

Abstract
Increased sympathetic activity is a well-known pathophysiological mechanism in insulin resistance (IR) and hypertension (HT). The carotid bodies (CB) are peripheral chemoreceptors that classically respond to hypoxia by increasing chemosensory activity in the carotid sinus nerve (CSN), causing hyperventilation and activation of the sympathoadrenal system. Besides its role in the control of ventilation, the CB has been proposed as a glucose sensor implicated in the control of energy homeostasis. However, to date no studies have anticipated its role in the development of IR. Herein, we propose that CB overstimulation is involved in the etiology of IR and HT, core metabolic and hemodynamic disturbances of highly prevalent diseases like the metabolic syndrome, type 2 diabetes, and obstructive sleep apnoea. We demonstrate that CB activity is increased in IR animal models and that CSN resection prevents CB overactivation and diet-induced IR and HT. Moreover, we show that insulin triggers CB, highlighting a new role for hyperinsulinemia as a stimulus for CB overactivation. We propose that CB is implicated in the pathogenesis of metabolic and hemodynamic disturbances through sympathoadrenal overactivation and may represent a novel therapeutic target in these diseases.
Insulin resistance (IR), arterial hypertension (HT), obesity, and dyslipidemia are core features of widespread diseases in Western societies such as the metabolic syndrome, type 2 diabetes, and obstructive sleep apnoea. Visceral obesity has been proposed to play a fundamental role in the simultaneous development of IR and HT that characterizes these diseases (1). Recent findings suggest that peripheral IR is also a common feature in lean obstructive sleep apnoea (2) as well as lean polycystic ovarian syndrome (3), despite its strong relationship with visceral obesity. Similarly, the association of HT with obstructive sleep apnoea is independent of obesity (4), as demonstrated by hypertensive lean sleep apnoea patients. Altogether, these findings point to the existence of an obesity-independent etiological factor that simultaneously causes IR and HT: the activation of the carotid bodies (CBs) has recently been suggested as a putative candidate (5).
The CBs are arterial chemoreceptors that sense changes in arterial blood O2, CO2, and pH levels. Hypoxia and acidosis/hypercapnia activate the CBs, which respond by increasing the action potential frequency in their sensory nerve, the carotid sinus nerve (CSN). CSN activity is integrated in the brain stem to induce a fan of respiratory reflexes aimed, primarily, at normalizing the altered blood gases via hyperventilation (6) and to regulate blood pressure and cardiac performance via sympathetic nervous system activation (7). The CB directly activates the adrenals via increased sympathetic drive and also increases sympathetic vasoconstrictor outflow to muscle, splanchnic, and renal beds (7,8). Enhanced sympathetic nerve activity is known to contribute to skeletal muscle IR and to impaired glucose tolerance, mainly due to sympathetic mediated lipolysis (9,10) and also to increased arterial pressure (9). Recently, the CB was proposed to be a glucose sensor (11) and implicated in energy homeostasis control (12).
The objective of this study was to investigate the role of the CB in the pathogenesis of metabolic and hemodynamic disturbances by testing the hypothesis that CB activity is increased in IR and HT animal models. Also, to clarify the role of obesity as an independent factor in CB activation, we compared CB function in both obese and lean models of IR.
The second hypothesis tested was that insulin is a trigger for CB activation. In vivo experiments have previously shown that intravenous infusion of insulin causes a CB-dependent increase in ventilation (13). The authors concluded that this effect was associated with the hypoglycemia caused by insulin administration; however, others have shown that low glucose is not a direct stimulus for rat CB chemoreceptors (14,15). These discordant results point toward insulin as a good alternative candidate to activate the CBs.
Finally, we performed chronic CSN bilateral resections to test the hypothesis that preventing the CBs from being overactivated averts the development of IR and HT and also the increase in sympathoadrenal activity, induced by hypercaloric diets in animals. The data presented herein clarify the role of the CB in the pathogenesis of diet-induced IR and HT and unveil a new promising target for intervention in type 2 diabetes, metabolic syndrome, and obstructive sleep apnoea.
RESEARCH DESIGN AND METHODS
Experiments were performed in Wistar rats (200–420 g) of both sexes, aged 3 months, obtained from the vivarium of Faculty of Medical Sciences. Two diet-induced IR and HT animal models were used: the rat submitted to a high-fat (HF) diet, a model that combines obesity, IR, and HT (16,17), and the rat submitted to a high-sucrose (HSu) diet, a lean model of combined IR and HT (16,18). Briefly, the control group was fed a sham diet (7.4% fat plus 75% carbohydrate [4% sugar] plus 17% protein; SDS diets RM1; Probiológica, Lisbon, Portugal). The HSu model was obtained by administration of 35% sucrose (Panlab, Lisbon, Portugal) in drinking water during 28 days. The HF model was fed a lipid-rich diet (45% fat plus 35% carbohydrate plus 20% protein; Mucedola, Milan, Italy) during 21 days. The HSu and HF animals are validated in the literature as animal models of the metabolic syndrome (19). To demonstrate that CB activity was increased in hypercaloric-fed animals, we compared HF and HSu with a control group.
To evaluate the contribution of CB to the genesis of IR and HT, bilateral resection of CSN was performed 5 days prior to submitting the animals to standard or hypercaloric diets. The carotid artery bifurcations were located bilaterally, and CSNs were identified and either sectioned bilaterally or left intact (sham). These procedures were performed in aseptic conditions under ketamine (30 mg/kg)/xylazine (4 mg/kg) anesthesia and buprenorphine (10 μg/kg) analgesia. Chronic resection of CSN was confirmed by absence of ischemic hypoxia–induced hyperventilation prior to experiments.
Rats fed with standard diet were used to investigate whether insulin triggers CB activation.
All test groups included an equal number of males and females. Display of an odd experimental number refers to the death of experimental units during the experimental procedure. Also, food and liquid intake was monitored during the treatments in all groups of animals. Body weight and animal behavioral changes were assessed twice per week.
All measurements were performed with animals under sodium pentobarbital (60 mg/kg i.p.) anesthesia, since pentobarbital was shown not to alter the metabolic parameters tested herein (constant rate for glucose disappearance [KITT], fasting glycemia, insulinemia, and free fatty acids) in comparison with conscious animals (20) or insulin responses to glucose (21). At the end of the experiments, the rats were killed by an intracardiac overdose of pentobarbital, except when heart puncture was performed to collect blood. Principles of laboratory care were followed in accordance with the European Union Directive for Protection of Vertebrates Used for Experimental and Other Scientific Ends (2010/63/EU). Experimental protocols were approved by the ethics committee of the Faculty of Medical Sciences.
Evaluation of basal ventilation and ischemic ventilatory responses in animal models of IR and HT.
A detailed description of these methods was previously published (22). Shortly, respiratory frequency and tidal volume were obtained by pneumotachograph (Hugo SACHS Elektronik; Harvard Apparatus, Madrid, Spain) in anesthetized and tracheostomized control rats and in rats submitted to hypercaloric diets. These respiratory parameters and blood pressure were continuously recorded in anesthetized and vagotomized rats breathing spontaneously and submitted to either bilateral occlusion (5–15 s) of common carotid artery. Bilateral midcervical vagotomy was performed to abolish the role of vagal afferents innervating the lungs and the aortic chemoreceptors with a major influence on respiratory activity (23). Control experiments were performed in animals submitted to bilateral cut of the CSN in order to distinguish central and peripherally mediated effects.
Effect of insulin on spontaneous ventilation in control animals.
Insulin effect on ventilation was assessed in control rats anesthetized, tracheostomized, and vagotomized. Briefly, an insulin bolus (1, 5, 10, 50, 100, and 200 mU/kg) was administered in external carotid artery and reaches the CB by being pushed by the blood flow of common carotid artery. Ventilatory parameters as described above were monitored. Euglycemic clamp was maintained through glucose (10 mg/kg/min) perfusion into the femoral vein. Confirmation of CB/insulin-mediated effect was done by measurement of ventilation after CSN cut.
Measurement of insulin sensitivity and mean arterial pressure.
Insulin tolerance test was used to measure insulin sensitivity (16,24). Mean arterial pressure (MAP) monitoring was measured (16). After insulin sensitivity and MAP evaluation, blood was collected by heart puncture and treated for quantification of soluble biomarkers (16). Visceral fat and adrenal medulla were collected after an abdominal laparotomy and weighted. Adrenal medullas were frozen in liquid nitrogen and stored placed at −80°C.
Measurement of plasma insulin, circulating free fatty acids, corticosterone and catecholamines levels, and adrenal medulla catecholamine content.
Plasma and serum were collected after heart puncture to EDTA-precoated tubes and to eppendorfs, respectively. Insulin concentrations and free fatty acids were determined in plasma, and corticosterone was determined in serum (16). Corticosterone determination was obtained with a DetectX corticosterone Immunoassay kit (Arbor Assays, Madrid, Spain). For catecholamine quantification in plasma, 400 μL plasma samples were purified and catecholamines were extracted and quantified as previously described (16). For quantification of catecholamine content in adrenal medulla, adrenal medullas previously frozen were homogenized in 0.6 N perchloric acid, and their endogenous catecholamine content was quantified as previously described (15).
CB dopamine and ATP release in response to hypoxia and to insulin.
CBs were cleaned free of CSN nearby connective tissues under dissection microscope and incubated in Tyrode solution (15,25). To evaluate CB activity in IR and HT animal models, we measured CB dopamine (plus 3,4-dihydroxy phenyl acetic acid [DOPAC], its major metabolite) release. CB ATP and dopamine release in response to insulin were monitored in control animals. In brief, CBs were incubated in 500 µL (250 µL for ATP and 250 µL for dopamine for insulin effects) Tyrode bicarbonate solution and cofactors for tyrosine hydroxylase and dopamine-β-hydroxylase (20 μmol/L tyrosine, 100 mmol/L ascorbic acid, and 500 nmol/L 6-methyl-tetrahydroptine) or Tyrode bicarbonate plus insulin (0.01–100 nmol/L). Solutions were kept at 37°C and continuously bubbled with normoxia (20% O2, 5% CO2, 75% N2), except when hypoxic stimuli were applied. Protocols for dopamine release in overfeeding rats include two 10-min normoxic incubations, followed by 10 min incubation in hypoxia (5% O2, 5% CO2, 75% N2) and 2 posthypoxic incubations in normoxia. Protocols for insulin effect on dopamine and ATP release include two 10-min incubations in normoxia, followed by three incubations with different insulin concentrations and two postinsulin incubations in normoxia. The solutions were renewed at each fixed time, and all fractions were collected and quantified as previously described (15).
Western blot analysis of insulin receptor, insulin receptor phosphorylation (phosphor-Tyr 1322), and tyrosine hydroxylase expression.
For evaluation of insulin receptor phosphorylation, CBs were isolated, cleaned, and incubated at 37°C during 30 min in Tyrode solution containing 1 and 100 nmol/L insulin and bubbled with 20% O2, 5% CO2, and 75% N2. After, CBs were immediately frozen in liquid nitrogen and placed at −80°C. For CB insulin receptor and tyrosine hydroxylase expression, CBs after cleaning were frozen in liquid nitrogen. CBs were homogenized in Zurich medium containing a cocktail of protease inhibitors (26). Proteins were separated in a 10 or 12% SDS PAGE gel electrophoresis and electroblotted on nitrocellulose membrane (0.2 µmol/L; BioRad, Madrid, Spain). To enhance detection sensitivity, we used a three-step Western blot protocol (27). After blocking, membranes were incubated with primary antibodies against insulin receptor (1:100; Santa Cruz Biotechnology, Madrid, Spain), insulin receptor phosphorylated (phosphor-Tyr 1322, 1:50; Assay Designs, Lisbon, Portugal), and tyrosine hydroxylase (1:10,000; Sigma, Madrid, Spain). The membranes were incubated in Tris-buffered saline with Tween (TBST) (0.1%) containing biotin-conjugated goat anti-mouse IgG (1:10,000; Millipore, Madrid, Spain) for 1 h, washed in TBST (0.02%), and incubated for 30 min in TBST (0.1%) containing horseradish peroxidase–conjugated streptavidin (1:10,000, Pierce, Madrid, Spain). Membranes were then washed in TBST (0.02%) and developed with enhanced chemiluminescence reagents (Immobilon Western; Millipore). Intensity of the signals was detected in a Chemidoc Molecular Imager (Chemidoc; BioRad, Madrid, Spain) and quantified using the Quantity-One software (BioRad). The membranes were reprobed and tested for β-actin immunoreactivity (bands in the 42 KDa region) to compare and normalize the expression of proteins with the amount of protein loaded.
Chemoreceptor cell culture and intracellular Ca2+ measurements.
Cleaned CBs were enzymatically dispersed, and dissociated cells were plated on poly-l-lysine–coated coverslips maintained in culture for up to 24 h as previously described (28). Coverslips were incubated with fura-2 acetoxymethyl ester and mounted in a perfusion chamber, and fura-2 fluorescence was measured as the ratio of the fluorescent emission at 340/380 nm of chemoreceptor cells (29). General protocol for Ca2+ measurements consisted of a sequential incubation hypoxia (N2; 1 min), 5 min normoxic incubation (20% O2), 3 min incubation with 1 nmol/L insulin, combination of both hypoxia (N2) and insulin (1 nmol/L), 5 min normoxia (20% O2), 1 min hypoxia (N2), and finally, 30 s high external KCl.
RESULTS
Administration of hypercaloric diets to Wistar rats produced changes in body weight, sympathetic nervous activity, blood pressure, and insulin sensitivity similar to the ones observed in humans (2,16,30). Liquid intake was similar in all animals tested (control group 101.21 ± 3.09 mL/kg/day, HF animals 89.50 ± 3.93 mL/kg/day, and HSu animals 93.22 ± 2.59 mL/kg/day). No significant differences were observed in food intake (control 57.78 ± 2.05 mg/kg/day, HF 62.56 ± 1.99 mg/kg/day, and HSu 51.22 ± 4.51 mg/kg/day). The daily caloric intake was 164.7 ± 5.8 kcal/kg/day in control animals, 299.0 ± 9.4 kcal/kg/day in HF animals (P < 0.001 vs. control), and 332.8 ± 12.8 kcal/kg/day for HSu animals (P < 0.001 vs. control). After CSN cut, the daily caloric intake was 179.6 ± 10.1 in the control group, 289.2 ± 6.5 in the HF group, and 327.6 ± 10.5 kcal/kg/day in the HSu group. The daily caloric intake was not changed by CSN cut, and there were no significant differences among the HF and HSu rats. IR and HT were confirmed by measurement of insulin sensitivity and blood pressure in HF and HSu animals. The HF diet caused a decrease in KITT from 4.69 ± 0.33% glucose/min in control animals to 2.98 ± 0.34% glucose/min (P < 0.01). The HSu diet decreased KITT to 2.68 ± 0.32% glucose/min (P < 0.01). HF and HSu diets caused a significant increase in MAP compared with controls (control 95.99 ± 3.21 mmHg, HF 142.31 ± 2.47 mmHg, and HSu 136.71 ± 4.51 mmHg). Fasting glycemia was not significantly different in control and HF groups, although the HSu diet significantly increased fasting glycemia in comparison with the control group (P < 0.001) (data not shown).
CB is overactivated in insulin-resistant and hypertensive rats.
Figure 1 demonstrates that CB activity is increased in animal models of IR and HT. Spontaneous ventilatory parameters (respiratory frequency, tidal volume, and the product of these two parameters, minute ventilation) were increased in both HF and HSu animals, with a more pronounced effect in HF animals (Fig. 1A and D). Surgical CSN cut completely abolished the increase in spontaneous ventilation induced by the diets (Fig. 1C and D), showing that this effect is mediated by the CB. In addition, ventilatory responses to ischemic hypoxia, assessed as the increase in ventilation produced by common carotid artery occlusions for periods of 5, 10, and 15 s, were augmented in HF animals (Fig. 1B and E). This increase in ventilation, which was proportional to the duration of the stimulus, was mediated through the CB, as it was abolished by CSN cut (Fig. 1C).
FIG. 1.

CB activity is increased in rat models of IR and HT. A and B: Typical recordings of respiratory rate (Resp. Rate) (bpm), tidal volume (mL), and blood pressure in basal conditions and in response to ischemic hypoxia, induced by occlusions of common carotid artery (CCO) in a control rat and in a rat submitted to an HF diet. C: Typical recording of ventilatory parameters after CSN cut in an HF rat. D: Mean minute ventilation (VE) (product of respiratory frequency and tidal volume) in control, HA, and HSu rats. E: Effect of common carotid occlusion of 5, 10, and 15 s on minute ventilation in control, HF, and HSu rats. F: Effect of hypercaloric diets on CB catecholamines (dopamine [DA] plus DOPAC) basal release (20% O2 plus 5% O2 balanced N2) (n = 5). G: Effect of hypercaloric diets on the release of catecholamines from CB evoked by hypoxia (5% O2 plus 5% CO2 balanced N2) (n = 5). H: Effect of HF and HSu diets in CB weight. Control n = 19, HF n = 27, HSu n = 24. I: Effect of HF and HSu diets on the inmmunoreactivity for tyrosine hydroxylase (TH) (60 KDa) expressed as the ratio tyrosine hydroxylase to β-actin (43 KDa) expression. Left panel: Representative immunoreactivity for tyrosine hydroxylase and β-actin in the CB in % of control, HF, and HSu animals. Bars (D–I) represent means ± SEM. One- and two-way ANOVA with Dunnett and Bonferroni multicomparison tests, respectively; *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05 vs. values within the same group.
In HSu animals, only the response to an ischemic hypoxia of 5 s was significantly increased, and as observed in the HF model this was also abolished by CSN cut. We concluded that both the HF and the HSu rat models of IR and HT present an overstimulated CB; however, the more pronounced increases in spontaneous ventilation and in ischemic hypoxia–induced hyperventilation observed in HF animals suggest that these animals hold a higher degree of CB activation. Catecholamines, namely dopamine, are the best characterized neurotransmitters in the CB (6), and its release in all mammalian species depends on extracellular Ca2+ and is proportional to stimulus intensity and to the increase in CSN activity and therefore to CB function (31,32). Thus, to confirm CB overactivation in HF and HSu animals, we measured both basal and hypoxia-evoked release of dopamine (plus DOPAC, the main metabolite of dopamine in the CB). We observed that basal release of dopamine was not significantly modified by hypercaloric diets (Fig. 1F); however, the release induced by hypoxia (5% O2) was increased 3.15-fold in HF and 2.12-fold in HSu rat models (Fig. 1G). Also, CB weight was significantly increased by 36.71 and 27.13% in HF and HSu models, respectively (Fig. 1H), which suggests that overactivation of CB is due to hyperplasia of the organ. In fact, Western blot analysis confirmed that the tyrosine hydroxylase expression, the rating enzyme for catecholamine biosynthesis, increased by 64.4% in HF (P < 0.01) and 30.8% in HSu animals (P = 0.12) (Fig. 1I), confirming CB overactivity in these pathological animal models.
Chronic CSN resection prevents IR and HT.
To test the involvement of the CB in the development of IR and HT, we performed a chronic CSN bilateral resection prior to hypercaloric diet administration, therefore blocking CB activity during the induction of IR. Rats submitted to CSN bilateral resection were compared with animals submitted to the same surgical procedure but in which CSN was left intact (sham). CSN bilateral resection was confirmed by the lack of increase in the ventilatory responses to ischemic hypoxia, assessed as common carotid artery occlusion (Fig. 2A). Sham procedure did not modify any of the parameters evaluated (insulin sensitivity, MAP, glycemia, insulinemia, free fatty acids, corticosterone, visceral fat, and plasma catecholamines) compared with control, HF, and HSu animals not submitted to any surgical procedure (16). Also, CSN bilateral resection did not alter liquid and food intake in any of the groups tested (data not shown).
FIG. 2.

CSN bilateral resection prevents IR and HT in HF and HSu animal models. A: Typical recording of respiratory rate (bpm) and tidal volume (mL) in response to ischemic hypoxia, induced by occlusion of common carotid artery in a rat submitted to CSN bilateral resection. The absence of increment in the ventilatory responses confirms CSN resection. B: Representative glucose excursion curve for insulin tolerance test in a control rat. Details on KITT calculation are described in research design and methods. A and C: Effect of CSN resection on insulin sensitivity determined by the insulin tolerance test, expressed as KITT in control, HF, and HSu rats. D: Effect of CSN resection on MAP in control, HF, and HSu rats. E: Absolute weight before and after hypercaloric diet administration and chronic sinus nerve resection. F: Increment in body weight, calculated as total weight variation during the experimental period, in control, HF, and HSu rats with and without CSN resection. G: Visceral fat, weighed postmortem and corrected to body weight in control, HF, and HSu rats with and without CSN resection. Bars represent means ± SEM. One- and two-way ANOVA with Dunnett and Bonferroni multicomparison tests, respectively; **P < 0.01, ***P < 0.001 vs. control; #P < 0.05; ##P < 0.01, ###P < 0.001 comparing values with and without CSN resection.
Figure 2B depicts a representative curve of a typical insulin tolerance test in a control rat. Insulin sensitivity was significantly decreased by 42.08 and 53.61% in HF and HSu rats, respectively (Fig. 2C). IR produced by hypercaloric diets was completely prevented by CSN resection (Fig. 2C), linking CB dysfunction with the development of IR. In addition, we observed that CSN resection in control animals decreased insulin sensitivity, suggesting that CB also contributes to maintain metabolic control in physiological conditions. MAP, as previously described (16), was increased by 38.79 and 35.70% in HF and HSu rats, respectively, and this effect was totally prevented by CSN chronic resection (Fig. 2D). Glucose homeostasis and insulin secretion became normalized, since fasting hyperglycemia and hyperinsulinemia returned to control values after CSN chronic denervation (Table 1). The increase in serum free fatty acids observed in HSu rats was abolished by CSN resection (Table 1). Neither HF and HSu diets nor CSN resection modified corticosterone levels (Table 1). Because of the strong association between obesity and visceral fat with IR and HT (1,11,12), we tested whether CSN resection could alter weight gain and visceral fat. In Fig. 2E, absolute weights before and after administration of hypercaloric diets and also before and after CSN resection are depicted. HF, but not control or HF, animals significantly gained weight during the experimental period (Fig. 2E and F). We found that CSN resection significantly decreases weight gain in HF animals (Fig. 2F) and avoids visceral fat deposition (Fig. 2G). Since IR, HT, and obesity are associated with sympathetic nervous system overactivity (1,11,12) and CB controls sympathetic outflow and sympathetic nerve activity (7,8), we also analyzed sympathoadrenal activity, measured both as circulating and adrenal medulla catecholamines in our animal models. Plasma norepinephrine significantly increased in both HF and HSu rats in relation to control animals (HF 48.40 ± 7.72 pmol/mL, HSu 71.32 ± 9.04 pmol/mL, and control 22.23 ± 2.98 pmol/mL) (Fig. 3A). Also, as depicted in Fig. 3B, plasma epinephrine increased by 151.52 and 178.31% in HF and HSu, respectively (control 30.80 ± 4.25 pmol/mL). These results suggest an increased sympathoadrenal activity (Fig. 3A and B) that was confirmed by the augmented catecholamine content in adrenal medulla of these animals (Fig. 3C and D). HF and HSu rats exhibited significant increases of 29.72 and 44.52% in adrenal medulla norepinephrine, respectively, and of 34.27 and 69.50% adrenal medulla epinephrine content compared with the controls (norepinephrine control 11.75 ± 0.58 nmol/mg tissue and epinephrine control 24.28 ± 2.62 nmol/mg tissue [Fig. 3C and D]). Chronic CSN cut did not affect sympathoadrenal activity in control animals; however, sympathoadrenal overactivation induced by hypercaloric diets was abolished in rats with CSN bilateral resection (Fig. 3A–D). These results demonstrate that CB plays a role in the genesis of IR and HT in animal models of type 2 diabetes and metabolic syndrome.
TABLE 1.
Effect of CSN chronic resection on fasting plasma glucose, plasma insulin, serum free fatty acids, and corticosterone levels in control, HF, and HSu rats
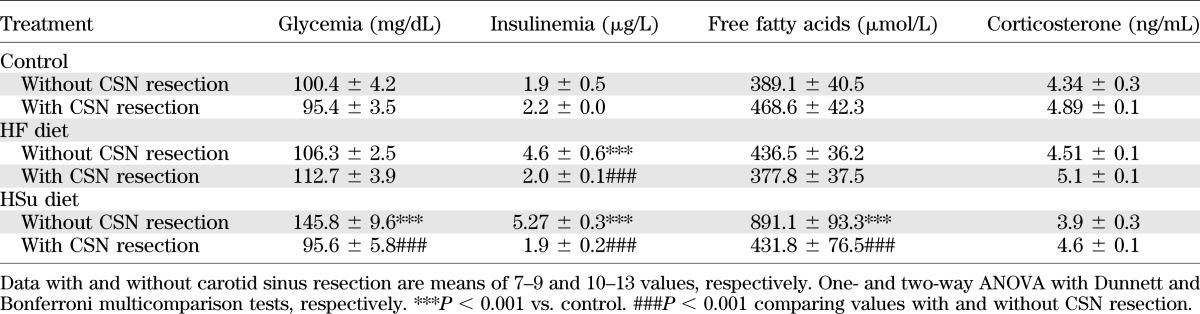
FIG. 3.

CSN bilateral resection prevents sympathoadrenal overactivation in HF and HSu animal models. A and B: Effect of CSN resection on circulating catecholamines, norepinephrine and epinephrine, respectively. C and D: Effect of CSN resection on adrenal medulla norepinephrine and epinephrine content, respectively. Bars represent means ± SEM. Two-way ANOVA with Bonferroni multicomparison tests, respectively; *P < 0.05, **P < 0.01, ***P < 0.001 vs. control; #P < 0.05; ##P < 0.01, ###P < 0.001 comparing values with and without CSN resection.
Insulin triggers CB activation.
In the present work, we propose that the stimulus for CB overactivation responsible for IR and HT is increased plasma insulin, and therefore we hypothesize that insulin is capable of triggering CB activation. We used a three-step Western blot approach (27) to examine the presence of the insulin receptor in the CB and its phosphorylation in response to insulin. Western blot analysis demonstrated that insulin receptors are present in the CB (Fig. 4A) and that their phosphorylation increases in the presence of 1 and 100 nmol/L insulin (Fig. 4A and B). Incubation of the CBs with 1 and 100 nmol/L insulin significantly increased insulin receptor phosphorylation by 98.6 and 47%, respectively (Fig. 4B). We also tested whether insulin receptor activation in the CB elicits a neurosecretory response by measuring intracellular Ca2+ and the release of catecholamines and ATP, two of the neurotransmitters released from CBs in response to hypoxia (6,14,25,31,32). Figure 5A depicts a bright-field image of a 20-h-old cell culture of dissociated CB and a typical recording of intracellular cell Ca2+, measured as the ratio of the fluorescent emission at 340/380 nm of chemoreceptor cells in basal conditions in response to hypoxia (N2), to 1 nmol/L insulin, and to 35 mmol/L K+ in the left and right panels, respectively. Hypoxia significantly increased [Ca2+]i by 15.97%. Also, 1 nmol/L insulin significantly increased [Ca2+]i by 6.53%. When applied simultaneously, insulin and hypoxia increased intracellular Ca2+ concentration by 21.53%, suggesting that the transduction mechanisms by which the two stimuli operate are different. To evoke a neurosecretory response, the increase in [Ca2+]i produced by insulin must be transduced into the release of neurotransmitters from the CB. Figure 5C and E shows that insulin (10 nmol/L) produced an increase in the basal release (black bars) of ATP and dopamine (plus DOPAC) from the whole CB in incubating solutions, and the effect was reversed after drug washout. The dose-response curves for the effect of insulin in neurotransmitter release in the whole CB are depicted in Fig. 5D and F. The curves fitted a sigmoid with EC50 of 0.552 and 6.17 nmol/L and maximal effects of 257.9 and 265.1% for CB ATP and dopamine release, respectively. Note that concentrations >400–500 pmol/L are already compatible with an hyperinsulinemic state (33,34) and that when insulin was applied at >10 nmol/L concentrations, it evoked the release of ATP and dopamine (plus DOPAC) from CB in a magnitude similar to that produced by hypoxia (5% O2) (Fig. 5D and F).
FIG. 4.

Insulin receptors are present in the CB, and their phosphorylation (Phosp) increases in response to insulin. A: Representative Western blot showing insulin receptor immunoreactivity in the CB and insulin receptor phosphorylation immunoreactivity in control CBs (CTR) and in response to 1 and 100 nmol/L insulin (30 min incubation), respectively, corresponding to the 97 KDa band. A reprobing of the membranes with an anti–β-actin antibody, corresponding to the 42 KDa band, is shown below the gels. B: Average insulin receptor phosphorylation in control and in CBs incubated with 1 and 100 nmol/L insulin in relation to β-actin immunoreactivity (n = 3–4). **P < 0.01, *P < 0.05. One-way ANOVA with Dunnett multicomparison test comparing the groups with the control. Data are means ± SEM.
FIG. 5.

Insulin increases the neurosecretory responses in the CBs. A: Microscope field of dissociated rat CB cell culture and the typical recording of intracellular cell Ca2+, measured as the ratio of the fluorescent emission at 340/380 nm of chemoreceptor cells in basal conditions, in response to hypoxia (N2), to 1 nmol/L insulin, and to 35 mmol/L K+. B: Effect of insulin on intracellular cell Ca2+, measured as means of the ΔRI in 179 chemoreceptor cells. In every cell, the fluorescence signal was integrated as a function of time (running integral [RI]). C and D: Time course for the release of ATP from CB in response to insulin (10 nmol/L) and dose-response curve for insulin action on ATP release and its comparison with the effect of hypoxia (5% O2 plus 5% CO2 balanced N2). Release protocol consisted of two incubations of CBs in normoxic solutions (20% O2 plus 5%CO2 balanced N2, 10 min), followed by insulin application during 30 min in normoxia and two final normoxic incubations. E and F: Group of experiments identical to C and D but measuring catecholamine (dopamine plus DOPAC) release from CB instead of ATP. ATP and catecholamine quantification in the CB are means of 4–6 data. Bars represent means ± SEM. One- and two-way ANOVA with Dunnett and Bonferroni multicomparison tests, respectively; *P < 0.05, **P < 0.01, *** P < 0.001 vs. control. Controls in the release experiments correspond to the period prior to insulin application. ins, insulin.
Knowing that stimuli-induced CB activation results in hyperventilation (6), we assessed the effects of insulin on ventilation. In vivo experiments have previously showed that intravenous infusion of insulin caused a CB-dependent increase in ventilation (13), an effect that was not due to hypoglycemia per se, since low glucose is not a direct stimulus for rat CB chemoreceptors (14,15). Therefore, we tested the effect of an intracarotid bolus of insulin on ventilation during a euglycemic clamp to avoid the confounding effects of systemic hypoglycemia. Figure 6A depicts a typical recording of pulmonary flow and tidal volume before and after an intracarotid administration of an insulin (50 mU/kg) bolus. Insulin increased respiratory rate, tidal volume (Fig. 6A and D), and the product of both parameters, minute ventilation (Fig. 6C) in a dose-dependent manner. The increase in ventilation induced by insulin is not immediate, showing a significant latency period (time to the onset of the response) comprised within the 106.0 ± 4.04 and 188.5 ± 3.51 s range (Fig. 6D). This observation is in accordance with the time scale necessary for the activation of tyrosine kinase receptors, namely, insulin receptors (35). Full dose-response curve for the effect of insulin in minute ventilation is depicted in Fig. 6C, fitting a sigmoid with an EC50 of 35 mU/kg and a maximal effect of 60.41%. Figure 6E depicts a typical euglycemic clamp after an intracarotid administration of an insulin bolus of 50 mU/kg. As expected, the amount of glucose infused to maintain euglycemia increased in an insulin dose–dependent manner (Fig. 6F). The effect of insulin on ventilation was totally mediated by the CB, since CSN cut completely abolished the increase in ventilation induced by insulin (Fig. 6D).
FIG. 6.

Insulin increases ventilation through a CB-mediated effect. A: Respiratory frequency and tidal volume (TV) recordings before and after administration of an intracarotid insulin (100 mg/kg) bolus. B: Mean basal ventilatory parameters, respiratory frequency (RR), tidal volume, and minute ventilation (VE) before insulin administration. C: Dose-response curve for the effect of insulin (1, 5, 10, 50, 100, and 200 mU/kg) on minute ventilation. For avoidance of the effect of hypoglycemia, the study of insulin effect on ventilation was performed in euglycemic conditions. Insulin effects on ventilation are means of 5–7 data. D: Typical respiratory frequency and tidal volume recordings due to the administration of an intracarotid insulin (100 mg/kg) bolus before and after CSN cut. E: Graph depicting a typical glucose perfusion curve to maintain euglycemia after insulin bolus and the levels of glycemia throughout the experiment. F: Total glucose concentrations perfused to maintain euglycemic clamp in response to the insulin concentrations (1, 5, 10, 50, 100, and 200 mU/kg) tested. Values represent means ± SEM. One-way ANOVA with Dunnett multicomparison test; *P < 0.05, **P < 0.01 vs. basal values. AUC, area under the curve; PulmFl, pulmonary flow.
DISCUSSION
This study represents a new conceptual framework regarding the pathogenesis of IR. Using a combination of neurochemical, physiological, and cellular biology techniques, we show that CB activity is increased in models of metabolic syndrome and type 2 diabetes and that CB dysfunction is involved in the development of IR and HT. In addition, we demonstrate for the first time that insulin triggers the peripheral chemoreceptors located in the CBs, suggesting that hyperinsulinemia may trigger CB-induced sympathoadrenal overactivity associated with metabolic disturbances.
Hyperinsulinemia is a known early pathological feature caused by increased secretory stress on the β-cell associated with peripheral IR caused by hypercaloric diets. Increased insulin levels trigger the CBs to activate the sympathetic nervous system, initiating a vicious cycle that worsens peripheral insulin action, impairs β-cell function, and causes systemic HT. In line with these results, the CB rises as a new therapeutic target for intervention in metabolic disturbances.
We show herein, and also for the first time, that CB activity is increased in diet-induced animal models of IR and HT. CB-mediated basal ventilation and ventilation in response to ischemic hypoxia were increased in the pathological models tested, as well as the CB chemoreceptor cell function—assessed both as hypoxia-induced release of dopamine and as tyrosine hydroxilase expression. The increase in CB cell function, together with increased CB weight observed in our experimental setting, is in agreement with the previous observations of Clarke et al. (36) showing that CB volume is increased in spontaneous insulin-dependent diabetic rats (strain BB/s)—an effect that could not be attributed to an increase in the vascular component of the organ. We have also observed that HF animals exhibited more pronounced increases in both spontaneous ventilation and ischemic hypoxia–induced hyperventilation than HSu animals, suggesting that the HF animal model is characterized by a higher degree of CB activation. Our results strongly suggest that there is an obesity-related factor that contributes to CB stimulation.
Although some authors have suggested that obesity does not enhance peripheral chemoreflex sensitivity (37), this topic remains controversial. It has been shown that chronic intermittent hypoxia increases expression of tumor necrosis factor-α and interleukin (IL)-1β within the CB (38) and that these proinflammatory cytokines may contribute directly to CB-mediated cardiorespiratory changes evoked by intermittent hypoxia. Obesity is also characterized by a subclinical proinflammatory condition with increased secretion of adipokines, including leptin, tumor necrosis factor-α, IL-1β, and IL-6 (39): the same cytokines proposed as having a role in chemoreceptor changes observed in sleep apnoea. On the other hand, obesity has been associated with increased sympathetic nervous system activity through a leptin-mediated mechanism that is still unclear (30). Recently, it was described that glomus cells in the CB express leptin receptors and that they are activated by intermittent hypoxia and by systemic leptin injections (40), which suggests that leptin may also be representative of an independent factor in CB activation.
Besides demonstrating that CB overactivity is present in animal models of IR and HT, we have also shown that CSN bilateral resection totally prevented diet-induced IR and HT, as well as increased fasting plasma glucose, fasting plasma insulin, free fatty acids, and systemic sympathoadrenal overactivity. In accordance with our results, it was previously observed by other authors that CB stimulation by corconium, a nicotinomimetic agent, causes a rise in circulating insulin that is reversed by CSN resection (41). We also found that CSN resection decreased insulin sensitivity in control animals, which suggests a role for CB in metabolic control, not only in pathological, but also in physiological conditions. This kind of mechanism is not novel in CB physiology, since it was recently proposed that the CB is involved in the counterregulatory response to hypoglycemia and in baroreflex control of blood pressure in humans (42).
Regarding the contribution of the CB to the development and maintenance of HT, our work agrees with previous results obtained by other groups in which it was observed that carotid sinus denervation prevented arterial pressure increase and decreased sympathetic activity in spontaneous hypertensive young rats (43). It is known that, apart from chemoreceptor activity, CSN carries information related with baroreceptor activity. However, we would like to emphasize that the results obtained herein, both in the common carotid occlusion experiments and in the carotid sinus denervation experiments, reflect a CB chemoreceptor-mediated effect. If there were a significant baroreceptor-mediated effect, the animals would have become hypotensive in response to acute ischemic hypoxia and hypertensive after CSN denervation (rev. in 44), which was not observed.
Our results show, for the first time, that insulin triggers CB activation and that high insulin doses are an effective stimulus for CB overactivation. It is generally accepted that insulin stimulates the sympathetic nervous system, with fasting hyperinsulinemia being one of the components of the sympathetic overactivation present in diabetes and the metabolic syndrome (45,46). However, insulin-induced sympathetic activity has been attributed to a central nervous system effect, since the infusion of insulin into the third cerebral ventricle increased sympathetic outflow without significantly increasing adrenal and renal sympathetic activity (47,48). Without contradicting these results, we show that insulin can also act on the CBs to increase sympathoadrenal outflow. We demonstrated that insulin receptors are present in the CB and that its phosphorylation increases in response to insulin. As depicted in Fig. 3B, 1 nmol/L produced a higher degree of insulin receptor phosphorylation than 100 nmol/L. We expected to find a concentration-dependent relationship in CB insulin receptor phosphorylation, which we did not observe at high insulin concentrations. At high insulin levels, insulin receptors are possibly saturated, inducing a functional desensitization either by decreasing tyrosine kinase activity or by promoting insulin receptor endocytosis and degradation as it happens in human HepG2 cell line (49) and in rat Fao cells (50). Also, we showed that insulin was capable of initiating a neurosecretory response measured as the increase in intracellular Ca2+ and the release of the neurotransmitters ATP and dopamine that is transduced into an increase in ventilation. The increase of ventilation induced by insulin is not novel (14); however, in Bin-Jaliah, Maskell, and Kumar’s work (13) insulin was administered intravenously, aiming to study the effects of insulin-induced hypoglycemia in ventilation. Herein, we administered insulin intracarotidally to guarantee that the first site of insulin action is the CB; also, we performed the experiments in euglycemic conditions to avoid the confounding effects of systemic hypoglycemia. These results together with the finding that the effect of insulin on ventilation disappears after CSN cut suggest that insulin action on ventilation is mediated by the CB.
In conclusion, we propose that insulin-triggered CB activation is responsible for increased sympathoadrenal activity and outflow, creating a vicious cycle that culminates in severe IR and arterial HT, the core features of the metabolic syndrome and type 2 diabetes.
ACKNOWLEDGMENTS
This study was supported by Portuguese Foundation for Science and Technology Grant PTDC/SAU-ORG/111417/2009, by 2009 L’Oreal/FCT/Unesco Medals of Honour for Women in Science, and by BFU2007-61848 (DGICYT), CIBER CB06/06/0050 (FISS-ICiii).
No potential conflicts of interest relevant to this article were reported.
M.J.R. performed the majority of the experiments and wrote research design and methods. J.F.S. helped in the experiments related to CB denervation and IR and in experiments related to CB overactivation in pathological rat models. Catecholamine quantification was performed in the laboratory of C.G., and C.G. also reviewed the manuscript. M.P.G. performed some of the experiments related to ATP quantification and insulin effects on ventilation and helped with the manuscript preparation. E.C.M. helped with the discussion. S.V.C. planned all the experiments, performed some of them, supervised the project, and wrote the manuscript. S.V.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors are grateful to Elena Gonzalez Muñoz from the laboratory of C.G. for catecholamine quantification.
Footnotes
See accompanying commentary, p. 2654.
REFERENCES
- 1.Katagiri H, Yamada T, Oka Y. Adiposity and cardiovascular disorders: disturbance of the regulatory system consisting of humoral and neuronal signals. Circ Res 2007;101:27–39 [DOI] [PubMed] [Google Scholar]
- 2.West SD, Nicoll DJ, Stradling JR. Prevalence of obstructive sleep apnoea in men with type 2 diabetes. Thorax 2006;61:945–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes 1989;38:1165–1174 [DOI] [PubMed] [Google Scholar]
- 4.Peppard PE, Young T, Palta M, Skatrud J. Prospective study of the association between sleep-disordered breathing and hypertension. N Engl J Med 2000;342:1378–1384 [DOI] [PubMed] [Google Scholar]
- 5.Iturriaga R, Rey S, Del Río R. Cardiovascular and ventilatory acclimatization induced by chronic intermittent hypoxia: a role for the carotid body in the pathophysiology of sleep apnea. Biol Res 2005;38:335–340 [DOI] [PubMed] [Google Scholar]
- 6.Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiol Rev 1994;74:829–898 [DOI] [PubMed] [Google Scholar]
- 7.Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev 1994;74:543–594 [DOI] [PubMed] [Google Scholar]
- 8.Cao WH, Morrison SF. Differential chemoreceptor reflex responses of adrenal preganglionic neurons. Am J Physiol Regul Integr Comp Physiol 2001;281:R1825–R1832 [DOI] [PubMed] [Google Scholar]
- 9.Esler M, Straznicky N, Eikelis N, Masuo K, Lambert G, Lambert E. Mechanisms of sympathetic activation in obesity-related hypertension. Hypertension 2006;48:787–796 [DOI] [PubMed] [Google Scholar]
- 10.Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Invest 2000;106:473–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pardal R, López-Barneo J. Low glucose-sensing cells in the carotid body. Nat Neurosci 2002;5:197–198 [DOI] [PubMed] [Google Scholar]
- 12.Koyama Y, Coker RH, Stone EE, et al. Evidence that carotid bodies play an important role in glucoregulation in vivo. Diabetes 2000;49:1434–1442 [DOI] [PubMed] [Google Scholar]
- 13.Bin-Jaliah I, Maskell PD, Kumar P. Indirect sensing of insulin-induced hypoglycaemia by the carotid body in the rat. J Physiol 2004;556:255–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Conde SV, Obeso A, Gonzalez C. Low glucose effects on rat carotid body chemoreceptor cells’ secretory responses and action potential frequency in the carotid sinus nerve. J Physiol 2007;585:721–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gallego-Martin T, Fernandez-Martinez S, Rigual R, Obeso A, Gonzalez C. Effects of low glucose on carotid body chemoreceptor cell activity studied in cultures of intact organs and in dissociated cells. Am J Physiol Cell Physiol 2012;302:C1128–C1140 [DOI] [PubMed] [Google Scholar]
- 16.Conde SV, Nunes da Silva T, Gonzalez C, Mota Carmo M, Monteiro EC, Guarino MP. Chronic caffeine intake decreases circulating catecholamines and prevents diet-induced insulin resistance and hypertension in rats. Br J Nutr 2012;107:86–95 [DOI] [PubMed] [Google Scholar]
- 17.Shearer J, Severson DL, Su L, Belardinelli L, Dhalla AK. Partial A1 adenosine receptor agonist regulates cardiac substrate utilization in insulin-resistant rats in vivo. J Pharmacol Exp Ther 2009;328:306–311 [DOI] [PubMed] [Google Scholar]
- 18.Ribeiro RT, Lautt WW, Legare DJ, Macedo MP. Insulin resistance induced by sucrose feeding in rats is due to an impairment of the hepatic parasympathetic nerves. Diabetologia 2005;48:976–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panchal SK, Brown L. Rodent models for metabolic syndrome research. J Biomed Biotechnol 2011;2011:351982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guarino MP, Santos AI, Mota-Carmo M, Costa PF. Effects of anaesthesia on insulin sensitivity and metabolic parameters in Wistar rats. In Vivo 2013;27:127–132 [PubMed] [Google Scholar]
- 21.Davidson MB. Studies on the mechanism of pentobarbital-induced glucose intolerance. Horm Metab Res 1971;3:243–247 [DOI] [PubMed] [Google Scholar]
- 22.Monteiro EC, Ribeiro JA. Inhibition by 1,3-dipropyl-8(p-sulfophenyl)xanthine of the respiratory stimulation induced by common carotid occlusion in rats. Life Sci 1989;45:939–945 [DOI] [PubMed] [Google Scholar]
- 23.Marek W, Muckenhoff K, Prabhakar NR. Significance of pulmonary vagal afferents for respiratory muscle activity in the cat. J Physiol Pharmacol 2008;59(Suppl. 6):407–420 [PubMed] [Google Scholar]
- 24.Monzillo LU, Hamdy O. Evaluation of insulin sensitivity in clinical practice and in research settings. Nutr Rev 2003;61:397–412 [DOI] [PubMed] [Google Scholar]
- 25.Conde SV, Monteiro EC, Rigual R, Obeso A, Gonzalez C. Hypoxic intensity: a determinant for the contribution of ATP and adenosine to the genesis of carotid body chemosensory activity. J Appl Physiol 2012;112:2002–2010 [DOI] [PubMed] [Google Scholar]
- 26.Caceres AI, Obeso A, Gonzalez C, Rocher A. Molecular identification and functional role of voltage-gated sodium channels in rat carotid body chemoreceptor cells. Regulation of expression by chronic hypoxia in vivo. J Neurochem 2007;102:231–245 [DOI] [PubMed] [Google Scholar]
- 27.Johnson RP, El-Yazbi AF, Takeya K, Walsh EJ, Walsh MP, Cole WC. Ca2+ sensitization via phosphorylation of myosin phosphatase targeting subunit at threonine-855 by Rho kinase contributes to the arterial myogenic response. J Physiol 2009;587:2537–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pérez-García MT, Obeso A, López-López JR, Herreros B, González C. Characterization of cultured chemoreceptor cells dissociated from adult rabbit carotid body. Am J Physiol 1992;263:C1152–C1159 [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Niño A, Obeso A, Baranda JA, Santo-Domingo J, Lopez-Lopez JR, Gonzalez C. MaxiK potassium channels in the function of chemoreceptor cells of the rat carotid body. Am J Physiol Cell Physiol 2009;297:C715–C722 [DOI] [PubMed] [Google Scholar]
- 30.Landsberg L, Young JB. Fasting, feeding and regulation of the sympathetic nervous system. N Engl J Med 1978;298:1295–1301 [DOI] [PubMed] [Google Scholar]
- 31.Obeso A, Almaraz L, Gonzalez C. Correlation between adenosine triphosphate levels, dopamine release and electrical activity in the carotid body: support for the metabolic hypothesis of chemoreception. Brain Res 1985;348:64–68 [DOI] [PubMed] [Google Scholar]
- 32.Vicario I, Rigual R, Obeso A, Gonzalez C. Characterization of the synthesis and release of catecholamine in the rat carotid body in vitro. Am J Physiol Cell Physiol 2000;278:C490–C499 [DOI] [PubMed] [Google Scholar]
- 33.Kronmal RA, Barzilay JI, Tracy RP, Savage PJ, Orchard TJ, Burke GL. The relationship of fasting serum radioimmune insulin levels to incident coronary heart disease in an insulin-treated diabetic cohort. J Clin Endocrinol Metab 2004;89:2852–2858 [DOI] [PubMed] [Google Scholar]
- 34.Stegenga ME, van der Crabben SN, Levi M, et al. Hyperglycemia stimulates coagulation, whereas hyperinsulinemia impairs fibrinolysis in healthy humans. Diabetes 2006;55:1807–1812 [DOI] [PubMed] [Google Scholar]
- 35.Czech MP. The nature and regulation of the insulin receptor: structure and function. Annu Rev Physiol 1985;47:357–381 [DOI] [PubMed] [Google Scholar]
- 36.Clarke JA, Daly Mde B, Ead HW, Hennessy EM. The carotid body of the spontaneous insulin-dependent diabetic rat. Braz J Med Biol Res 1999;32:85–91 [DOI] [PubMed] [Google Scholar]
- 37.Narkiewicz K, van de Borne PJ, Pesek CA, Dyken ME, Montano N, Somers VK. Selective potentiation of peripheral chemoreflex sensitivity in obstructive sleep apnea. Circulation 1999;99:1183–1189 [DOI] [PubMed] [Google Scholar]
- 38.Del Rio R, Moya EA, Iturriaga R. Contribution of inflammation on carotid body chemosensory potentiation induced by intermittent hypoxia. Adv Exp Med Biol 2012;758:199–205 [DOI] [PubMed] [Google Scholar]
- 39.Trayhurn P, Wood IS. Signalling role of adipose tissue: adipokines and inflammation in obesity. Biochem Soc Trans 2005;33:1078–1081 [DOI] [PubMed] [Google Scholar]
- 40.Messenger SA, Ciriello J. Effects of intermittent hypoxia on leptin signalling in the carotid body. Neuroscience. 29 November 2012 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 41.Anichkov SV, Tomilina TN. Reflexes from carotid chemoreceptors upon insulin level in blood. Arch Int Pharmacodyn Ther 1962;139:53–59 [PubMed] [Google Scholar]
- 42.Wehrwein EA, Basu R, Basu A, Curry TB, Rizza RA, Joyner MJ. Hyperoxia blunts counterregulation during hypoglycaemia in humans: possible role for the carotid bodies? J Physiol 2010;588:4593–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abdala AP, McBryde FD, Marina N, et al. Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. J Physiol 2012;590:4269–4277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheffers IJ, Kroon AA, de Leeuw PW. Carotid baroreflex activation: past, present, and future. Curr Hypertens Rep 2010;12:61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes 1988;37:1595–1607 [DOI] [PubMed] [Google Scholar]
- 46.Landsberg L. Insulin resistance and the metabolic syndrome. Diabetologia 2005;48:1244–1246 [DOI] [PubMed] [Google Scholar]
- 47.Muntzel MS, Anderson EA, Johnson AK, Mark AL. Mechanisms of insulin action on sympathetic nerve activity. Clin Exp Hypertens 1995;17:39–50 [DOI] [PubMed] [Google Scholar]
- 48.Muntzel MS, Morgan DA, Mark AL, Johnson AK. Intracerebroventricular insulin produces nonuniform regional increases in sympathetic nerve activity. Am J Physiol 1994;267:R1350–R1355 [DOI] [PubMed] [Google Scholar]
- 49.Blake AD, Hayes NS, Slater EE, Strader CD. Insulin receptor desensitization correlates with attenuation of tyrosine kinase activity, but not of receptor endocytosis. Biochem J 1987;245:357–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crettaz M, Jialal I, Kasuga M, Kahn CR. Insulin receptor regulation and desensitization in rat hepatoma cells. The loss of the oligomeric forms of the receptor correlates with the change in receptor affinity. J Biol Chem 1984;259:11543–11549 [PubMed] [Google Scholar]