples collected via cardiac puncture 6 weeks after surgery. Plasma glucose was measured via the glucose-oxidase (Trinder) method (Genzyme Diagnostics). HbA1c levels were measured with a Siemens DCA 200 Vantage Analyzer (Siemens Healthcare Diagnostics, Tarrytown, NY) from whole blood collected from the saphenous vein with EDTA as an anticoagulant.
Immunofluorescent image quantification.
Pancreata were harvested from mice at 6 weeks after surgery, postfixed overnight in 4% paraformaldehyde at 4°C, rinsed in 70% ethanol, embedded in paraffin, and processed for sectioning by Wax-it Histology Services (Vancouver, Canada). Three sections separated by 200 µm per mouse were immunostained for insulin (guinea pig anti-insulin antibody, Cat# I8510, 1:1,000; Sigma-Aldrich) and glucagon (mouse antiglucagon antibody, Cat# G2654, 1:1,000; Sigma-Aldrich) overnight at 4°C. Slides were then incubated with AlexaFluor-conjugated secondary antibodies (Life Technologies, Burlington, Ontario, Canada) for 1 h at room temperature. Whole sections were scanned and quantified as previously described (14). Total insulin-positive area or glucagon-positive area was expressed relative to whole pancreas area and averaged across three pancreas sections per mouse.
Oral glucose tolerance test and glucose-stimulated insulin secretion test.
Mice were fasted for 6 h and then administered an oral gavage of 30% glucose dissolved in water (2 g glucose/kg body weight). Blood glucose was monitored and blood was collected for plasma insulin measurement prior to gavage (time 0) and at the indicated times after gavage.
Statistical analysis.
Unless otherwise stated, statistical analyses were performed using a one-way ANOVA with Tukey post hoc test (GraphPad Prism; GraphPad Software, La Jolla, CA). Statistical significance was set at P < 0.05.
RESULTS
Leptin administration dose-dependently reverses STZ-induced hyperglycemia.
To examine whether leptin administration can enhance islet transplant efficacy, we first determined a dose of leptin that alone was insufficient to achieve normoglycemia. We performed a dose-response experiment in which mice were injected with STZ on day −6 to induce diabetes; subsequently, mice were implanted with osmotic pumps infusing 1, 3, 5, or 10 µg/day leptin continuously for 4 weeks. One group of diabetic mice received pump implants delivering vehicle only (STZ-vehicle), and mice that were not STZ-injected (nondiabetic) served as controls.
Blood glucose was measured for 4 weeks after surgery. As we previously reported (8), 10 µg/day leptin administration normalized blood glucose levels within 5 days (Fig. 1A), as did 5 µg/day leptin. Treatment with 3 µg/day leptin decreased blood glucose moderately within this timeframe, eventually normalizing blood glucose levels by day 15. Mice receiving 1 µg/day leptin had a trend toward decreased blood glucose, but values never significantly decreased compared with STZ-vehicle controls. Unexpectedly, the ability of 10 µg/day leptin to normalize blood glucose waned after 12 days of treatment, and hyperglycemia returned (24.7 ± 6.4 mmol/L on day 29). Blood glucose increased similarly in 5 µg/day–treated mice, but to a lesser extent (14.1 ± 2.5 mmol/L on day 29). Regardless, area under the curve (AUC) analysis of blood glucose tracking from days 5–12 revealed that blood glucose levels were dose-proportionally lowered by leptin, and it was clear that 1 μg/day leptin was a suitable dose to test the effect of leptin on islet transplantation because it did not significantly lower blood glucose levels compared with STZ-vehicle controls.
FIG. 1.
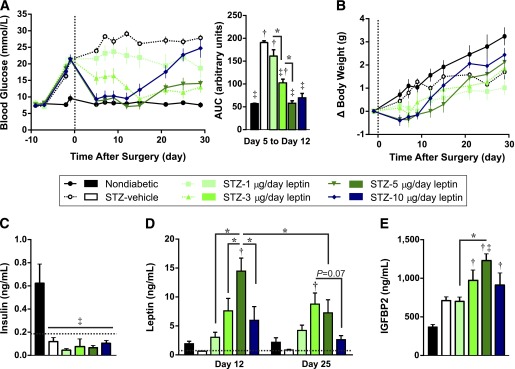
Leptin reverses STZ-induced diabetes in a dose-dependent manner. Mice were treated with STZ and 6 days later received implantation of osmotic pumps (day 0, vertical broken line) delivering doses of either leptin ranging from 1 to 10 µg/day (STZ and 1, 3, 5, or 10 µg/day) or vehicle only (STZ-vehicle). Nondiabetic mice served as controls. Fasting blood glucose (A) and change in body weight (B) relative to day −1. AUC analysis of blood glucose levels from day 5 to day 12 is presented (A). AUC analysis of body weight change from day 5 to day 12 revealed no significant differences and is not shown. C: The 4-h fasted plasma insulin levels on day 25. Limit of detection is shown by broken horizontal line. D: The 4-h fasted plasma leptin levels on days 12 and 25. Four of five STZ-vehicle mice had leptin levels below the detection limit (indicated by broken line) on both days, and this group was not included in statistical analyses. Statistical analyses were performed by two-way ANOVA with Tukey post hoc test. E: The 4-h fasted plasma IGFBP2 levels on day 12. Data are presented as mean ± SEM, n = 4–5. *P < 0.05. †P < 0.05 vs. nondiabetic controls. ‡P < 0.05 vs. STZ-vehicle controls.
We also examined the effect of leptin on body weight in STZ-induced diabetic mice (Fig. 1B). Prior to pump implantation, body weight was significantly reduced from 24.1 ± 0.3 g in nondiabetic controls to an average of 23.0 ± 0.2 g in STZ-injected mice (P = 0.04, t test). Body weight change was not significantly altered between STZ-injected groups in response to leptin or vehicle administration. However, mice treated with 5 or 10 μg/day leptin initially tended to lose weight, and subsequently gained weight after day 12. This correlates with the diminished long-term blood glucose response to the highest leptin doses.
We and others previously have reported that high-dose leptin lowers blood glucose in type 1 diabetic rodent models without increasing circulating insulin levels, β-cell area, or pancreatic insulin content (4–6,8,15–17). In agreement with this, plasma insulin was significantly reduced by STZ, and leptin did not increase insulin levels compared with STZ-vehicle mice (Fig. 1C). This is not surprising given the numerous studies showing that leptin inhibits insulin secretion (7,18–22).
We next measured the concentration of plasma leptin 12 and 25 days after implantation (Fig. 1D). Leptin levels on day 12 were reduced below the limit of detection in STZ-vehicle mice, consistent with the expected decline in leptin after STZ administration (23). Plasma leptin levels were restored to nondiabetic levels by 1 µg/day treatment, and increased dose-proportionally with 5 µg/day treatment. Notably, the lowest dose of leptin that achieved normoglycemia at this time point (5 µg/day) produced circulating leptin levels significantly higher than those of nondiabetic mice (P = 0.0001), revealing that supraphysiological leptin levels are needed to achieve normoglycemia in STZ-diabetic mice. Treatment with 10 µg/day produced a lower plasma leptin concentration on day 12 than 5 µg/day leptin. Given that both of these doses initially yielded similar glucose-lowering activity, and that at day 12 the efficacy of 10 µg/day leptin began to wane, we postulate that plasma leptin levels were initially higher in the 10 µg/day group but had declined by the time of measurement. Supporting this, plasma leptin declined from day 12 to day 25 in the group treated with 5 µg/day (P = 0.0095), corresponding to the loss of efficacy of this dose at day 21. In contrast, the leptin doses of 1 and 3 µg/day, which maintained constant efficacy for the duration of the study, did not show a reduction of plasma leptin over time.
To determine whether plasma leptin concentrations corresponded to biologically active leptin, we measured plasma concentrations of IGFBP2 on day 12 (Fig. 1E). IGFBP2 is a recently identified target of leptin, and circulating levels are dose-dependently elevated by leptin (13,24). STZ administration caused a 1.9-fold increase in IGFBP2 levels compared with nondiabetic mice, supporting previous reports that STZ increases hepatic Igfbp2 expression in rats (25). Although 1 µg/day leptin did not further change IGFBP2 levels, 3 and 5 µg/day leptin resulted in dose-dependent increases in plasma IGFPB2. Mice receiving 10 µg/day leptin had a nonsignificant decline in plasma IGFBP2 compared with mice treated with 5 µg/day. Thus, plasma IGFBP2 levels corresponded with plasma leptin concentrations on day 12, confirming that plasma leptin levels correlated with biologically active leptin. We postulate that the return to hyperglycemia in mice treated with supraphysiological doses of leptin was attributable to a decrease in circulating biologically active leptin levels.
Leptin-treated mice have improved glucose tolerance and hypoglycemia during an oral glucose tolerance test.
We next assessed whether leptin could dose-dependently lower glucose excursions in STZ-diabetic mice by performing oral glucose tolerance tests (OGTTs) on day 19 after pump implantation (Fig. 2A). Overall glucose excursions were assessed by AUC (Fig. 2B). All 5 STZ-vehicle mice and 4 of 5 mice treated with STZ and 1 µg/day leptin had blood glucose levels above the limit of detection (33.3 mmol/L) at one or more time points after gavage. Values of 33.3 mmol/L were assigned for these time points, and these groups were not included in statistical analyses. In agreement with their increasing fasted blood glucose levels prior to the day of OGTT (Fig. 1A), mice treated with 10 µg/day leptin showed a trend toward glucose intolerance compared with nondiabetic controls and mice treated with 3 or 5 µg/day leptin. Treatment with 3 and 5 µg/day leptin nearly normalized glucose tolerance, but trended toward hyperglycemia during the glucose peak and toward hypoglycemia during the recovery phase of the test. In fact, 2 of 5 mice receiving 5 µg/day leptin and 1 of 5 mice receiving 3 µg/day leptin had blood glucose <2.9 mmol/L at 90 min and had to be excluded from the remainder of the test.
FIG. 2.
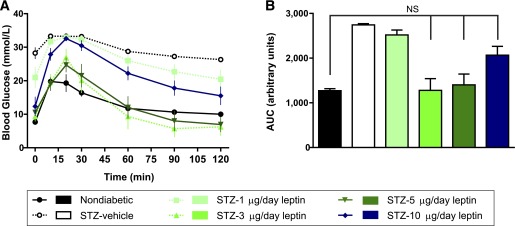
Leptin improves glucose tolerance in STZ-diabetic mice in a dose-dependent manner. OGTTs were performed in nondiabetic controls and STZ-induced diabetic mice receiving different doses of leptin (STZ and 1, 3, 5, or 10 µg/day) or vehicle only (STZ-vehicle) after a 6-h fast on day 19 after surgery. Mice were gavaged with 2 g/kg glucose at time 0. Time course of blood glucose tracking (A) and AUC from 0–90 min (B). Four of five mice treated with 1 µg/day and all STZ-vehicle–treated mice had blood glucose above the detection limit (33.3 mmol/L) at one or more time points in which case values of 33.3 mmol/L were assigned. These groups were not included in statistical analyses. Two mice treated with STZ and 5 µg/day (STZ-5 µg/day) and one mouse treated with STZ and 3 µg/day (STZ-3 µg/day) had blood glucose <2.9 mmol/L at 90 min and were rescued with exogenous glucose. Data from these mice are omitted from the 120-min time point; n = 5 for 0–90 min and n = 3–5 for 120 min. Data are presented as mean ± SEM. NS, not significant.
Collectively, our leptin dose study revealed two key points: 1 µg/day leptin does not reverse hyperglycemia or glucose intolerance, making it an appropriate dose to test whether leptin can improve islet transplant performance; and lack of long-term leptin efficacy and risk of hypoglycemia may be limitations associated with high-dose leptin monotherapy for diabetes, thus strengthening the argument to combine low-dose leptin therapy with a regulated cell-based source of insulin for achievement of well-controlled blood glucose levels.
Low-dose leptin administration enhances islet transplant efficacy.
We subsequently examined whether leptin administration could improve the ability of islet transplantation to ameliorate STZ-induced diabetes. Six days after STZ administration (day 0), all mice underwent two surgeries simultaneously: transplantation of syngeneic islets under the left kidney capsule (or sham surgery) and subcutaneous implantation of 6-week osmotic pumps infusing 1 µg/day leptin (or vehicle). Previous reports (10), as well as data from our group, indicate that transplantation of <200 islets under the kidney capsule is typically insufficient to reverse hyperglycemia in STZ-diabetic mice. Therefore, we transplanted diabetic mice with suboptimal doses of either 50 or 125 islets, along with infusion of either leptin (50 or 125 islets-leptin) or vehicle (50 or 125 islets-vehicle). These groups were compared with diabetic mice administered an optimal islet transplant dose of 300 islets (300 islets-vehicle), mice receiving leptin only (sham-leptin), untreated diabetic mice (sham-vehicle), and healthy nondiabetic controls that received sham surgery and vehicle infusion.
We first examined the effect of leptin and islet cotherapy on 4-h fasted blood glucose levels (Fig. 3A). Untreated diabetic mice maintained blood glucose levels >21 mmol/L for the study duration. For statistical analyses, AUC was calculated using fasting blood glucose data from days 5–43 after surgery (Fig. 3B). Transplantation of 300 islets rapidly restored normoglycemia, revealing that the donor islets were functional and viable. As expected, neither 50 islets nor 125 islets alone significantly lowered blood glucose compared with sham-vehicle mice, nor did leptin treatment alone. However, the combination of leptin with either 50 or 125 islets led to a robust and significant lowering of blood glucose levels compared with sham-vehicle mice (P = 0.026 and 0.0019, respectively). In fact, mice treated with 125 islets in the presence of leptin had mean fasting blood glucose levels nearly as low as mice receiving 300 islets (11.5 ± 0.4 mmol/L vs. 7.6 ± 1.2 mmol/L).
FIG. 3.
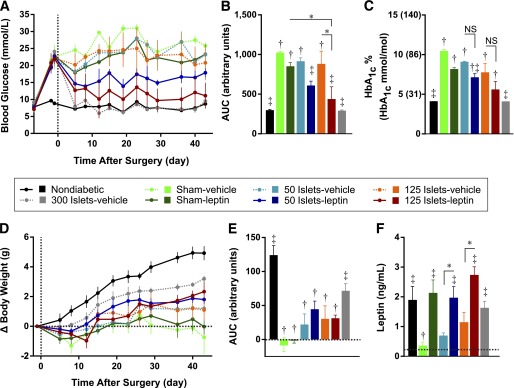
Leptin administration enhances the efficacy of islet transplantation for treatment of STZ-induced diabetes. Mice were treated with STZ on day −6 and subsequently received transplants of 50, 125, or 300 islets or sham surgery (day 0) and simultaneous osmotic pump implants delivering 1 µg/day leptin or vehicle for 6 weeks. A: The 4-h fasted blood glucose levels. B: Blood glucose data from days 5 to 43 analyzed by AUC. C: The 4-h fasted HbA1c levels in whole blood are presented as percents and mmol/mol equivalents are in parentheses. The 4-h fasted body weight gain normalized to day −1 (D) and net AUC from days 5 to 43 (E). F: The 4-h fasted plasma leptin measured from cardiac puncture samples collected 6 weeks after surgery. Data are presented as mean ± SEM (n = 3–5). *P < 0.05. †P < 0.05 vs. nondiabetic controls. ‡P < 0.05 vs. sham-vehicle controls. NS, not significant.
HbA1c levels in mice 6 weeks after surgery (Fig. 3C) revealed similar effects on long-term glycemic control. As expected, there was a dramatic increase in HbA1c in sham-vehicle mice compared with nondiabetic controls (P < 0.0001), and transplantation of 300 islets normalized HbA1c levels to nondiabetic levels. Mice treated with leptin alone and with 50 or 125 islets alone showed nonsignificant trends toward reduced HbA1c levels compared with sham-vehicle mice. In contrast, combination of leptin with either 50 or 125 islets led to a substantial reduction in HbA1c compared with sham-vehicle mice (P = 0.006 and 0.002, respectively).
We also tracked the effect of islet transplantation with leptin cotherapy on body weight (Fig. 3D). Prior to implantation, body weight was significantly reduced to 24.3 ± 0.4 g in STZ-injected mice compared with 26.4 ± 0.4 g in nondiabetic controls (P = 0.025, t test). Cumulative weight gain over the 6 weeks after surgery was determined by net AUC analysis (Fig. 3E). With the exception of the 300 islets-vehicle group, cumulative weight gain was lower in STZ-injected groups compared with nondiabetic controls. Neither suboptimal islet transplant nor leptin nor combination therapy significantly altered cumulative weight gain compared with sham-vehicle controls, although we noted that all transplant recipient groups exhibited a positive cumulative weight gain, whereas mice treated with sham-vehicle and sham-leptin had no cumulative weight gain.
Plasma leptin levels were measured at 6 weeks after surgery (Fig. 3F). Leptin levels were significantly reduced in sham-vehicle mice compared with nondiabetic controls, and administration of 1 µg/day leptin restored plasma leptin concentrations to nondiabetic levels. Mice that underwent islets transplantation had a trend toward increased leptin concentrations, compared with sham-vehicle mice, that was proportional to the amount of islets transplanted. Transplantation of 300 islets yielded leptin levels similar to those of nondiabetic controls (1.6 ± 0.3 ng/mL), consistent with stimulation of endogenous leptin production by graft-derived insulin. Mice treated with islet and leptin cotherapy had similar plasma leptin concentrations as sham-leptin and nondiabetic mice. Therefore, although unable to reverse hyperglycemia alone, a replacement dose of leptin can robustly improve islet graft performance.
Leptin enhances islet transplant efficacy without increasing circulating insulin or endogenous β-cell recovery.
We next determined whether the improvement of islet transplant efficacy in the presence of leptin cotherapy was attributable to an increase in circulating insulin levels (Fig. 4A). Leptin alone had no effect on plasma insulin levels, whereas transplantation of islets alone significantly increased plasma insulin levels above those of sham-vehicle controls. Interestingly, despite the observed glucose-lowering effect of leptin cotherapy, leptin did not further increase plasma insulin concentrations compared with mice receiving islet transplants without leptin. If anything, there was a nonsignificant decrease in recipients of 125 islets cotreated with leptin compared with recipients of 125 islets alone. Measurement of pancreatic β-cell area revealed an expected ∼85% decrease in β-cell area in sham-vehicle mice compared with nondiabetic controls (Fig. 4B and C). Transplantation of 125 islets alone or with leptin did not increase β-cell area compared with sham-vehicle controls, supporting that the increase in plasma insulin after islet transplantation was from graft-derived insulin. Representative pancreatic sections from nondiabetic and STZ-injected mice demonstrate the extreme loss of β-cells (Fig. 4C). In contrast, pancreatic α-cell area did not differ between nondiabetic controls and sham-vehicle mice (Fig. 4B and C). Increased α-cell area as a proportion of islet area has been interpreted as α-cell expansion after STZ administration (26). However, when α-cell area is normalized to total pancreatic area, there is clearly no expansion of α-cells in STZ-injected mice compared with nondiabetic mice. Islet transplantation with or without leptin had no effect on α-cell area.
FIG. 4.

Leptin cotherapy does not alter circulating insulin levels or β-cell recovery. A: The 4-h fasted plasma insulin levels were measured 6 weeks after transplant in all groups by ultrasensitive insulin ELISA. Limit of detection (0.019 ng/mL) is shown as horizontal broken line (n = 3–5). B: Immunofluorescent quantifications of β-cell and α-cell areas were performed in nondiabetic controls and STZ-induced diabetic mice treated with 125 islets (125 islets-vehicle), with 125 islets and leptin (125 islets-leptin), or left untreated (sham-vehicle) (n = 3–4). Data are presented as mean ± SEM. †P < 0.05 vs. nondiabetic controls. ‡P < 0.05 vs. sham-vehicle controls. C: Representative images of pancreata from nondiabetic and sham-vehicle groups costained for insulin (red), glucagon (green), and DAPI (white) (n = 3–4). Scale bars represent 1,000 µm (left top, left bottom) and 100 µm (right top, right bottom). NS, not significant.
Leptin and islet cotherapy improve glucose tolerance in STZ-diabetic mice.
To assess whether leptin and islet cotherapy also improved glucose excursions, we performed OGTTs on day 20 after surgery (Fig. 5A). As expected, sham-vehicle mice had severely impaired glucose tolerance compared with nondiabetic controls, and transplantation of 300 islets nearly normalized glucose excursion. At the glycemic peak, some mice had blood glucose concentrations >33.3 mmol/L limit of detection; thus, we collected blood from all mice at 10 and 20 min after gavage and measured plasma glucose by the glucose oxidase method (Fig. 5B). Plasma glucose concentrations in mice treated with suboptimal islet doses or leptin alone were similar to sham-vehicle controls at 10 and 20 min. In contrast, the combination of leptin and 125 islets significantly lowered plasma glucose at 10 min, whereas there was a nonsignificant trend toward lower plasma glucose in mice treated with leptin and 50 islets. During this test, we also measured glucose-stimulated plasma insulin levels (Fig. 5C). Only nondiabetic controls and mice transplanted with 300 islets had detectable glucose-stimulated insulin levels, further supporting that leptin and islet cotherapy achieves metabolic control similar to that of recipients of an optimal islet transplant, without restoring plasma insulin levels.
FIG. 5.

Leptin and islet cotherapy improves glucose tolerance. A: OGTT on day 20 after a 6-h fast in mice transplanted with islets with or without leptin and untreated diabetic (sham-vehicle) mice, leptin-treated diabetic (sham-leptin) mice, and nondiabetic controls. B: Because some mice had blood glucose levels over the limit of detection after gavage, blood was collected at 10 and 20 min for plasma glucose measurement. C: Plasma insulin levels after gavage. The limit of detection (0.188 ng/mL) is indicated by a broken horizontal line. Only nondiabetic and 300 islets-vehicle groups had detectable plasma insulin levels at any given time. The remainder of mice were assigned values of 0.188 ng/mL. Data are presented as mean ± SEM (n = 3–5). *P < 0.05. †P < 0.05 vs. nondiabetic controls. ‡P < 0.05 vs. sham-vehicle controls. NS, not significant.
Low-dose leptin administration reverses dyslipidemia in STZ-diabetic mice.
Because leptin clearly enhanced the ability of islet transplantation to improve glycemic control, we subsequently examined whether leptin and islet cotherapy also improved lipid homeostasis. We measured plasma triglycerides (Fig. 6A), FFA (Fig. 6B), and β-hydroxybutyrate levels (Fig. 6C) on day 12 after surgery. As expected, STZ administration caused elevations in plasma triglycerides, FFA, and β-hydroxybutyrate levels from nondiabetic values. Transplantation of 300 islets normalized all three metabolites in diabetic mice, whereas transplantation of 50 or 125 islets without leptin led to intermediate values between those of nondiabetic and sham-vehicle controls. Interestingly, despite having a minimal impact on glucose metabolism, 1 µg/day leptin alone completely normalized all three of these metabolites. This is consistent with the profound lipogenic actions of leptin, which occur in part through stimulating lipid oxidation in multiple tissues and through inhibiting hepatic triglyceride synthesis and secretion (6,27–31). Consequently, combination of leptin with 50 or 125 islets led to plasma triglyceride, FFA, and β-hydroxybutyrate concentrations similar to those of healthy nondiabetic controls.
FIG. 6.

Low-dose leptin administration normalizes lipid levels in STZ-diabetic mice. The 4-h fasted plasma triglycerides (A), FFA (B), and β-hydroxybutyrate (C) were measured in mice transplanted with islets with or without leptin and in untreated diabetic (sham-vehicle) mice, leptin-treated diabetic (sham-leptin) mice, and nondiabetic controls on day 12 after surgery. Data are presented as mean ± SEM (n = 3–5). *P < 0.05. †P < 0.05 vs. nondiabetic controls. ‡P < 0.05 vs. STZ-vehicle controls.
DISCUSSION
We and others have found that high-dose leptin administration reverses hyperglycemia in rodent models of type 1 diabetes (4–8). However, the dose-response profile of glucose-lowering by leptin in type 1 diabetic models is not well defined. The current study reveals that leptin lowers blood glucose in STZ-diabetic mice in a dose-dependent manner, and that a supraphysiological level of leptin was necessary to achieve normoglycemia. This supports a previous report that a physiological leptin dose in STZ-diabetic rats only modestly reduced blood glucose levels (25).
There is some rationale to support the use of leptin as a monotherapy. First, type 1 diabetes, at least in rodents, results in relative leptin deficiency (23), a state in which leptin therapy is most effective. Second, leptin has beneficial effects on lipid metabolism that oppose the potentially deleterious lipogenic action of insulin (6). Furthermore, unlike insulin, there is no weight gain associated with leptin-induced normalization of glucose levels in type 1 diabetic rodents (5,6,8). However, our study revealed key aspects of high-dose leptin therapy that may make it inappropriate for use as a monotherapy. With the doses of leptin sufficient to achieve rapid normoglycemia (5 and 10 µg/day), the efficacy of leptin was transient. We postulate that the transient nature of high-dose leptin could be attributable to a loss of leptin bioactivity within long-term implanted pumps, or to a physiological response in mice to counteract high-dose leptin treatment. Additionally, the lowest leptin doses that achieved fasting normoglycemia produced severe hypoglycemia during an OGTT in ∼30% of the mice. Previously, we observed that injection of an insulin dose that only modestly lowered glucose levels in diabetic mice resulted in severe hypoglycemia when combined with high-dose leptin therapy (8). Consequently, the addition of leptin to unregulated high insulin doses may confer increased hypoglycemic risk; thus, we propose that combination of leptin with either low-dose insulin or a regulated source of insulin such as islets could be a safer and more effective method to lower blood glucose. In the current study, administration of low-dose leptin, although insufficient to ameliorate diabetes alone, robustly augmented the ability of a suboptimal dose of syngeneic islets to improve fasting glycemia and glucose tolerance in STZ-diabetic mice. When combined with leptin, 125 islets functioned almost as well as a transplant of 300 islets in regard to achieving glycemic control, whereas 125 islets alone failed to ameliorate diabetes. Furthermore, although suboptimal islet transplantation alone did not reverse dyslipidemia, low-dose leptin alone normalized lipid metabolism and ketone levels in diabetic mice.
A potential mechanism by which leptin could improve the ability of islet transplantation to lower blood glucose is through increased insulin secretion from transplanted or endogenous β-cells. However, leptin and islet cotherapy did not affect pancreatic β-cell area in our study. Moreover, although leptin can protect islets against lipotoxicity and β-cell apoptosis (32,33), we found that leptin cotherapy did not increase circulating insulin levels in islet transplant recipients. Rather, in mice receiving a transplant of 125 islets, administration of leptin produced a nonsignificant decrease in circulating insulin. This supports multiple studies showing that leptin has an inhibitory action on insulin secretion (7,18–22). The finding that low-dose leptin alone had no glucose-lowering ability indicates that the mechanism of leptin cotherapy is dependent on the islet graft providing a source of insulin. Alternatively, leptin therapy has been shown to enhance insulin sensitivity in diabetic rodents (4,8,25,34). In our study, low-dose leptin alone was effective in reversing the dyslipidemia of uncontrolled diabetes, which could contribute to enhanced insulin sensitivity in peripheral tissues. Collectively, by providing a more lipopenic insulin-sensitive environment for islet grafts in a transplant recipient, leptin administration may allow graft-derived insulin to have more profound metabolic effects.
Our data reveal that as few as half the number of islets normally required to restore euglycemia in mice can achieve glycemic control when combined with low-dose leptin administration. This suggests that the number of diabetic patients treated with cadaveric islet transplantation could be virtually doubled by simply adding leptin to their therapeutic regimen. Furthermore, the profound lipopenic action of low-dose leptin may provide a particular advantage by protecting against cardiovascular complications associated with dyslipidemia (35,36). However, in our opinion, the potentially deleterious effects of leptin administration on the underlying autoimmune aspect of type 1 diabetes should be assessed with caution. High-dose leptin administration in NOD mice has been shown to accelerate autoimmune β-cell destruction (37), whereas mutations in the leptin receptor are protective in NOD mice (38,39). However, Kruger et al. (7) found that pretreatment of biobreeding rats with leptin prevented immune destruction of both endogenous and transplanted β-cells. Likewise, leptin treatment confers clinical benefit to patients with autoimmune lipodystrophy and type 1 diabetes despite the underlying autoimmune disorder (40). Thus, although the impact of leptin on autoimmune destruction of β-cells should be carefully assessed, the potential therapeutic benefit of leptin coadministration with islet transplantation for type 1 diabetes warrants consideration.
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research (CIHR). T.J.K. received a senior scholarship from the Michael Smith Foundation for Health Research. H.C.D. is supported by an Alexander Graham Bell Canada Graduate Scholarship from the Natural Sciences and Engineering Research Council of Canada, and by the Anne and John Brown Fellowship in Diabetes and Obesity-Related Research. J.E.B. is supported by a Juvenile Diabetes Research Foundation postdoctoral fellowship, L’Oreal Canada for Women in Science Research Excellence Fellowship, and the CIHR Transplantation Training Program. E.T. is supported by the Canadian Diabetes Association.
No potential conflicts of interest relevant to this article were reported.
H.C.D. wrote the manuscript. H.C.D. and W.L.Q. researched and analyzed data. J.E.B., E.T., A.A., M.M.G., and J.K.F. contributed to data research, analysis, discussion, and manuscript review. T.J.K. contributed to discussion and reviewed the manuscript. T.J.K. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
REFERENCES
- 1.Shapiro AM, Lakey JR, Ryan EA, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000;343:230–238 [DOI] [PubMed] [Google Scholar]
- 2.Ryan EA, Paty BW, Senior PA, et al. Five-year follow-up after clinical islet transplantation. Diabetes 2005;54:2060–2069 [DOI] [PubMed] [Google Scholar]
- 3.Denroche HC, Huynh FK, Kieffer TJ. The role of leptin in glucose homeostasis. J Diabetes Invest 2012;3:115–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chinookoswong N, Wang JL, Shi ZQ. Leptin restores euglycemia and normalizes glucose turnover in insulin-deficient diabetes in the rat. Diabetes 1999;48:1487–1492 [DOI] [PubMed] [Google Scholar]
- 5.Yu X, Park BH, Wang MY, Wang ZV, Unger RH. Making insulin-deficient type 1 diabetic rodents thrive without insulin. Proc Natl Acad Sci USA 2008;105:14070–14075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang MY, Chen L, Clark GO, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA 2010;107:4813–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kruger AJ, Yang C, Lipson KL, et al. Leptin treatment confers clinical benefit at multiple stages of virally induced type 1 diabetes in BB rats. Autoimmunity 2011;44:137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Denroche HC, Levi J, Wideman RD, et al. Leptin therapy reverses hyperglycemia in mice with streptozotocin-induced diabetes, independent of hepatic leptin signaling. Diabetes 2011;60:1414–1423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miyanaga F, Ogawa Y, Ebihara K, et al. Leptin as an adjunct of insulin therapy in insulin-deficient diabetes. Diabetologia 2003;46:1329–1337 [DOI] [PubMed] [Google Scholar]
- 10.Juang JH, Bonner-Weir S, Wu YJ, Weir GC. Beneficial influence of glycemic control upon the growth and function of transplanted islets. Diabetes 1994;43:1334–1339 [DOI] [PubMed] [Google Scholar]
- 11.Ferrer-Garcia JC, Merino-Torres JF, Pérez Bermejo G, Herrera-Vela C, Ponce-Marco JL, Piñon-Selles F. Insulin-induced normoglycemia reduces islet number needed to achieve normoglycemia after allogeneic islet transplantation in diabetic mice. Cell Transplant 2003;12:849–857 [PubMed] [Google Scholar]
- 12.Wideman RD, Yu IL, Webber TD, et al. Improving function and survival of pancreatic islets by endogenous production of glucagon-like peptide 1 (GLP-1). Proc Natl Acad Sci USA 2006;103:13468–13473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levi J, Huynh FK, Denroche HC, et al. Hepatic leptin signalling and subdiaphragmatic vagal efferents are not required for leptin-induced increases of plasma IGF binding protein-2 (IGFBP-2) in ob/ob mice. Diabetologia 2012;55:752–762 [DOI] [PubMed] [Google Scholar]
- 14.Rezania A, Bruin JE, Riedel MJ, et al. Maturation of human embryonic stem cell-derived pancreatic progenitors into functional islets capable of treating pre-existing diabetes in mice. Diabetes 2012;61:2016–2029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.German JP, Thaler JP, Wisse BE, et al. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology 2011;152:394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA 2010;107:17391–17396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hidaka S, Yoshimatsu H, Kondou S, et al. Chronic central leptin infusion restores hyperglycemia independent of food intake and insulin level in streptozotocin-induced diabetic rats. FASEB J 2002;16:509–518 [DOI] [PubMed] [Google Scholar]
- 18.Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic beta-cells. Diabetes 1997;46:1087–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seufert J, Kieffer TJ, Habener JF. Leptin inhibits insulin gene transcription and reverses hyperinsulinemia in leptin-deficient ob/ob mice. Proc Natl Acad Sci USA 1999;96:674–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seufert J, Kieffer TJ, Leech CA, et al. Leptin suppression of insulin secretion and gene expression in human pancreatic islets: implications for the development of adipogenic diabetes mellitus. J Clin Endocrinol Metab 1999;84:670–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weigle DS, Bukowski TR, Foster DC, et al. Recombinant ob protein reduces feeding and body weight in the ob/ob mouse. J Clin Invest 1995;96:2065–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kulkarni RN, Wang ZL, Wang RM, et al. Leptin rapidly suppresses insulin release from insulinoma cells, rat and human islets and, in vivo, in mice. J Clin Invest 1997;100:2729–2736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Havel PJ, Uriu-Hare JY, Liu T, et al. Marked and rapid decreases of circulating leptin in streptozotocin diabetic rats: reversal by insulin. Am J Physiol 1998;274:R1482–R1491 [DOI] [PubMed] [Google Scholar]
- 24.Hedbacker K, Birsoy K, Wysocki RW, et al. Antidiabetic effects of IGFBP2, a leptin-regulated gene. Cell Metab 2010;11:11–22 [DOI] [PubMed] [Google Scholar]
- 25.German JP, Wisse BE, Thaler JP, et al. Leptin deficiency causes insulin resistance induced by uncontrolled diabetes. Diabetes 2010;59:1626–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Zhang Y, Bone RN, et al. Regeneration of pancreatic non-β endocrine cells in adult mice following a single diabetes-inducing dose of streptozotocin. PLoS ONE 2012;7:e36675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang MY, Lee Y, Unger RH. Novel form of lipolysis induced by leptin. J Biol Chem 1999;274:17541–17544 [DOI] [PubMed] [Google Scholar]
- 28.Orci L, Cook WS, Ravazzola M, et al. Rapid transformation of white adipocytes into fat-oxidizing machines. Proc Natl Acad Sci USA 2004;101:2058–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang W, Dedousis N, Bandi A, Lopaschuk GD, O’Doherty RM. Liver triglyceride secretion and lipid oxidative metabolism are rapidly altered by leptin in vivo. Endocrinology 2006;147:1480–1487 [DOI] [PubMed] [Google Scholar]
- 30.Huynh FK, Neumann UH, Wang Y, Rodrigues B, Kieffer TJ, Covey SD. A role for hepatic leptin signaling in lipid metabolism via altered very low density lipoprotein composition and liver lipase activity in mice. Hepatology 2013;57:543–554 [DOI] [PubMed] [Google Scholar]
- 31.Cohen P, Miyazaki M, Socci ND, et al. Role for stearoyl-CoA desaturase-1 in leptin-mediated weight loss. Science 2002;297:240–243 [DOI] [PubMed] [Google Scholar]
- 32.Okuya S, Tanabe K, Tanizawa Y, Oka Y. Leptin increases the viability of isolated rat pancreatic islets by suppressing apoptosis. Endocrinology 2001;142:4827–4830 [DOI] [PubMed] [Google Scholar]
- 33.Lee Y, Ravazzola M, Park BH, Bashmakov YK, Orci L, Unger RH. Metabolic mechanisms of failure of intraportally transplanted pancreatic beta-cells in rats: role of lipotoxicity and prevention by leptin. Diabetes 2007;56:2295–2301 [DOI] [PubMed] [Google Scholar]
- 34.Lin CY, Higginbotham DA, Judd RL, White BD. Central leptin increases insulin sensitivity in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab 2002;282:E1084–E1091 [DOI] [PubMed] [Google Scholar]
- 35.Hamaguchi M, Kojima T, Takeda N, et al. Nonalcoholic fatty liver disease is a novel predictor of cardiovascular disease. World J Gastroenterol 2007;13:1579–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onat A, Sari I, Yazici M, Can G, Hergenç G, Avci GS. Plasma triglycerides, an independent predictor of cardiovascular disease in men: a prospective study based on a population with prevalent metabolic syndrome. Int J Cardiol 2006;108:89–95 [DOI] [PubMed] [Google Scholar]
- 37.Matarese G, Sanna V, Lechler RI, et al. Leptin accelerates autoimmune diabetes in female NOD mice. Diabetes 2002;51:1356–1361 [DOI] [PubMed] [Google Scholar]
- 38.Lee CH, Reifsnyder PC, Naggert JK, et al. Novel leptin receptor mutation in NOD/LtJ mice suppresses type 1 diabetes progression: I. Pathophysiological analysis. Diabetes 2005;54:2525–2532 [DOI] [PubMed] [Google Scholar]
- 39.Lee CH, Chen YG, Chen J, et al. Novel leptin receptor mutation in NOD/LtJ mice suppresses type 1 diabetes progression: II. Immunologic analysis. Diabetes 2006;55:171–178 [PubMed] [Google Scholar]
- 40.Park JY, Chong AY, Cochran EK, et al. Type 1 diabetes associated with acquired generalized lipodystrophy and insulin resistance: the effect of long-term leptin therapy. J Clin Endocrinol Metab 2008;93:26–31 [DOI] [PMC free article] [PubMed] [Google Scholar]