

Abstract
We recently designed a group of novel exosite 2-directed, sulfated, small, allosteric inhibitors of thrombin. To develop more potent inhibitors, monosulfated benzofuran tri- and tetrameric homologs of the parent designed dimers were synthesized in 7–8 steps and found to exhibit a wide range of potencies. Among these, trimer 9a was found to be nearly 10-fold more potent than the first generation molecules. Michaelis-Menten studies indicated an allosteric mechanism of inhibition. Competitive studies using a hirudin peptide (exosite 1 ligand) and, unfractionated heparin, heparin octasaccharide and γ′-fibrinogen peptide (exosite 2 ligands), demonstrated exosite 2 recognition in a manner different from the parent dimers. Alanine scanning mutagenesis of 12 Arg/Lys residues of exosite 2 revealed a defect in 9a potency for Arg233Ala thrombin only confirming the major difference in site of recognition between the two structurally related sulfated benzofurans. The results suggest that multiple avenues are available within exosite 2 for inducing thrombin inhibition.
Introduction
The coagulation cascade is set of sequential, and yet highly inter-woven, proteolytic reactions that operate efficiently to prevent excessive loss of blood and ingestion of microbes. It can also be triggered by an aberrant intravascular signal, which may lead to an internal clot that can be catastrophic. Several anticoagulants have been approved for treatment of such conditions including unfractionated heparin (UFH), low molecular weight heparins (LMWHs), warfarin, hirudin and its analogs, argatroban, dabigatran, rivaroxaban and apixaban. These agents either directly or indirectly target thrombin and/or factor Xa, two key enzymes of the coagulation cascade.1–3
Thrombin is a trypsin-like serine protease that is formed rapidly upon initiation of coagulation and has been the primary target for development of novel anticoagulant therapy.2,4,5 Yet, plasma thrombin is an unusual protease that displays both pro- as well as anti-coagulant roles. Whereas it cleaves fibrinogen to stem the flow of blood, its specificity changes to the cleavage of protein C in the presence of thrombomodulin to induce blood patency.6–8 The characteristic thermodynamic feature of thrombin that achieves this manuever is its conformational plasticity. Thrombin exhibits an ensemble of conformations that can rapidly interconvert, especially in the presence of appropriate ligands. In fact, nature appears to have engineered thrombin as a pivot to rapidly alter the flux down either the pro- or anti- coagulant pathways. Thus, regulating thrombin is critical and challenging.
A specific approach exploited by nature to regulate thrombin is allosteric modulation of its active site. Three distinct allosteric sites are known on thrombin including the sodium binding site and anion-binding exosites 1 and 2.8–10 Each of these are located approximately 10–30 Å away from the active site. Sodium binding converts thrombin from the ‘slow’ form to the ‘fast’ form, which has been inferred as a switch for transforming the anticoagulant flux to the procoagulant one,11 although the physiologic significance of this has been questioned recently.12 Exosites 1 and 2 are electropositive domains that engage several physiologic ligands including glycosaminoglycans (GAGs), thrombomodulin, fibrinogen, glycoprotein Ibα and protease activated receptor–1.6–9 Both exosites 1 and 2 are energetically linked to the active site as demonstrated by altered rates of cleavage of substrates in the presence of different ligands. For example, exosite 1 ligand hirugen significantly increases the catalytic efficiency of thrombin for small chromogenic substrates,13,14 while exosite 2 ligand fragment 1.2 induces better recognition of thrombin’s active site by a small fluorophore.12 In fact, the conformational plasticity of thrombin appears to present a series of states along the monotonous path between the zymogen-like and proteinase-like forms that can be stabilized by an allosteric ligand.12 Thus, an appropriately designed ligand can select and stabilize a distinct thrombin state with its unique characteristics of substrate specificity and catalytic activity.
Nearly all allosteric regulators of thrombin discovered to date are polymeric molecules, i.e., proteins and sulfated polysaccharides. Some time ago, we reasoned that such interactions, especially of sulfated polysaccharides called GAGs, could serve as reasonable starting points for the design of medicinally relevant small molecules. Thus, sulfated low molecular weight lignins (LMWLs) were designed as oligomeric mimetics of sulfated GAGs and found to inhibit thrombin with nanomolar potency by utilizing exosite 2 (Figure 1).15,16 To transform the heterogeneous, sulfated LMWLs into homogeneous small molecules, we developed sulfated benzofuran monomers, which were found to retain exosite 2-mediated thrombin inhibition potential of the parent oligomers.17 Homologation of the monomers to sulfated benzofuran dimers increased the inhibitory potency 100–1000-fold and also displayed good human plasma anticoagulant effect.18 Further, the dimeric scaffold exhibited high selectivity for thrombin due to their recognition of a specific site in exosite 2.19
Figure 1.

Rationale for the study of monosulfated benzofuran trimers. Sulfated low molecular weight lignins were designed to mimic the interactions of sulfated glycosaminoglycans and found to directly and allosterically inhibit human α-thrombin.15,16 The heterogeneity of these macromolecules was eliminated in the design of sulfated benzofuran monomers, which also inhibited thrombin.17 The potency of inhibition increased by orders of magnitude following homologation to the dimeric scaffold.18,19 This led to the rationale that the trimeric and tetrameric scaffold would be more potent and allosteric inhibitors. R represents different functional groups.
The medicinal potential of the sulfated benzofurans is high. The molecules are not only small, but also present a combination of anionic and hydrophobic forces, which afford interesting protein recognition and physicochemical properties.20 Sulfated benzofuran dimers are highly water soluble, readily synthesized, and essentially nontoxic to mammalian cells.19 Their most interesting property – allosteric inhibition of thrombin – is different from all known clinically used thrombin inhibitors (competitive inhibition). Thus, to discover more potent allosteric sulfated benzofurans, we designed a library of higher analogs. Herein, we report on the synthesis, biological activity, mechanism of action and site of binding of sulfated benzofuran oligomers. A specific trimer (9a) was found to display much higher potency than the earlier generation inhibitors. Interestingly, 9a recognized a site within exosite 2 that was different from that recognized by sulfated benzofuran dimers. The result suggests that multiple avenues within exosite 2 can be exploited to modulate the highly dynamic conformational equilibria displayed by thrombin. This work puts forward a highly potent, small molecule as a direct allosteric inhibitor of thrombin and presents a new sub-site in exosite 2 for drug discovery.
Results
Rationale Behind the Study of Sulfated Benzofuran Trimers/Tetramer
Allosteric targeting of enzymes can afford major advantages of higher selectivity and tunable efficacy in comparison to the traditional orthosteric targeting. Higher selectivity arises from much less conserved allosteric sites as compared to catalytic sites, while tunable efficacy arises from the dependence of energetic coupling between the ligand and substrate binding sites on the structure of the ligand. Both of these properties are critical for regulating thrombin so as to maintain hemostatic balance. Our previous work with sulfated benzofuran dimers established that these unique small molecules have the dual property of high selectivity and efficacy expected of allosteric inhibitors.17–19 For example, a group of sulfated benzofuran dimers inhibited thrombin, but not the closely related factor Xa, and displayed variable efficacies in the range of 60 to 95%.19 The potency of inhibition was found to be 5–50 μM, which was reasonable considering the molecular size and the exposed nature of Arg173, the residue involved in their recognition. Structure-activity relationship (SAR) studies revealed that a sulfate group at the 5-position, a hydrophobic ester at the 3-position and an alkyl ether at the 6-position were optimal for potency.18
Despite their reasonable potency, the sulfated benzofuran dimers are still ~200-fold less potent than sulfated LMWLs,16 which were the parent oligomers for the design of small molecules. Whereas sulfated LMWLs are heterogeneous and polydisperse structures with an average chain length of 5 – 13 monomers,16 sulfated benzofuran dimers contain only two monomers. Hence, we sought to assess whether homologation to a trimeric or tetrameric length would enhance the potency of inhibition with retention of exosite 2-dependent allosteric inhibition of thrombin. Thus, a focused library of ten sulfated benzofuran trimers and one tetramer was designed considering the previous SAR results. Two of the three benzofuran units were maintained essentially invariant as found in the parent sulfated benzofuran dimer.18 Variations were introduced at the available 3-, 5-, and 6-positions of the terminal benzofuran unit, which included hydrophilic to bulky hydrophobic groups at the 3-ester, hydrophobic groups at the 6-position, sulfate group at either 5- or 6-position, a carboxylic acid group at the 3-position, and an unsulfated 5-OH group (see Schemes 1 – 4).
Scheme 1.

Synthesis of sulfated benzofuran trimers 9a – 9g and 13 – 14.
Synthesis of Sulfated Benzofuran Trimers and a Tetramer
The synthesis of benzofurans 1a – 1e from catechol in one step followed by dimerization and functionalization (Scheme 1) to provide several sulfated derivatives has been described in detail in our earlier work.18 Briefly, mono-alkylation of 1a – 1e with three different alkyl halides primarily afforded 2a – 2g with 6-substitution. Intermediates 2a – 2g were protected and brominated to give reactive handles 4a – 4g. Coupling of allylic bromide 4a with benzofurans 2a or 2e followed by deprotection resulted in dimers 6a and 6e in reasonably good overall yield (Scheme 1, 25 – 30 % overall isolated yield).
The synthesis of eight sulfated benzofuran trimers 9a – 9g and 13 – 14 was achieved by coupling reactive handles 4a – 4g with either 6a or 6e (Scheme 1). Unlike relatively easy coupling of two monomers 4a and 2a, the coupling of a dimer 6a with 4a – 4g did not proceed well under normal conditions, perhaps because of enhanced steric hindrance. Microwave-based coupling afforded the intermediate trimers 7a – 7g (see Supplementary Material for structures and details) in 80 min with nearly 50% yield. These were then deprotected using potassium fluoride and sulfated using triethylamine-sulfur trioxide under microwave conditions.21 The triethylammonium cation was exchanged for sodium using a weak cation exchange resin to yield monosulfated benzofuran trimers 9a – 9g (30 – 40% overall from 6a). Likewise, coupling of 6e, which contains an allylic group, with 4a followed by deprotection gave trimer 11, which was selectively hydrolyzed using catalytic amount of Pd(PPh3)4 in the presence of triethyl amine to give 12 in good yields.22 Both 11 and 12 were then chemically sulfated to their respective monosulfated derivatives 13 and 14 in good overall yields from dimeric starting point. It is interesting that sulfation of 12 proceeded smoothly despite the presence of carboxylic acid group, which has been known to disrupt the formation of the intermediate sulfo-ester.18
To assess the chain length dependence of thrombin inhibition, monosulfated benzofuran tetramer 17 was also synthesized (Scheme 2). Trimer intermediate 8a was processed through base-catalyzed coupling, desilylation and sulfation to give 17 in an overall yield of 36%. Yet, inhibition results described below restricted our synthetic efforts to only one tetramer member. Finally, a ‘flipped’ sulfated benzofuran trimer 23 (Scheme 3) was synthesized to assess the specificity of recognition of parent trimers 9a – 9g. The ‘flipped’ trimer 23 contains a sulfate group at the 6-position, whereas trimers 9a – 9g and 13 – 14 (see Scheme 1) have a sulfate at the 5-position. Trimer 23 could be prepared by exploiting the 5-substituted regio isomer 18 formed in small amounts (~10 %) in the methylation of benzofuran 1a. Coupling of 18 with 2a followed by the established series of reactions described above resulted in the ‘flipped’ trimer 23 (Scheme 3).
Scheme 2.

Synthesis of sulfated benzofuran tetramer 17.
Scheme 3.

Synthesis of ‘flipped’ sulfated benzofuran trimer 23.
Thrombin Inhibition Potential of Sulfated Benzofuran Trimers and Tetramer
The focused library of 11 potential inhibitors displays structural variations in the terminal benzofuran unit containing the 5-sulfate at the 6-position, which include –CH3, –C2H5, and –CH(CH3)2 groups, and at the 3-position, which include –CH3, –C2H5, –CH(CH3)2 and –CH2CH2OCH3 groups (trimers 9a – 9g). Other structural variations include 3-allylic ester (13) or 3-carboxylic acid (14) in the benzofuran unit remote from the sulfated benzofuran unit and the ‘flipped’ benzofuran (23). In addition to tetramer 17, trimer 12 was also included in the study because it displayed fairly good water solubility despite being devoid of any sulfate group.
Thrombin inhibition was studied using Spectrozyme TH hydrolysis assay, as described previously for dimers.16 Of the 11 molecules, nine molecules inhibited thrombin at pH 7.4 and 25 °C (Figure 2, Table 1). Trimer 9a was found to be the most potent inhibitor with an IC50 of 670 nM and an efficacy of approximately 80%. In comparison to the corresponding parent dimer,18 this represents a nine-fold increase in potency for the trimer with no difference in efficacy. Other inhibitors displayed potencies in the range of 2 to 165 μM, except for two molecules which were essentially inactive. Comparison of IC50s of 9a – 9d shows an interesting relationship in terms of the nature of the 3-ester (Table 1). The potency decreases precipitously (>500-fold) as the substituent changes from ethyl/methyl (9a/9b) to t-butyl (9c) to methoxyethyl (9d) indicating stringent steric and/or polar requirements. This possibly suggests recognition of a rather constrained binding site by sulfated benzofuran trimers in the vicinity of the terminal 3-ester position. Likewise, the potency decreases 8.6- and 148-fold upon changing the 6-substituent from methyl (9a) to ethyl (9f) and to isopropyl (9g), respectively. This implies a significant size constraint on the opposite face of the same benzofuran unit too.
Figure 2.
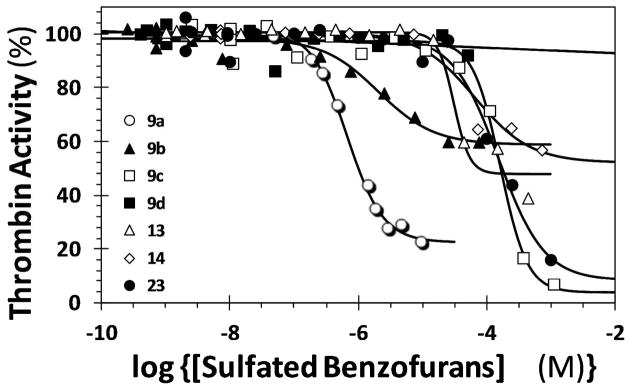
Direct inhibition of human α-thrombin by designed sulfated benzofuran trimers and a tetramer. The inhibition of thrombin was measured spectrophotometrically through hydrolysis of Spectrozyme TH in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000 at 25 °C. Solid lines represent fits to the data using equation 1 to obtain IC50, HS, YM, and YO, as described in ‘Experimental Procedures’.
Table 1.
Inhibition of human α-thrombin by sulfated benzofurans.a
| logIC50 | IC50 (μM) | HS | ΔYa (%) | |
|---|---|---|---|---|
| 9a | −6.17 ± 0.03 | 0.67 ± 0.04b | 1.5 ± 0.2 | 79 ± 9 |
| 9b | −5.69 ± 0.13 | 2.0 ± 0.6 | 1.0c | 39 ± 2 |
| 9c | −3.78 ± 0.07 | 165 ± 25 | 2.1 ± 0.5 | ~94d |
| 9d | – | >500 | NAe | NA |
| 9f | −5.23 ± 0.11 | 5.8 ± 1.5 | 2.3 ± 1.2 | 43 ± 3 |
| 9g | ~ −4.0d | ~ 99d | UDf | ~ 26d |
| 12 | – | >500 | NA | NA |
| 13 | −4.51 ± 0.06 | 31 ± 4 | 3.5 ± 1.2 | 53 ± 4 |
| 14 | −4.18 ± 0.17 | 66 ± 25 | 1.0c | 49 ± 5 |
| 17 | −3.80 ± 0.15 | 158 ± 55 | 1.0c | 62 ± 16 |
| 23 | −3.81 ± 0.13 | 154 ± 45 | 1.2 ± 0.3 | ~ 91d |
Logistic equation 1 was used to fit the dose-dependence of the residual thrombin activity to obtain logIC50, HS, Y0, and YM values for inhibition in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2, and 0.1% PEG8000 at 25 °C (see ‘Experimental Procedures’). ΔY = YM − Y0.
Errors represent ± 1 S. E.
HS was fixed to 1.0 to define the inhibition parameters better.
Approximate value because either HS, YM or Y0 was not defined well.
Not applicable.
Undefined.
In contrast, the 3-allyl ester (13) present on the unit most away from the sulfated benzofuran unit introduces a modest 46-fold reduction in potency from 9a. This potency reduces further only 2-fold upon hydrolysis of the allyl group (13) to a carboxylic acid (14) (Table 1) suggesting that the benzofuran monomer most away from the sulfated benzofuran unit is binding to a site that is much less sterically and/or electronically constrained. This is in striking contrast to the complete lack of inhibition observed with the corresponding sulfated benzofuran dimer containing an equivalent carboxylic acid group.18 Elimination of the 5-sulfate from 14, as in trimer 12, eliminated thrombin inhibition potential suggesting the central significance of the lone sulfate group. The trimer scaffold was found to be optimal for activity as suggested by the loss of nearly 236-fold in potency with tetramer 17. This is a bit surprising considering that sulfated LMWLs with chain lengths of 5 – 13 units inhibit thrombin with high potency (~30 nM).16 It is possible that this difference in activity arises from fundamental differences in the structure between sulfated LMWLs and sulfated benzofurans. Whereas the former contain dihydrobenzofuran units, the latter contain fully aromatic units. The dihydrobenzofuran units perhaps afford greater flexibility resulting in better engagement of the thrombin surface. The results suggest that the binding site on thrombin accommodates a few trimeric benzofurans, i.e., 9a or 9b, well and deviation from these structures is detrimental. This conclusion is also supported by results with the 6-sulfated regio isomer 23, which displays an IC50 as weak as the tetramer 17.
Sulfated benzofuran trimers display an impressive 26 – 91% range of efficacy of inhibition (Table 1). Such a broad range arising from this small library is striking. Interestingly, the efficacies for most potent inhibitors are also considerably different. It is difficult to understand the structural basis of the efficacy of inhibition without the crystal structure of the co-complex, however it is possible that the stringent interactions made by the benzofuran unit containing the 5-sulfate is the primary driver of the energetic coupling that induces inhibition. This hypothesis will have to be tested through detailed biophysical and/or computational studies.
A final aspect of thrombin inhibition induced by sulfated benzofuran trimers is the specificity of inhibition. When the molecules were screened against human factor Xa, which is a closely related coagulation enzyme, trimers 9a – 15 did not display any significant inhibition at concentration as high as 365 μM (see Table S1 in Supporting Information).
Kinetics of Thrombin Inhibition By Trimer 9a
To identify the mechanism of thrombin inhibition induced by sulfated benzofuran trimers, the kinetics of substrate hydrolysis by thrombin was studied in the presence of 9a under two conditions. Considering that the inhibitors being studied have unique structures and are likely to display allosteric properties in the manner of their parent designs,19 the inhibitors were incubated with thrombin either for 2 h at 37 °C or for overnight (16 h) at 25 °C. The initial rate of Spectrozyme TH amidolysis displayed the characteristic hyperbolic profile (Figure 3), as expected on the basis of Michaelis – Menten kinetics from which KM and VMAX were calculated. The KM for Spectrozyme TH decreased from 9.5 μM in the absence of 9a to 3.1 μM in the presence of 3.6 μM 9a at 25 °C (Table 2), a decrease of nearly 3.1-fold. A similar 4.1-fold decrease in KM was observed at 37 °C in the presence of 9.9 μM 9a. Simultaneously the VMAX of hydrolysis also decreased by 4.3- and 5.7-fold at 25 °C and 37 °C, respectively. This suggests that under both conditions the presence of 9a affects the binding of the chromogenic substrate to the active site of the enzyme, while simultaneously reducing its forward rate constant. More importantly, the reduction in KM and VMAX at each temperature is essentially equivalent (either 0.86–1.2-fold per μM at 25 °C or 0.4–0.6-fold per μM at 37 °C). The simultaneous and equivalent decrease in KM and VMAX under both conditions indicates a primarily uncompetitive mechanism of thrombin inhibition by 9a. This mechanism is interesting and different from the noncompetitive mechanism displayed by sulfated benzofuran dimers studied earlier.19 Thus, the trimers appear to be unexpectedly different from the dimers.
Figure 3.

Michaelis-Menten kinetics of Spectrozyme TH hydrolysis by human α-thrombin in presence of trimer 9a. The initial rate of hydrolysis at various inhibitor concentrations (◆ = 0 μM; △ = 0.36 μM; ● = 0.63 μM; □ = 1.8 μM; ○ = 3.6 μM) was measured spectrophotometrically in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000 following overnight incubation at 25 °C. Solid lines represent non-linear regressional fits to the data by the standard Michaelis-Menten equation to yield KM and VMAX.
Table 2.
Michaelis-Menten kinetics of Spectrozyme TH hydrolysis by α-thrombin in the presence of the trimer 9a.a
| [9a] (μM) | Overnight incubation at 25°C | 2 hr incubation at 37°C | ||
|---|---|---|---|---|
| KM (μM) | VMAX (mAU/min) | KM (μM) | VMAX (mAU/min) | |
|
|
|
|||
| 0 | 9.5 ± 1.8b | 81 ± 5 | 7.7 ± 2.0 | 86 ± 7 |
| 0.36 | 11.1 ± 2.5 | 80 ± 6 | –c | – |
| 0.63 | 7.7 ± 1.3 | 69 ± 3 | 6.7 ± 2.3 | 72 ± 8 |
| 1.53 | – | – | 2.2 ± 0.8 | 30 ± 2 |
| 1.80 | 3.2 ± 0.5 | 35 ± 1 | – | – |
| 3.60 | 3.1 ± 0.60 | 19 ± 1 | 2.7 ± 0.6 | 25 ± 1 |
| 9.90 | – | – | 1.9 ± 0.6 | 15 ± 0.8 |
KM and VMAX were measured by monitoring the initial rate of thrombin hydrolysis of Spectrozyme TH (2 – 80 μM) from the linear increase in absorbance at 405 nm in the presence of fixed concentration of 9a that was incubated either overnight at 25°C or for two hours at 37°C in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000. The data was fitted by the standard Michaelis – Menten equation to obtain KM and VMAX, as described in ‘Experimental Procedures’.
Error represents ±1 S.E.
Not measured
The uncompetitive mechanism implies that thrombin is inhibited with the formation of enzyme – substrate – inhibitor ternary complex. This suggests that the trimers may be inducing a change in the catalytic triad that disfavors catalysis even in the presence of substrate. This does not however, imply that the catalytic triad is made completely dysfunctional. Thrombin retains partial catalytic activity, which alludes to regulatory features of sulfated benzofuran trimers. Thus, a question arises whether thrombin can be completely inhibited by an active-site directed inhibitor, e.g., D-Phe-Pro-Arg-chloromethylketone (PPACK) following its inhibition by 9a. Figure S10 (Supplementary Material) shows the thrombin activity profile of such a sequential inhibition by 9a and PPACK. As expected, PPACK reduces thrombin’s activity over and above that of 9a and renders it totally inactive. Thus, if necessary, competitive and uncompetitive inhibitors can be simultaneously used to induce total inhibition.
Trimer 9a Does Not Bind in Exosite 1 of Thrombin
To identify the site of interaction on thrombin, we measured the effect of a 12-residue hirugen analog, HirP, on the IC50 of 9a. HirP binding to thrombin has been extensively studied earlier.16,23 It interacts with exosite 1 and enhances the kCAT of substrate hydrolysis.16 The apparent IC50 of 9a in the presence of HirP was measured in a manner similar to that used for direct inhibition studies. The presence of HirP did not significantly affect the inhibition profile of 9a (Table 3, see Figure S1 in Supplementary Material). Using the logistic equation 1, the apparent IC50s calculated in the presence of the three HirP concentrations tested were 0.9, 0.8 and 0.9 μM indicating that 9a does not appear to bind in the exosite 1 of thrombin.
Table 3.
Inhibition of α-thrombin by trimer 9a in presence of exosite 1 or exosite 2 ligands.a
| logIC50 | IC50 (μM) | HS | ΔYa (%) | |
|---|---|---|---|---|
| 0 μM | −6.17 ± 0.03 | 0.67 ± 0.04b | 1.5 ± 0.2 | 79 ± 9 |
| [Hirugen peptide] | ||||
| 3 nM | −6.07 ± 0.03 | 0.9 ± 0.1 | 2.5 ± 0.5 | 44 ± 2 |
| 5 nM | −6.10 ± 0.03 | 0.8 ± 0.1 | 2.4 ± 0.3 | 46 ± 2 |
| 15 nM | −6.04 ± 0.03 | 0.9 ± 0.1 | 3.0 ± 0.7 | 43 ± 1 |
| [Heparin] | ||||
| 4.2 μM | −6.00 ± 0.04 | 1.0 ± 0.1 | 2.2 ± 0.4 | 71 ± 7 |
| 12.8 μM | −5.68 ± 0.03 | 2.1 ± 0.1 | 1.8 ± 0.2 | 80 ± 11 |
| 25.0 μM | −5.42 ± 0.02 | 3.8 ± 0.2 | 1.7 ± 0.2 | 81 ± 10 |
| 42.6 μM | −5.26 ± 0.06 | 5.6 ± 0.8 | 1.2 ± 0.2 | ~90c |
| [Octasaccharide H8] | ||||
| 1.0 μM | −5.87 ± 0.02 | 1.3 ± 0.1 | 2.3 ± 0.3 | 58 ± 3 |
| 3.0 μM | −5.87 ± 0.04 | 1.4 ± 0.1 | 2.6 ± 0.6 | 54 ± 4 |
| 10 μM | −5.78 ± 0.02 | 1.6 ± 0.1 | 2.8 ± 0.4 | 50 ± 2 |
| 20 μM | −5.75 ± 0.03 | 1.8 ± 0.1 | 2.6 ± 0.5 | 49 ± 2 |
| [γ′-Fibrinogen peptide] | ||||
| 0.2 μM | −5.94 ± 0.02 | 1.2 ± 0.1 | 2.4 ± 0.3 | 73 ± 8 |
| 0.65 μM | −5.95 ± 0.03 | 1.1 ± 0.1 | 2.1 ± 0.3 | 71 ± 8 |
| 2.0 μM | −5.92 ± 0.02 | 1.2 ± 0.1 | 3.3 ± 0.6 | 67 ± 4 |
| 6.5 μM | −5.88 ± 0.06 | 1.3 ± 0.2 | 2.1 ± 0.6 | 65 ± 7 |
Inhibition studies were performed in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG 8000 in PEG20000-coated acrylic cuvettes. S-2366 was used as thrombin substrate and trimer 9a was incubated overnight with thrombin at 25 °C. Logistic equation 1 was used to fit the dose-dependence of the residual thrombin activity to obtain log IC50, HS, Y0, and YM. ΔY = YM − Y0. See ‘Experimental Procedures’ for details.
Represents 1 ± S.E.
Approximate value because Y0 was not well defined.
Trimer 9a Interacts With Exosite 2 of Thrombin
To identify whether sulfated benzofurans bind in exosite 2 of thrombin, we measured the apparent IC50 of 9a in the presence of three different exosite 2 ligands including UFH, H8, and FibP. UFH, a heterogeneous polysaccharide with an average molecular weight of ~13000, is approximately 5-fold longer than octasaccharide H8, while FibP is a 20-amino acid peptide sequence derived from fibrinogen. All three ligands bind in exosite 2, but span slightly different amino acid residues and, therefore, could serve as good probes for localizing 9a binding site. Whereas the crystal structure of H8 – thrombin complex indicates ionic interactions with Arg93, Arg101, Arg126, Arg165, and Lys240 residues,24 the interaction of UFH with thrombin is considered to also involve Arg236.25 FibP, on the other hand, interacts with H8 binding residues26 and also Arg97, Arg173 and Arg175.27
Figure 4A shows the dose-response profiles of 9a inhibition of thrombin in the presence of UFH as a competitor. A characteristic shift in the profiles is observed as function of the concentration of UFH suggesting competition between the two ligands for thrombin binding. The apparent IC50 increases from 0.68 μM in the absence of UFH to 5.6 μM in the presence of 42.6 μM UFH (Table 3). To evaluate whether this competition is essentially ideal, the predicted IC50 in the presence of UFH was calculated using Dixon – Webb equation 2 (Kcomp = dissociation constant of the competitor) using the measured affinity of 6.8 μM for UFH – thrombin interaction in the laboratory.24 The measured and Dixon – Webb predicted IC50s compare favorably at each concentration of UFH (Figure 4B) indicating that 9a and UFH compete with each other essentially ideally for binding in exosite 2 of thrombin.
Figure 4.

Competition by exosite 2 ligands and Dixon-Webb analysis of the competitive effects. Competitive effect of UFH (A) and H8 (C) on the inhibition of human α-thrombin by 9a in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000 at 25 °C. Solid lines represent fits using the logistic equation 1 to obtain the apparent IC50 as described in ‘Experimental Procedures’. Comparisons of the predicted and experimentally measured IC50 values in the presence of UFH and H8 at various competitor concentrations are shown in B and D, respectively. Open bars represent the measured values, whereas closed bars are those predicted using Dixon-Webb equation 2.
| (Eq. 2) |
To test whether 9a competes with H8, similar studies were performed in the presence of 1 to 20 μM H8 (KH8 was found to be 2.7 μM). Figure 4C shows the dose-response curves and Table 3 lists the calculated apparent IC50 values. The potency increases to 1.8 μM at 20 μM H8, which represents a small increase that does not compare well with the predicted increase based on Dixon – Webb analysis (Figure 4D). This suggests that H8 does not compete with 9a, although a marginal overlap in interactions between the two cannot be excluded.
To assess whether 9a engages thrombin residues that interact with FibP, competition studies were performed in the presence of 0.2 to 6.5 μM FibP (KFibP = 0.6 μM).27 No shift in the dose-response profile was observed in the presence of this competitor and the apparent IC50 did not increase suggesting no competition between the two ligands (Table 3 and see Figure S2).
Taken together, these results indicate that 9a binds in exosite 2, but its interaction is different from that identified for sulfated benzofuran dimers.19 Whereas the dimers compete fully with FibP and partially with UFH, the trimers compete well with UFH, show a very weak, if at all, competition with H8 and do not compete with FibP. Thus, despite an obvious similarity in structure between the dimers and trimers, their interaction at a molecular level with thrombin is dramatically different.
Sulfated Benzofuran Trimer 9a Interacts with a Specific Arginine in Exosite 2
To further narrow down the site of binding, we studied the inhibition of 12 recombinant thrombins (a wild-type and other mutants) containing single replacement of Arg and Lys residues of exosite 2 to Ala. These recombinant thrombins were expressed in baby hamster kidney cells and have been studied earlier by the Rezaie laboratory.28,29 Each mutant was screened for direct inhibition by 9a using S2238, which is a relatively more sensitive thrombin substrate. The recombinant wild-type thrombin displayed an IC50 of 0.26 μM, which is nearly 2.5-fold lower than that for human plasma α-thrombin. Such differences are not uncommon and may arise due to variations in the glycosylation pattern between recombinant and plasma proteins. Despite this difference in potency, we proceeded with the study of 9a inhibition of thrombin mutants containing alanine replacement for nearly all positively charged residues of exosite 2. These residues are known to interact with UFH, H8 and FibP (see above). In addition, Lys169Ala, Arg233Ala and Arg235Ala thrombins were also studied.
The dose–response profiles were a bit challenging because the recombinant proteins tended to display a slightly higher Hill slope than that for human plasma thrombin. A higher HS requires careful selection of inhibitor concentrations to define the dose-response profiles adequately, which introduce an experimental challenge due to limited availability of recombinant mutants. Yet, at least 9 – 10 data points were used to define the dose-response profiles. Figure 5A shows the profiles for the Arg233Ala and wild-type thrombins and the measured IC50s for all thrombins studied are listed in Table 4. The potencies for all recombinant thrombins ranged from 0.19 μM to 0.43 μM, except for the Arg233Ala mutant that showed an IC50 of 0.98 μM (Table 4). Figure 5B shows a comparison of the change in IC50 for each mutant from that of the wild-type. The Arg233Ala thrombin is the only enzyme with a significantly higher defect in 9a recognition (3.8-fold loss in potency) as compared to mutants that display at best a 1.5-fold defect. This indicates that Arg233 of exosite 2 is the primary residue of contact for 9a.
Figure 5.

Identification of residues that contribute to the potency of sulfated benzofuran trimer 9a using recombinant site-directed mutants of human thrombin. Thrombin mutants were expressed in the Rezaie laboratory and their inhibition by 9a studied in a manner identical to the plasma α-thrombin. Data shown in A is the dose-response profile for recombinant wild-type and R233A thrombins. Solid lines in A represent sigmoidal dose-response fits using equation 1. The ratio of IC50 values of the recombinant mutant to that of recombinant wild-type thrombin is plotted in B. See text for details.
Table 4.
Inhibition of recombinant wild-type and mutant thrombins by trimer 9a.a
| Thrombin | log IC50 | HS | IC50 (μM) | ΔYa (%) |
|---|---|---|---|---|
| rWT | −6.58 ± 0.02 | 3.4 ± 0.5b | 0.26 ± 0.01 | 49 ± 2 |
| R93A | −6.73 ± 0.04 | 1.8 ± 0.3 | 0.19 ± 0.02 | 66 ± 6 |
| R97A | −6.44 ± 0.04 | 2.6 ± 0.5 | 0.37 ± 0.03 | 47 ± 3 |
| R101A | −6.41 ± 0.07 | 2.1 ± 0.6 | 0.39 ± 0.06 | 55 ± 6 |
| R126A | −6.55 ± 0.07 | 1.8 ± 0.5 | 0.29 ± 0.05 | 65 ± 11 |
| R165A | −6.59 ± 0.11 | 1.1 ± 0.3 | 0.26 ± 0.07 | 61 ± 8 |
| K169A | −6.48 ± 0.03 | 2.1 ± 0.2 | 0.33 ± 0.02 | 59 ± 3 |
| R173A | −6.59 ± 0.04 | 2.4 ± 0.7 | 0.26 ± 0.03 | 62 ± 8 |
| R175A | −6.363 ± 0.04 | 5.2 ± 2.1 | 0.43 ± 0.04 | 37 ± 3 |
| R233A | −6.01 ± 0.04 | 3.1 ± 0.6 | 0.98 ± 0.10 | 44 ± 2 |
| K235A | −6.57 ± 0.02 | 2.0 ±0.1 | 0.27 ± 0.01 | 50 ± 1 |
| K236A | −6.40 ± 0.01 | 3.0 ± 0.3 | 0.40 ± 0.04 | 52 ± 1 |
| K240A | −6.57 ± 0.04 | 2.3 ± 0.4 | 0.27 ± 0.03 | 49 ± 3 |
Inhibition studies were performed in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG 8000 in PEG20000-coated acrylic cuvettes. S-2366 was used as thrombin substrate and trimer 9a was incubated overnight with the enzyme at 25 °C. Logistic equation 1 was used to fit the dose-dependence of the residual thrombin activity to obtain log IC50, HS, Y0, and YM. ΔY = YM − Y0. See ‘Experimental Procedures’ for details.
Represents 1 ± S.E.
Plausible Binding Mode of Trimer 9a in Exosite 2
To gain some insight into plausible mode of exosite 2 recognition by sulfated benzofuran trimer 9a, we utilized genetic algorithm-based docking and scoring program GOLD, which had been used successfully earlier for a sulfated benzofuran dimer.19 Trimer 9a was docked into exosite 2 near Arg233. Following an exhaustive search, GOLD identified a small group of favorable binding modes that display an ionic interaction between the guanidine of Arg233 and 5-O-sulfate of trimer 9a (Figure 6). Four of the top eight binding poses displayed similar orientation (RMSD < 2.5 Å). The preferred binding mode shows that the benzofuran ring orients in the shallow groove of exosite 2 such that the 3-ester side chain (Me- or Et-) can interact well with the narrow hydrophobic channel form by Phe232 and other backbone atoms (Arg233, Leu234, Lys235 and Lys236). The size of this channel most probably cannot accommodate larger groups, thus explaining the observed loss of inhibitory activity in 9c and 9d (>300-fold loss, Table 1). Interestingly, the trimer 14 containing a carboxylic acid group at 3-position of a terminal benzofuran inhibits thrombin, while the corresponding dimer was found to be completely inactive in our earlier work.18 This model of trimer 9a binding in exosite 2 provides a plausible explanation as the terminal benzofuran containing the carboxylic acid side chains is found to be placed in a much less constrained region with significant exposure to solvent. This also explains why trimer 13 with an allyl ester inhibits with an essentially similar potency as trimer 14 with a carboxylic acid group (Table 1). Support for this model also comes in the form of tetramer 17, which binds with a 225-fold weaker affinity. Although 9a binding model shows room for an extra benzofuran ring at the terminus opposite to the 5-O-sulfate group, the inter-monomer bond will have to underogo considerable bending while encountering the ridge formed by Glu97, Asp100, Arg101, Lys169, Arg175, Ile176, Thr177 and Asp178 residues, which reduces its affinity.
Figure 6.

Proposed mode of binding of sulfated benzofuran trimer 9a in exosite 2 of thrombin. Utilizing a 100 GA runs, GOLD-based docking of 9a led to four nearly equivalent poses of the top eight solutions. Trimer 9a (stick representation in yellow) was found to bind to Arg233 in a region distant from that of a sulfated benzofuran dimer (sticks, cyan), which engages Arg173.19 Thrombin surface is shown in light grey with positively charged amino acids of exosite 2 in blue.
Yet, the 9a model should be interpreted with caution. The bond between Arg233 and 5-O-sulfate is primarily electrostatic and not strong. This results in the weaker defect noted upon mutation of Arg233 (~3-fold). A stronger bond, such as a linear hydrogen-bond, would have engineered a much more powerful defect, as observed with Arg173Ala mutation on the affinity of sulfated benzofuran dimer.19 A significant portion of 9a binding energy is likely to arise from hydrophobic forces, which are driven by entropically favorable displacement of water molecules.30 Considering that 9a is only the best molecule in a small library, considerable structural modifications may need to be introduced for engineering a more specific inhibitor.
Trimer 9a Prolongs Human Plasma Clotting Time
To assess the anticoagulant potential of 9a, we performed two human plasma clotting assays that are routinely used to screen new molecules in vitro. These are PT and APTT assays that attempt to measure the effect of an anticoagulant on the extrinsic and intrinsic flux of the coagulation cascade, respectively. Trimer 9a displayed the characteristic increase in clotting times confirming its anticoagulant potential (Figure 7). The anticoagulant effect is better in APTT conditions than in PT conditions. The concentrations of 9a required to double the PT and APTT were calculated as described earlier for sulfated benzofuran dimers and other compounds.18 A 2-fold increase in APTT and PT required 139 μM and 568 μM 9a. These results also suggest that the sulfated benzofuran trimers are approximately 10-fold more potent in human plasma than the dimers.18 This follows the improvement in IC50 of trimers as compared to the dimers and validates the rationale behind homologation. Yet, trimer 9a is still 10 times less potent in plasma anticoagulation than the parent polymeric sulfated LMWLs.15 This implies that further modifications on the trimeric scaffold, and not the tetrameric scaffold, would be necessary to reach the potency of the longer scaffold.
Figure 7.
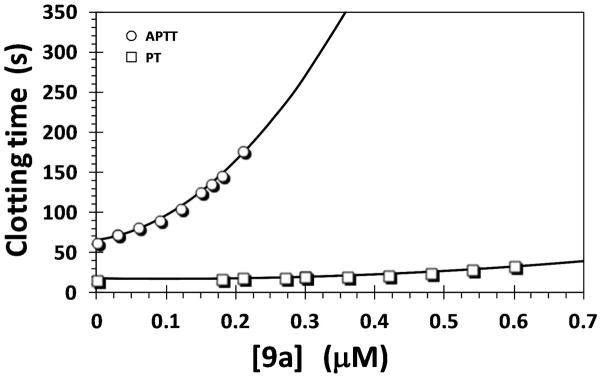
Prolongation of human plasma clotting time as a function of concentration of sulfated benzofuran trimer 9a in either APTT assay (○) or PT assay (□). Solid lines are trend lines (and not non-linear regressional fits). APTT and PT assays were performed in duplicate, as described in the experimental section.
Discussion
This work shows that monosulfated benzofuran trimers can modulate thrombin’s catalytic activity with high potency and variable efficacy by interacting with the anion-binding exosite 2. A specific trimer 9a was at least 9-fold more potent than the corresponding dimer in both direct enzyme inhibition assay and human plasma clotting assay.18 These results are in line with the expectation derived from the activity of sulfated LMWLs.15 The polymeric sulfated LMWLs were designed to be heterogeneous and polydisperse so as to mimic the nature of the natural anticoagulant, heparin, but also suffer from the limitations arising from the presence of millions of sequences. The sulfated benzofuran trimers are synthetically prepared, homogeneous and well-characterized. Trimer 9a has good anticoagulant potential; however, much work is needed to assess its wider pharmacological activity before establishing its firm clinical relevance. This would include anticoagulation studies in human blood, effect on thrombin’s macromolecular targets including protein C, glycoprotein Ibα, and protease activated receptor-1.6–8,11 At the present time, trimer 9a represents an exciting lead toward the discovery of the first clinically relevant, allosteric regulator of human plasma thrombin.
A novel aspect of trimer 9a is that it inhibits thrombin with an efficacy of approximately 80%, and not 100%, which alludes to the possibility of allosteric regulation. It has been proposed that regulators, and not inhibitors, of coagulation, are likely to exhibit reduced bleeding complications. A regulator of coagulation will ensure a balance between pro-coagulation and anti-coagulation states, while an inhibitor may tip the balance to anti-coagulation, especially with 100% inhibition efficacy. Thus, sulfated benzofuran trimers exhibit considerable promise as safer anticoagulants.
An interesting finding of this work is that 9a binds in a site different from that of the sulfated benzofuran dimers (see Figure 6). Whereas 9a recognizes Arg233 in exosite 2, the corresponding sulfated dimer recognizes Arg173.19 Arg233 is also important for recognition of sulfated LMWLs, although two additional residues were found to be more important.31 Replacement of Arg233 for Ala reduced the potency of sulfated LMWLs by ~2.5-fold, which is similar to the defect found for 9a. Thus, sulfated benzofuran trimers appear to mimic certain oligomeric sequences of sulfated LMWLs. Although both Arg233 and Arg173 are located in exosite 2, they are approximately 20 – 25 Å apart. This suggests two different modes of binding for the two structurally related allosteric inhibitors. Thus, there are two distinct sub-sites within exosite 2 that bind to the sulfated benzofuran scaffold and modulate the conformation of the catalytic site so as to attenuate its activity. This allosteric phenomenon most probably arises from the highly plastic nature of thrombin that allows selective stabilization of a conformational state driven by ligand binding.10,12 The observation that molecules as small as sulfated benzofuran dimers and trimers are able to function as allosteric regulators is exciting.
An interesting corollary of the two different binding modes for sulfated benzofuran dimer and its higher oligomer is that the two inhibitors could generate synergistic effects. However, the combination experiment did not display any synergism (not shown). We hypothesize that the lack of synergism between the two binding sites probably arises from the binding of trimer first to thrombin, which induces a conformational change in exosite 2 as well as the active site that disfavors binding of the dimer to an altered exosite 2. This result alludes to the highly pliable nature of allosteric regulation of thrombin, as also reported recently for other ligands.32
Despite the lack of synergistic effects, the two different binding sites presents two independent avenues as well as one co-dependent opportunity for further rational drug design. The two independent avenues would entail structure-based design of analogs of the dimer and trimer scaffolds so as to enhance potency and specificity as well as tailor efficacy. The co-dependent opportunity would entail designing a larger, new molecule that ties the two oligomers so as to significantly enhance affinity and specificity. This approach would mimic the fragment-based drug design strategy, wherein the two fragments would be the trimer and the dimer. Although interesting, the strategy is not necessarily straightforward and guaranteed to work. An example of the challenge is our results with tetramer 17, which was found to be much less active. Yet, the nanomolar potency of longer oligomers present in sulfated low molecular weight lignins15,16 alludes to the feasibility of the approach.
Overall, the homogenous sulfated benzofuran trimer scaffold is the first scaffold that affords exosite 2 driven allosteric regulators of thrombin with sub-micromolar potency. These molecules may yield true regulators of thrombin considering that they display variable efficacy of inhibition. Further studies with macromolecular substrates of thrombin are needed to better assess allosteric regulation.
Experimental Procedures
Chemicals, Reagents, Proteins and Analytical Chemistry
Reagents, chemicals and solvents were purchased either from Sigma-Aldrich (Milwaukee, WI) or Fisher (Pittsburgh, PA) and were used as received. Reagents and chemicals were handled under inert nitrogen atmosphere using syringe techniques for liquid and dried spatula for solids. Glassware was oven dried for 12 h before use. Anhydrous sodium sulfate was used for drying organic extracts following work-up. Reactions were monitored by analytical TLC on UNIPLATE™ silica gel GHLF 250 μm pre-coated plates (ANALTECH, Newark, DE) with UV detection (254 nm). Column chromatography was performed using Teledyne ISCO (Lincoln, NE) Comb flash RF system and disposable normal silica cartridges of 30–50 μ particle size, 230–400 mesh size and 60 Å pore size. Sodium exchange chromatography was performed using SP Sephadex C-25 sodium cation exchange resin from GE Healthcare Life Sciences (Piscataway, NJ). Sulfation and coupling reactions under microwave conditions were performed using a CEM-Discover synthesizer (Matthews, NC). The purity of sulfated benzofuran trimers and intermediates along their synthesis was assessed using Shimadzu VP HPLC system. The purity of compounds synthesized in this work was assessed by reversed-phase HPLC and ascertained to be >95% before proceeding for subsequent reaction or biological analysis.
1H and 13C NMR spectra were recorded on either 300 or 400 MHz (Varian Mercury or Bruker Ultrashield™ Plus) in appropriate deuterated solvents including CDCl3, DMSO-d6 or D2O. NMR signals are reported in ppm with the internal solvent signals as standards. The NMR data is reported as chemical shift (ppm) followed by the splitting pattern (s = singlet, d = doublet, t = triplet, m = multiplet), coupling constant (Hz), and integration. Mass spectrometry was performed on using Micromass ZMD4000 single quadrupole mass spectrometer with ESI ionization probe operating in negative ion mode (Waters Corp., Milford, MA). The samples were dissolved in acetonitrile-containing formic acid (5% v/v) and infused at 10 μL/min. The source block temperature and the probe temperature were typically held at 100 and 120 °C, respectively, while corona and cone voltages were selected through manual optimization. The desolvation nitrogen flow was 500 L/hour. The MS profiles were acquired in the mass range from 110 to 1000 daltons at 400 amu/sec.
Human plasma α-thrombin was from Haematologic Technologies (Essex Junction, VT). Stock solutions of α-thrombin were prepared in 20 mM sodium phosphate buffer, pH 7.4, containing 100 mM NaCl. Chromogenic substrates Spectrozyme TH (H-D-cyclohexylalanyl-alanyl-arginine para-nitroanilide diacetate) was purchased from American Diagnostica (Greenwich, CT) and S2238 (H-D-Phe-Pip-Arg–p-nitroanilide) was purchased from ANASPEC (Fremont, CA). UFH and PPACK were purchased from Sigma (St. Louis, MO). Heparin octasaccharide (H8) was purchased from V-Labs (Covington, LA). Tyr63-sulfated hirudin-(54–65) (HirP) was a generous gift from Dr. Paul Bock. A peptide corresponding to the 20 amino acids at the carboxyl terminal of γ′-fibrinogen chain (VRPEHPAETE-Y(PO3)-DSL-Y(PO3)-PEDDL) (FibP) was obtained from Dr. David Farrell. All other chemicals were analytical reagent grade from either Sigma Chemicals (St. Louis, MO) or Fisher (Pittsburgh, PA) and used as such.
Recombinant Thrombin Mutants
Recombinant wild-type and mutant thrombins were prepared in Dr. Rezaie’s laboratory, as described earlier.28,29 Briefly, Arg93Ala, Arg97Ala, Arg101Ala, Arg126Ala, Arg165Ala, Lys169Ala, Arg173Ala, Arg175Ala, Arg233Ala, Lys235Ala, Lys236Ala and Lys240Ala thrombins were prepared in prothrombin-1 form by PCR mutagenesis and expression in baby hamster kidney cells (BHK) using the pNUT-PL2 expression/purification vector system. The mutants were purified to homogeneity by immunoaffinity chromatography using the Ca2+-dependent monoclonal antibody, HPC4, and activated to thrombin. The active-site concentrations of thrombin mutants were determined by an amidolytic activity assay and stoichiometric titrations with antithrombin. These concentrations were within 90–100% of those expected on the basis of their absorbance at 280 nm.
Synthesis of Sulfated Benzofuran Trimers 9a – 9g
To the solution of 6a (0.3 mmol) and 4a – 4g (0.4 mmol) in ethyl acetate (3 mL) was added cesium carbonate (0.3 mmoles) in anhydrous DMF (0.5 mL) in a microwave tube (10 mL). The reaction mixture was exposed to microwaves at 90 °C for 80 min, quenched with methylene chloride, and extracted with brine. The organic layer was dried over magnesium sulfate, concentrated, purified using flash chromatography on silica gel (20–60 % ethyl acetate in hexanes) resulting in 7a – 7g (40–60 % yield). The purity of these products was assessed by reversed phase HPLC and found to be greater than 95% (see Supplementary Material for details on spectral characterization). A solution of 7a – 7g (0.3 mmol) in DMF (3 mL) was then reacted with KF (4 mmoles) and acetic acid (10 μL) at RT for 5 h. The reaction was quenched by methylene chloride and extracted with brine solution. The aqueous layer was extracted with a fresh batch of methylene chloride. The organic layer was combined, dried over magnesium sulfate and concentrated in vacuum. The solid so obtained was precipitated using methanol to obtain products 8a – 8g in ~90% yield. These were characterized by spectroscopic/spectrometric techniques (see Supplementary Material) and sulfated using trimethylamine-sulfur trioxide complex, as described earlier.21 In this reaction, to a solution of 8a – 8g (50 mg) in acetonitrile:DMF (4:1, 0.45 mL) in a microwave tube was added triethylamine (10 equiv) and trimethylamine-sulfur trioxide complex (15 equiv). The reaction mixture was exposed to microwaves (50 W) for 40 min at 100 °C followed by vacuum concentration to remove all solvent. The solid so obtained was directly loaded on a Sephadex C-25 cation exchange resin and eluted with water. Fractions containing the sulfated product were lyophilized to obtain a solid residue, which was further purified on flash chromatography using 0–20% methanol in methylene chloride. For cation exchange, approximately 100 mg sulfated sample was loaded onto 10 g of the cation exchanger in 10×460 mm column and eluted with deionized, distilled water at 0.5 mL/min. The purity of these compounds was found to be >95% by reversed phase HPLC. Characterization of the sulfated trimers 9a – 9g was performed using 1H and 13C NMR spectroscopy and ESI-MS (see Supplementary Material).
Synthesis of Sulfated Benzofuran Trimers 13 and 14
The synthesis of these began with alkylation reaction using cesium carbonate, as described above for trimers 9a – 9g. Thus, 6a (0.3 mmol) was reacted with 4a (0.4 mmol) in ethyl acetate (4 mL) and anhydrous DMF (1 mL) in the presence of cesium carbonate (0.3 mmoles), followed by work and purification (see Supplementary Material for details) to obtain 10 in 60 % yield. Desilylation of 10 (0.3 mmol) in DMF (3 mL) with KF (4 mmoles) and acetic acid (10 μL) gave 11 (see Supplementary Material). To the solution of 11 (0.19 mmol) in methylene chloride (1.5 mL) was added Pd(PPh3)4 (0.009 mmol) and PPh3 (0.018 mmol), and the mixture was stirred for 10 min in ice bath. To this mixture was then added pyrrolidine (0.21 mmol) and stirred for 30 min. The reaction was quenched, extracted with brine, dried, and concentrated in vacuum to obtain a solid, which was purified by flash chromatography to yield 12. Trimers 11 and 12 were sulfated to 13 and 14, respectively, under microwave conditions with trimethylamine-sulfur trioxide complex, as described above, purified, and characterized (see Supplementary Material for details).
Synthesis of Sulfated Benzofuran Tetramer 17 and ‘Flipped’ Sulfated Benzofuran Trimer 23
The synthesis of these compounds using intermediates 2a, 6a, 7a and 18 involved standard reaction conditions established in the synthesis of other sulfated benzofuran trimers. These reactions included Cs2CO3-based alkylation, KF-acetic acid-based desilylation, and MeN:SO3-based microwave sulfation. Each of these have been described above as well as in Supplementary Material, which also provides the spectral characterization data.
Inhibition of Thrombin Using Chromogenic Substrate Hydrolysis Assay
Thrombin activity in the presence of sulfated benzofuran inhibitors was studied using a chromogenic substrate hydrolysis assay in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000 in PEG20000-coated acrylic cuvettes. Spectrozyme TH was used as substrate and the residual thrombin activity was quantified, as reported earlier.16 Briefly, a solution of 10 μL of an inhibitor to give 1–900 μM final concentration was diluted with 970 μL of the buffer and 5 μL of thrombin (5 nM final concentration). Following incubation for 10 min, 15 μL of 2 mM Spectrozyme TH was added (30 μM final concentration) and the initial rate of hydrolysis from the linear increase in absorbance at 405 nm as a function of time was rapidly measured. Logistic equation 1 was used to fit the dose-dependence of residual thrombin activity to obtain IC50. In this equation, Y is the ratio of residual thrombin activity in the presence of the inhibitor to that in its absence; YM and Y0 are the maximum and minimum possible values of the residual percentage of thrombin activity, respectively; and IC50 is the concentration of inhibitors that results in 50% inhibition of enzyme activity. Sigmaplot 8.0 (SPSS, Inc. Chicago, IL) was used to perform non-linear curve fitting in which YM, YO, HS (Hill Slope), and IC50 were allowed to float.
| (Eq. 1) |
Michaelis-Menten Kinetics of Substrate Hydrolysis by Thrombin
The initial rate of Spectrozyme TH hydrolysis by 5 nM thrombin was monitored from the linear increase in absorbance at 405 nm corresponding to less than 10% consumption of the substrate. The initial rate was measured as a function of various concentrations of the substrate (2 – 80 μM) in the presence of fixed concentration of sulfated benzofuran trimer, which was incubated with thrombin either overnight at 25°C or for 2 h at 37°C in 20 mM Tris-HCl buffer, pH 7.4 containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG8000. The data was fitted using the standard Michaelis-Menten equation to determine KM and VMAX.
Competitive Binding Studies with Exosite 1 or Exosite 2 Ligands
Thrombin inhibition by 9a was studied in the presence of either exosite 1 competitor HirP or exosite 2 competitors including UFH, H8 and FibP in a manner similar to that described for direct thrombin inhibition. Briefly, a solution of 9a (0.045–22.5 μM) and thrombin (5–10 nM) was incubated overnight at 25 °C with either HirP (0–15 nM), UFH (0–42.6 μM), H8 (0–20 μM) or FibP (0.2–6.5 μM) in 20 mM Tris-HCl buffer, pH 7.4, containing 100 mM NaCl, 2.5 mM CaCl2 and 0.1 % PEG 8000. Following overnight incubation, the residual thrombin activity was measured through Spectrozyme TH hydrolysis assay. The dose-dependence of the fractional residual thrombin activity at each concentration of 9a was fitted using equation 1 to obtain the apparent IC50, HS, Y0 and YM.
Inhibition of Recombinant Thrombins
Direct inhibition of recombinant wild-type and mutant thrombins by 9a was measured through the chromogenic substrate hydrolysis assay described above, except for the use of a more sensitive substrate, S2238, in these experiments. Solutions of 9a (0.5 – 150 μL) of 90 μM were diluted with the assay buffer in PEG20000-coated acrylic cuvette following by addition of recombinant thrombins (1.5–7.5 nM final concentration) and overnight incubation at 25 °C. S2238 (20 μL of 2 mM stock) was added to measure the residual enzyme activity from the initial rate of increase in absorbance at 405 nm. Logistic equation 1 was used to fit the dose-response profile to obtain the apparent IC50, HS, Y0 and YM.
Human Plasma Clotting Assays
Prothrombin (PT) and activated partial thromboplastin (APTT) times were measured using a BBL Fibrometer (Becton-Dickinson, Sparles, MD) in a standard 1-stage recalcification assay. For the APTT assay, 90 μL citrated human plasma, 10 μL inhibitor (or vehicle for blank test), and 100 μL of pre-warmed 0.2% ellagic acid were mixed and incubated for 4 minutes. Clotting was initiated by adding 100 μL of 25 mM CaCl2 (37 °C) and time necessary for the plasma to clot recorded. For PT assays, 10 μL sample of the inhibitor (or vehicle for blank test), and 90 μL of citrated human plasma were mixed and incubated for 30 s at 37 °C followed by addition of 200 μL pre-warmed thromboplastin (reconstituted as per direction provided by manufacturer). Each clotting assay was performed in duplicate. The data were fit to a quadratic trend line, which was used to determine the concentration of the inhibitor necessary to double the clotting time, 2×APTT or 2×PT.
Molecular Modeling of Sulfated Benzofuran Trimer 9a Binding to Thrombin
The crystal structure of thrombin (PDB code 3uwj, resolution 1.5 Å) was obtained from the RCSB protein data bank and prepared in Sybyl-X v. 2.0 (www.tripos.com/sybyl). Cocrystallized ligands and water molecules were removed using the “prepare protein” module. The missing side chains of Lys236 and Lys240 were engineered in silico by implementing the Lovell rotamer library33 in Sybyl-X. His91 and His230, which were close to Arg233, were set to the epsilon protonated state to maximize hydrogen bonding and avoid steric clashes. Hydrogen atoms were added and the protein minimized for 1000 steps with a gradient of 0.05 kcal/molÅ using the Powell method.34 Trimer 9a was docked using GOLD v. 5.135 utilizing all residues within 22 Å of this basic residue. An experiment consisting of 100 GA runs was employed to ensure sufficient conformational and spatial sampling. A hydrogen bond constraint was used to encourage interaction between Arg233 and the sulfate group of trimer 9a. Docked poses were scored using GOLD score, from which the top 10 poses were selected for detailed analysis.
Supplementary Material
Acknowledgments
This work was supported by grants HL090586 and HL107152 from the National Institutes of Health to URD. We thank Professors Paul E. Bock of Vanderbilt University, Alireza Rezaie of St. Louis University, and David Farrell of Oregon Health & Science University for the generous supply of HirP, thrombin mutants and FibP, respectively, used in this work.
Abbreviations
- APTT
activated partial thromboplastin time
- BHK
baby hamster kidney
- FibP
gamma prime fibrinogen peptide
- GAG
Glycosaminoglycan
- H8
heparin octasaccharide
- HS
Hill slope
- LMWH
low molecular weight heparin
- LMWL
low molecular weight lignin
- PEG
Polyethylene glycol
- PT
prothrombin time
- SAR
structure-activity relationship
- UFH
unfractionated heparin
Footnotes
General procedures for the synthesis of sulfated benzofurans and other intermediate compounds are described in Supplementary Information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Henry BL, Desai UR. Anticoagulants. In: Abraham DJ, Rotella DP, editors. Burger’s Medicinal Chemistry, Drug Discovery and Development. 7. John Wiley; Hoboken: 2010. pp. 365–408. [Google Scholar]
- 2.Straub A, Roehrig S, Hillisch A. Oral, direct thrombin and factor Xa inhibitors: The replacement of warfarin, leeches and pig intestines? Angew Chem Int Ed. 2011;50:4574–4590. doi: 10.1002/anie.201004575. [DOI] [PubMed] [Google Scholar]
- 3.Gresele P, Busti C, Paganelli G. Heparin in the prophylaxis and treatment of venous thromboembolism and other thrombotic diseases. Handb Exp Pharmacol. 2012:179–209. doi: 10.1007/978-3-642-23056-1_9. [DOI] [PubMed] [Google Scholar]
- 4.Vacca JP. New advances in the discovery of thrombin and factor Xa inhibitors. Curr Op Chem Biol. 2000;4:394–400. doi: 10.1016/s1367-5931(00)00112-5. [DOI] [PubMed] [Google Scholar]
- 5.Nar H. The role of structural information in the discovery of direct thrombin and factor Xa inhibitors. Trends Pharmacol Sci. 2012;33:279–288. doi: 10.1016/j.tips.2012.03.004. [DOI] [PubMed] [Google Scholar]
- 6.Di Cera E, Dang QD, Ayala YM. Molecular mechanisms of thrombin function. Cell Mol Life Sci. 1997;53:701–730. doi: 10.1007/s000180050091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lane DA, Philippou H, Huntington JA. Directing thrombin. Blood. 2005;106:2605–2612. doi: 10.1182/blood-2005-04-1710. [DOI] [PubMed] [Google Scholar]
- 8.Huntington JA. Thrombin plasticity. Biochim Biophys Acta. 2012;1824:246–252. doi: 10.1016/j.bbapap.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 9.Bode W. The structure of thrombin: A janus-headed proteinase. Sem Thromb Hemost. 2006;32:16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- 10.Lechtenberg BC, Freund SMV, Huntington JA. An ensemble view of thrombin allostery. Biol Chem. 2012;393:889–898. doi: 10.1515/hsz-2012-0178. [DOI] [PubMed] [Google Scholar]
- 11.Di Cera E. Thrombin. Mol Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamath P, Huntington JA, Krishnaswamy S. Ligand binding shuttles thrombin along a continuum of zymogen- and proteinase-like states. J Biol Chem. 2010;285:28651–28658. doi: 10.1074/jbc.M110.154914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye J, Liu LW, Esmon CT, Johnson AE. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion-binding exosite of thrombin and alter its specificity. J Biol Chem. 1992;267:11023–11028. [PubMed] [Google Scholar]
- 14.Hortin GL, Trimpe BL. Allosteric changes in thrombin’s activity produced by peptides corresponding to segments of natural inhibitors and substrates. J Biol Chem. 1991;266:6866–6871. [PubMed] [Google Scholar]
- 15.Monien BH, Henry BL, Raghuraman A, Hindle M, Desai UR. Novel chemo-enzymatic oligomers of cinnamic acids as direct and indirect inhibitors of coagulation proteinases. Bioorg Med Chem. 2006;14:7988–7998. doi: 10.1016/j.bmc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 16.Henry BL, Monien BH, Bock PE, Desai UR. A novel allosteric pathway of thrombin inhibition. Exosite II mediated potent inhibition of thrombin by chemo-enzymatic, sulfated dehydropolymers of 4-hydroxycinnamic acids. J Biol Chem. 2007;282:31891–31899. doi: 10.1074/jbc.M704257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Verghese J, Liang A, Sidhu PS, Hindle M, Zhou Q, Desai UR. First steps in the direction of synthetic, allosteric, direct inhibitors of thrombin and factor Xa. Bioorg Med Chem Lett. 2009;19:4126–4129. doi: 10.1016/j.bmcl.2009.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sidhu PS, Liang A, Mehta AY, Abdel Aziz MH, Zhou Q, Desai UR. Rational design of potent, small, synthetic allosteric inhibitors of thrombin. J Med Chem. 2011;54:5522–5531. doi: 10.1021/jm2005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdel Aziz MH, Sidhu PS, Liang A, Kim JY, Mosier PD, Zhou Q, Farrell DH, Desai UR. Designing allosteric regulators of thrombin. Monosulfated benzofuran dimers selectively interact with Arg173 of exosite 2 to induce inhibition. J Med Chem. 2012;55:6888–6897. doi: 10.1021/jm300670q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liang A, Thakkar JN, Desai UR. Study of physico-chemical properties of novel highly sulfated, aromatic, mimetics of heparin and heparan sulfate. J Pharm Sci. 2010;99:1207–1216. doi: 10.1002/jps.21908. [DOI] [PubMed] [Google Scholar]
- 21.Raghuraman A, Riaz M, Hindle M, Desai UR. Rapid, high-yielding microwave-assisted per-sulfation of organic scaffolds. Tetrahedron Lett. 2007;48:6754–6758. doi: 10.1016/j.tetlet.2007.07.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deziel R. Mild palladium(0)-catalyzed deprotection of allyl esters. A useful application in the synthesis of carbapenems and other β-lactam derivatives. Tetrahedron Lett. 1987;28:4371–4372. [Google Scholar]
- 23.Bock PE, Olson ST, Bjork I. Inactivation of thrombin by antithrombin is accompanied by inactivation of regulatory exosite I. J Biol Chem. 1997;272:19837–19845. doi: 10.1074/jbc.272.32.19837. [DOI] [PubMed] [Google Scholar]
- 24.Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J Biol Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 25.Sheehan JP, Sadler JE. Molecular mapping of the heparin-binding exosite of thrombin. Proc Natl Acad Sci USA. 1994;91:5518–5522. doi: 10.1073/pnas.91.12.5518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pineda AO, Chen ZW, Marino F, Mathews FS, Mosesson MW, Di Cera E. Crystal structure of thrombin in complex with fibrinogen gamma’ peptide. Biophys Chem. 2007;125:556–559. doi: 10.1016/j.bpc.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 27.Sabo TM, Farrell DH, Maurer MC. Conformational analysis of gamma’ peptide (410–427) interactions with thrombin anion binding exosite II. Biochemistry. 2006;45:7434–7445. doi: 10.1021/bi060360k. [DOI] [PubMed] [Google Scholar]
- 28.He X, Ye J, Esmon CT, Rezaie AR. Influence of arginines 93, 97, and 101 of thrombin to its functional specificity. Biochemistry. 1997;36:8969–8976. doi: 10.1021/bi9704717. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Rezaie AR. Calcium-binding sites of the thrombin-thrombomodulin-protein C complex: Possible implications for the effect of platelet factor 4 on the activation of vitamin K-dependent coagulation factors. Thromb Haemost. 2007;97:899–906. doi: 10.1160/th06-12-0697. [DOI] [PubMed] [Google Scholar]
- 30.Sarkar A, Kellogg GE. Hydrophobicity--shake flasks, protein folding and drug discovery. Curr Top Med Chem. 2010;10:67–83. doi: 10.2174/156802610790232233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdel Aziz MH, Mosier PD, Desai UR. Identification of the site of binding of sulfated, low molecular weight lignins on thrombin. Biochem Biophys Res Commun. 2011;413:348–352. doi: 10.1016/j.bbrc.2011.08.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Malovichko MV, Sabo TM, Maurer MC. Ligand binding to anion-binding exosites regulates conformational properties of thrombin. J Biol Chem. 2013;288:8667–8678. doi: 10.1074/jbc.M112.410829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins. 2000;40:389–408. [PubMed] [Google Scholar]
- 34.Powell MJD. Restart procedures for the conjugate gradient method. Math Program. 1977;12:241–254. [Google Scholar]
- 35.Jones G, Willett P, Glen RC, Leach AR, Taylor R. Development and validation of a genetic algorithm for flexible docking. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.