

Abstract
Purpose
Agents that target the epigenome demonstrate activity in breast cancer models. In preclinical studies, the histone deacetylase inhibitor vorinostat induces cell cycle arrest, apoptosis and differentiation. We evaluated biomarker modulation in breast cancer tissues obtained from women with newly-diagnosed invasive disease who received vorinostat and those who did not.
Experimental Design
Tumor specimens were collected from 25 women who received up to 6 doses of oral vorinostat 300 mg twice daily and from 25 untreated controls in a non-randomized study. Candidate gene expression was analyzed by RT-PCR using the Oncotype DX® 21-gene assay, and by immunohistochemistry for Ki-67 and cleaved caspase-3. Matched samples from treated women were analyzed for gene methylation by QM-MSP. Wilcoxon non-parametric tests were used to compare changes in quantitative gene expression levels pre- and post-vorinostat with changes in expression in untreated controls, and changes in gene methylation between pre- and post-vorinostat samples.
Results
Vorinostat was well-tolerated and there were no study-related delays in treatment. Compared to untreated controls, there were statistically significant decreases in the expression of proliferation-associated genes Ki-67 (p=0.003), STK15 (p=0.005), and Cyclin B1 (p=0.03) following vorinostat, but not in other genes by the Oncotype DX® assay, or in expression of Ki-67 or cleaved caspase-3 by immunohistochemistry. Changes in methylation were not observed.
Conclusions
Short term vorinostat administration is associated with a significant decrease in expression of proliferation-associated genes in untreated breast cancers. This demonstration of biological activity supports investigation of vorinostat in combination with other agents for the management of breast cancer.
Keywords: Breast cancer, epigenetics, histone deacetylase inhibitors, vorinostat, predictive biomarkers
Introduction
Most women with early breast cancer survive the disease, but others suffer recurrence despite completion of recommended local and systemic therapy. Once metastatic, breast cancer is seldom curable, emphasizing the need for new therapeutic options. Carcinogenesis, including cancer initiation and progression, is a multi-step process. Ample evidence demonstrates that, in addition to the role that inherited or sporadic mutations play, epigenetic alterations can lead to abnormal gene expression and subsequent tumorigenesis (1). Epigenetic alterations can include histone modifications such as acetylation or methylation as well as abnormal methylation of DNA in the promoter region of important genes (2). Epigenetic alterations are observed in virtually all breast cancers and may be reversible; their modulation through histone deacetylase (HDAC) or DNA methyltransferase (DNMT) inhibitors, which have been shown to reverse such alterations, has become an attractive area of new drug investigation.
Several classes of HDAC inhibitors have been developed and are currently under investigation or in clinical use. Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA, NSC 701852) is a potent HDAC inhibitor that can be administered orally with excellent bioavailability. Oral vorinostat was available for clinical investigation through the National Cancer Institute’s (NCI) Cancer Therapy Evaluation Program (CTEP), and the maximum tolerated doses identified for further study included 400 mg daily, 200 mg twice a day (bid), an intermittent dose schedule of 300 mg bid for 3 days per week or 250 mg three times daily for 14 days followed by 7 days of rest (3, 4). Vorinostat 400 mg daily was approved in 2006 by the United States Food and Drug Administration for the treatment of progressive, persistent or recurrent cutaneous T-cell lymphoma following two systemic therapies.
Several preclinical studies supported a role for HDAC inhibitors and, in particular vorinostat, in breast cancer. In a NMU-induced rat mammary tumorigenesis model, vorinostat reduced tumor incidence by 40% (5). In vitro studies demonstrated that vorinostat inhibited clonogenic growth of the breast cancer cell lines MCF-7, MDA-231, and MDA-435 by inducing G1 and G2/M cell cycle arrest and subsequent apoptosis within 24–72 hours following drug administration (6). Vorinostat-induced apoptosis is reversed by the administration of a caspase inhibitor, suggesting that caspases are involved in vorinostat-induced apoptosis. Importantly, the effects on cell growth and death were not as pronounced in the normal breast epithelial MCF-10 cells or fibroblasts, suggesting a therapeutic window (7). Vorinostat also induced morphological and other changes consistent with differentiation in tumor cells with different properties, including estrogen receptor (ER)-negative, HER-2/neu-amplified and EGFR-amplified cell lines (8). Of note, the effects of vorinostat on the cells were reversible upon drug discontinuation, supporting the need for chronic administration.
Together, these preclinical studies suggested that HDAC inhibitors may have anti-tumor activity in breast cancer, but the effects of single agent vorinostat on human breast tumors were unknown. We initiated a prospective clinical study to evaluate the safety, tolerability, and biomarker modulation associated with short term administration of vorinostat to women with primary breast cancer prior to definitive breast surgery or other primary treatment. A non-randomized control cohort was also recruited to understand the variation in biomarker marker expression in the same tumor over time in the absence of any intervention. The results can be used to design future studies of the combination of vorinostat and other standard or novel agents.
Materials and Methods
Patients
Women, age 18 years or older, awaiting definitive surgery or preoperative therapy for a histologically-confirmed invasive breast cancer of clinical tumor size ≥ 1 cm were eligible for the study. Additional inclusion criteria included Eastern Cooperative Oncology Group performance status 0–2 and adequate blood counts and organ function, including leukocytes ≥3,000/mm3, absolute neutrophil count ≥1,500/mm3, platelets ≥100,000/mm3, bilirubin within normal limits, AST/ALT within 2.5 times the upper limit of normal and PT ≤14 seconds. Major exclusion criteria included hormone contraceptive and replacement therapy use within 30 days of diagnostic biopsy (vaginal preparations were allowed), prior or concomitant treatment for the current cancer, history of prior irradiation to the involved breast, uncontrolled intercurrent illness that could limit compliance, pregnancy, or known HIV-positive status due to the potential for drug interaction between vorinostat and antiretroviral therapy. A non-randomized control cohort of women meeting the same inclusion criteria who were unwilling to receive vorinostat but willing to donate tissue for the study analyses was also assembled.
Study Design
The study, including all amendments and revisions, was approved by CTEP and by the Johns Hopkins Institutional Review Board. An Investigational New Drug (IND) application was filed and held by CTEP. Women enrolled from two sites, Johns Hopkins Medical Institutes and Anne Arundel Medical Center. Informed consent was obtained from all participants in the vorinostat and control groups. Women in the vorinostat group were scheduled to receive 6 doses of oral vorinostat at 300 mg twice daily (bid), with the last dose administered by study personnel approximately 2 hours before the scheduled breast surgery (or biopsy). No additional systemic therapy was allowed between pre- and post- treatment biopsies. The schedule was selected based on the phase I study in solid malignancies recommending a maximum tolerated dose of vorinostat 300 mg bid for 3 out of 7 days (4). Pre- and post-vorinostat samples were evaluated for candidate biomarkers that may predict response to vorinostat. Women who declined vorinostat, but agreed to donate tissues for biomarker assessment, signed a separate informed consent and were enrolled as controls.
Because vorinostat’s dose limiting toxicities include anorexia, dehydration, diarrhea, and fatigue, patients were instructed to maintain adequate fluid and food intake. Patients who suffered grade 2 or greater dehydration, diarrhea, or anorexia were evaluated by a member of the study staff and treated appropriately. To assess for toxicity, a study team member contacted each treated participant following the second or third anticipated vorinostat dose. Treatment-related adverse events (AEs) were reported utilizing NCI Common Terminology Criteria for Adverse Events (CTCAE) version 3.0.
On the day of tissue collection, study personnel assessed vital signs and toxicities, and collected study blood samples. Laboratory tests collected following the last dose to ensure safety included a complete blood count, chemistry panel, and PT/PTT. The subjects were also seen during the post-operative clinic visit to assess toxicities, obtain vital signs, and recheck any previous abnormal values. Subjects were contacted approximately 30 days following the final dose of vorinostat to monitor for adverse events and to evaluate whether postoperative or other primary treatment complications may have resulted from the study drug.
Biomarker Analysis
Paraffin-embedded tissue was obtained from the diagnostic biopsy and from the definitive surgical specimens, and subjected to biomarker analysis. Pre-specified markers included expression, methylation, and histone acetylation of candidate genes.
We performed immunohistochemistry (IHC) for Ki-67 and cleaved caspase-3 using commercially available monoclonal antibodies under the direction of the study pathologist (EG) who was blinded to patient data during analyses. We prepared slides using Target Retrieval Solution S1699 (DAKO, Carpinteria, CA) and incubated slides with antibodies to Ki-67 (DAKO; 1:100 dilution) or cleaved caspase 3 (Cell Signaling, Danvers, MA; 1:200 dilution), followed by incubation with secondary antibody and development using the DAKO LASB/HRP system according to manufacturer’s instructions. Normal lymph node tissues were also stained and examined for expected staining patterns as controls (positive and negative) for both antibodies. At least 100 cells were counted from at least two separate fields of tumor (200 cells total) for samples to be evaluable. Percent positive cells were calculated by counting and dividing the numbers of apoptotic cells or cells labeling with Ki-67 by the total number of cells scored.
Formalin-fixed paraffin-embedded tumor blocks or slides containing at least 30 μm of tissue were sent to Genomic Health, Inc. for reverse transcription polymerase chain reaction (RT-PCR) analysis. Briefly, RNA was extracted (MasterPure™ kit, EpicentreTechnologies, Inc. (Madison, WI), total RNA content was measured and the absence of DNA contamination was verified by a quantitative TaqMan® PCR assay for β-actin DNA, which includes both positive and negative controls. Gene expression profiling was performed according to standardized operating procedures for quantitative RT-PCR for the Oncotype DX® 21-gene assay (9). Reference-normalized expression measurements of 16 individual cancer-related genes are expressed on the log2 scale and typically range from 2 to 15, where each 1-unit increase reflects approximately a two-fold increase in RNA (9).
Peripheral blood mononuclear cells (PBMC) were isolated from blood samples (10–30 ml collected in heparinized tubes) and frozen (cell pellet) for future extraction. PBMCs were to be isolated by centrifugion and nuclei from mononuclear cells isolated in lysis buffer, with histones then isolated, as previously described (10). Quantitative multiplex-methylation-specific PCR (QM-MSP) was used to evaluate candidate gene methylation. Details of this method and primer sequences have been reported previously (11, 12).
Pharmacokinetics
Blood samples (5 ml collected in serum tubes) for vorinostat concentrations were collected before dosing and on the day of tissue collection. Initially, samples were obtained as a trough (minimal concentration) and later switched to the presumed maximal concentration time (approximately 30 minutes after dose administration). Samples were allowed to clot at 4C for 20 to 30 minutes, and then centrifuged at 2,000 × g for 15 minutes at 4°C. The resulting serum was transferred to polypropylene cryotubes and stored at −70°C until analyzed for vorinostat concentrations over the range of 3 to 1000 ng/mL with a validated LC-MS/MS assay (13).
Statistical Consideration
The primary study objectives were to evaluate the safety and tolerability of three days of oral vorinostat 300 mg bid in women with primary breast cancer before definitive breast surgery, or other primary treatment, and to evaluate baseline and change in proliferation and apoptosis by IHC in pre- and post-treatment tumor specimens in women who received vorinostat compared to untreated controls. Exploratory objectives included baseline and change in gene methylation silencing and expression of candidate genes in vorinostat-treated women.
Wilcoxon signed rank tests were used to compare pre- and post-treatment pharmacodynamic values within each group, and Wilcoxon rank-sum tests were used to compare the differences (post – pre) between groups. Exact two-sided p-values were calculated. Since this analysis is primarily exploratory, adjustments for multiple comparisons were not made. Pharmacokinetic parameters were summarized using descriptive statistics. Vorinostat concentrations were correlated with the percent change in each biomarker using the Pearson’s correlation coefficient. The a priori level of significance was P<0.05.
For each methylated gene ; the cumulative methylation index (CMI) was reported as the sum of all %M for all genes. To compare CMI between pre- and post-treatment samples, Wilcoxon signed rank tests (two-tailed) were performed using GraphPad Prism version 5.04 for Windows, GraphPad Software, San Diego California USA, www.graphpad.com.
Results
Patient Characteristics
From March 2006 to October 2008, 25 women enrolled in the study and received at least one dose of vorinostat, and 29 additional women enrolled as untreated controls (Figure 1). Of the women receiving vorinostat, 22 received all 6 doses, and 3 women received 1, 4, and 5 doses, respectively (Figure 1). Twenty-four vorinostat-treated women underwent surgery or a biopsy as scheduled and surgery was delayed for one patient for reasons unrelated to the study. Patient characteristics are summarized in Table 1. All 29 enrolled controls proceeded to surgery as planned, however, matched tissues from diagnostic biopsy and definitive surgical specimen were available for biomarker studies from 25 women who are included in the analysis. Four additional samples were not released for research purposes due to a final tumor pathological size <1 cm.
Figure 1.

Study Enrollment and Samples Evaluable for Biomarker Analysis.
*Demographics comparable to the entire group
IHC: immunohistochemistry, N: number, NA: not applicable, QM-MSP: Quantitative multiplex-methylation-specific PCR, RT-PCR: Reverse transcription polymerase chain reaction
Table 1.
Patient Characteristics
| Characteristic | Vorinostat (N=25) | Controls (N=25)* |
|---|---|---|
|
| ||
| Median Age (range) | 55 (34–71) | 52 (34–79) |
|
| ||
| Median Tumor size (range) # | 2 cm (1.1–5.3) | 1.9 cm (1.0–4.8) |
|
| ||
| Tumor size # | ||
| 0–2 | 13 (52%) | 13 (52%) |
| >2–5 | 11 (44%) | 12 (48%) |
| >5 | 1 (4%) | 0 (0) |
|
| ||
| Nodal Status | ||
| Node-negative | 11 (44%) | 14 (56%) |
| Node-positive | 14 (56%) | 11 (44%) |
|
| ||
| ER/PR Status | ||
| ER+/PR+ | 14 (56%) | 15 (60%) |
| ER+/PR− | 6 (24%) | 0 (0%) |
| ER−/PR− | 5 (20%) | 10 (40%) |
|
| ||
| HER2 Status | ||
| HER2+ | 3 (12%) | 4 (16%) |
| HER2− | 21 (84%) | 20 (80%) |
| Unknown | 1 (4%) | 1 (4%) |
|
| ||
| Triple Negative | ||
| Yes | 4 (16%) | 7 (28%) |
| No | 20 (80%) | 17 (68%) |
| Unknown | 1 (4%) | 1 (4%) |
|
| ||
| Type of Specimen Collected | ||
| Lumpectomy | 17 (68%) | 13 (52%) |
| Mastectomy | 5 (20%) | 12 (48%) |
| Core biopsy | 2 (8%) | 0 (0) |
| None | 1 (4%) | 0 (0) |
ER- estrogen receptor, PR- progesterone receptor.
29 women enrolled but only 25 had evaluable matched samples
Pathological tumor size was recorded in 23 women, and clinical measurements on 2 women undergoing neoadjuvant chemotherapy.
Safety and Tolerability
Oral vorinostat was well-tolerated. Grade 1 non-hematological toxicities included diarrhea (28%), fatigue (16%), taste changes (16%), anorexia (12%), nausea (16%), and headaches (4%). Grade 1 leukopenia was seen in 24% of patients but there were no significant changes in hemoglobin, platelet count, electrolytes, renal and liver function laboratory tests, or coagulation factors. All surgical procedures were performed without study-related delays, change in surgical plans, or complications. Likewise, two women were able to initiate recommended preoperative chemotherapy without delays.
Biomarker Analysis
In the vorinostat-treated group, the last dose was administered by a study team member prior to tissue collection in 23 women. Of those, subsequent tissue collection time was not recorded in one case. The average time to tissue sample collection was 4.2 hours (1.2–7.9 hours). One additional woman had a non study-related delay in surgery, where no tissue was collected, and another had discontinued the study drug following the first dose and the collection was delayed (time to surgery 62.2 hours).
Matched tumor tissue from baseline and surgery was successfully collected for biomarker analysis from 24 treated women and 25 controls and subjected to IHC and RT-PCR analysis. However not all matched specimens were evaluable for prespecified marker analysis as described below. Only two women agreed to an optional pre-vorinostat breast tumor biopsy that was placed in RNAlater® Solution. Most women declined the optional biopsy due to the required intensity of clinical and study procedures in the presurgical period, while others declined due to concerns of the associated discomfort for a non-clinically-indicated biopsy.
Matched samples for Ki-67 by IHC were available from 22 (92%) treated and from 15 (60%) controls. There was no change in Ki-67 by IHC compared to baseline in the treated or untreated patients, and there were no differences between groups in the changes from baseline (P=0.42, Figure 2A). Likewise, there was no differences in cleaved caspase-3 by IHC within or between groups (P=0.50, Figure 2B); however, matched samples from only 19 (71%) treated and 12 (48%) controls were evaluable for analysis of the marker. Fewer samples were evaluable for the caspase-3 analysis because of a low total number of cells which did not allow for a reliable reading.
Figure 2.
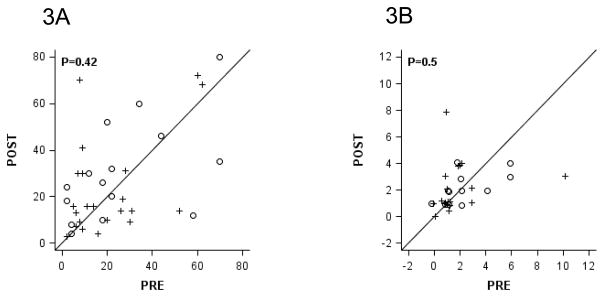
Pre- and Post- Vorinostat Values of Gene Expression by Immunohistochemistry.
Ki67 (A) and Cleaved Caspase 3 (B) in Vorinostat-Treated Women (+) versus Control (○). Exact two-sided p-values are from the 2-sample Wilcoxon rank-sum test.
Candidate gene mRNA expression was assessed by the Oncotype DX® assay. Only 14 (58%) and 11 (44%) paired samples from treated and controls, respectively, were available for the Oncotype DX® assay. The samples evaluable for this assay were from participants with similar characteristics to the entire cohort (data not shown). The smaller proportion of evaluable matched samples for the assay was due primarily to the requirement for several consecutive slides which were often not available from the initial core biopsy. Despite the small sample size, we observed statistically significant greater reductions in the mRNA expression of the proliferation-associated genes, Ki-67 (Figure 3A: P=0.003), STK15 (Figure 3B: P=0.005), and cyclin B1 (Figure 3D: P=0.03), in the samples from vorinostat-treated women as compared to samples from untreated women. Directionally consistent patterns were observed in the other two proliferation-associated genes included in the assay (Figure 3C: MYBL2, P=0.20, and Figure 3E: survivin, P=0.11). Overall, a statistically significant larger reduction was observed in the proliferation axis, which includes the 5 proliferation-associated genes (Figure 3F: P=0.01), in tissues derived from vorinostat-treated women when compared to control women.
Figure 3.

Pre- and Post- Vorinostat Values of Proliferation-Associated Gene Expression using the Oncotype DX® 21-gene assay.
Vorinostat-Treated Women (+, N=14) versus Control (○, N=11) for Ki-67 (A), STK15 (B), MYBL2 (C), Cyclin B1 (D), Survivin (E), and the Proliferation Axis (F). Values are on a log2 scale. Exact two-sided p-values are based upon the 2-sample Wilcoxon rank-sum test.
For 25 evaluable matched sample sets for both IHC and RT-PCR, 76% had ER positive tumors by both assays (100% concordance). There was no statistically significant difference in ER expression between the treated and untreated groups (Figure 4A). The differences in expression of progesterone receptor (PR) that were observed (Figure 4B) were due to an increased expression in the untreated group which may be due to chance and is also reflected in the change in ER-axis which includes ER and PR (Figure 4C). There were no consistent changes among the pairs for genes in the Invasion or HER-2 groups included in the Oncotype DX® assay (Supplementary Figure 1).
Figure 4.
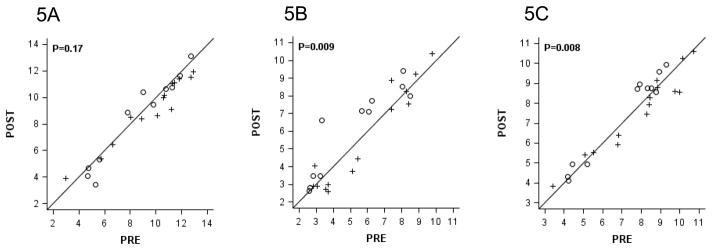
Pre- and Post- Vorinostat Values of Estrogen Related Gene Expression Using the Oncotype DX® 21-gene Assay.
Vorinostat-Treated Women (+, N=14) versus Control (○, N=11) for ER (A), PR (B), and the ER Axis (C).
Values are on a log2 scale. Exact two-sided p-values are from the 2-sample Wilcoxon rank-sum test.
Changes in methylation of candidate genes as assessed by QM-MSP were compared in 19 evaluable matched pre- and post-vorinostat samples. Pre- and post-treatment samples were associated with very similar methylation profiles (Figure 5). As hypothesized, no change in cumulative methylation index was seen (P=0.24, Wilcoxon signed rank test) after vorinostat treatment.
Figure 5.
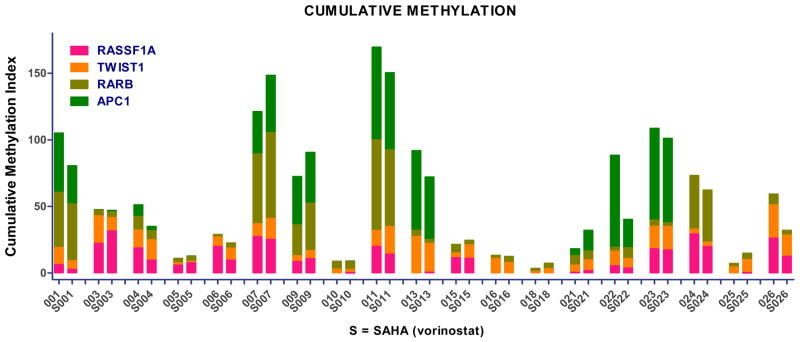
Baseline and Change in Cumulative Methylation Index in Vorinostat-Treated Women N=19, P=0.24).
Pharmacokinetics
Samples for trough vorinostat concentration were drawn immediately prior to the last dose in the first 13 patients. Vorinostat trough concentrations were 71.7 ± 90.7 ng/mL in 7 of the 13 patients, and below the limits of quantification in the rest of the patients. Therefore, the sampling schema was altered to capture the maximal exposure (Cmax) in the remaining 11 patients (i.e., samples drawn 30–60 minutes following the last dose of vorinostat). Vorinostat Cmax were detectable in all patients (234.0 ± 180.1 ng/mL). No association was observed between vorinostat concentrations and biomarker expression (P>0.05). While PBMC were isolated from blood samples and cell pellets frozen, we were unfortunately not able to successfully dissolve the pellet in lysis buffer and the samples were not subjected to histone acetylation analysis.
Discussion
Breast cancer is a consequence of the accumulation of multiple genetic and epigenetic alterations. We hypothesized that epigenetic modifications which, unlike genetic alterations, are potentially reversible and represent valid clinical targets that can be exploited in combination strategies. To set the foundation for such combination studies, we performed a “window of opportunity” study of preoperative vorinostat to provide evidence for single agent activity. In this study, short term vorinostat was safe, adverse events were modest, and were not associated with delays in surgical plans or outcomes. Although only 44–92% of samples were available for biomarker analysis, we observed a significant reduction in expression of proliferation-associated genes using the RT-PCR assay, suggesting that vorinostat is biologically active.
The main strengths of our study include the enrollment of women who have not received prior treatment for their current cancer and the collection of all samples from treated individuals at a uniform time, 2 hours following administration of an oral agent with a short half-life, though the collection time may not have been optimal to capture the maximal pharmacodynamic effect or account for intra-individual differences in vorinostat pharmacokinetics. Another important strength of our trial is the inclusion of a parallel untreated control group to allow evaluation of biomarker expression from the same tumor at two different times.
The main limitation of the trial is the unexpectedly low proportion of matched evaluable samples available for the biomarkers studied. The goal of obtaining usable matched specimens from ≥80% of participants was not achieved for all pre-specified biomarkers. Our results suggest that women with tumors ≥1.5–2 cm may be better candidates for window of opportunity studies and our recent studies have been amended accordingly. As has been recommended by others, a randomized placebo-controlled trial should be considered when conducting window trials, although the design is often unappealing for patients (14). It is possible that we would have had a higher proportion of evaluable control samples should the study have been randomized. We also recommend a close collaboration between members of the multidisciplinary team including oncologists, surgeons, and pathologists to allow optimal presentation of the study objectives and plan.
In addition, the large number of parameters evaluated increases the potential for false discoveries. We selected biomarkers based on our preclinical studies and the breast cancer clinical trial field to guide selection of endpoints (e.g. Ki-67). Nonetheless the biomarker analyses should be regarded as exploratory and will be used to design studies of combinations of vorinostat with other agents.
Compared to controls, samples from vorinostat-treated women showed a significant reduction in proliferation-related genes despite the short treatment duration. Thus we achieved our goal of demonstrating evidence of biological activity with an HDAC inhibitor in newly diagnosed untreated breast cancers. While we have not observed a reduction in Ki-67 by IHC, previous studies have demonstrated poor correlation between IHC and RT-PCR determination of the marker (15). There were no significant changes in expression of other candidate genes in the Oncotype DX assay including ER or HER-2 although the number of patients with ER-negative or HER-2 positive tumors was low because many of these women with these tumor characteristics presented with Stage II-III cancer and received preoperative chemotherapy. It is possible that novel agents may also be tested in this setting, but, because effective systemic treatments are available and required it is difficult to perform a long term study of a novel drug administered as a single agent.
Other potential confounding factors may be that the dose or schedule chosen was insufficient to achieve adequate exposure. The selected candidate markers may were not have been optimal. For example, others utilize a 400 mg daily vorinostat dose for 2 or 3 consecutive weeks. In vitro studies have demonstrated that HDAC inhibitors may lead to either up- or down-regulation of ER, depending on the tumor subtype, and down-regulation of EGFR or HER-2. Since the antitumor effects are likely reversible upon drug discontinuation, our goal was not to develop the drug for single agent administration but to confirm and define its effects on breast tumors to help design future studies in which the agent is administered in combination with other drugs.
Our laboratory data suggest that HDAC inhibitors do not result in demethylation of methylated gene promoters of candidate genes, even genes like ER whose mRNA expression is reinduced in ER-negative human breast cancer cell lines (16). We therefore hypothesized that HDAC inhibition will not be associated with reversal of gene methylation in candidate genes in human tumors. Our results show concordance of candidate gene promoter methylation between the pre- and post-vorinostat samples, yet lack of reversal of methylation with vorinostat exposure, which may be due to either biological reality or inadequate vorinostat exposure. Indeed, the vorinostat exposure observed in the current study ranged from trough concentrations that were undetectable to 72 ng/mL (~0.3 μM) and Cmax values of 234 ng/mL (~0.9 μM). These concentrations were lower than the 1.25–3 μM typically utilized preclinically to achieve gene re-expression (7). In the context of this limitation, we did not observe correlations between vorinostat concentrations and changes in gene expression or other biomarkers.
Substantial preclinical data demonstrate the activity of vorinostat or other HDAC inhibitors with endocrine interventions, cytotoxic agents, anti HER2 agents, or novel agents including other epigenetic modifiers and have been reviewed recently (17). Importantly, both the efficacy of HDAC inhibitors and re-expression of candidate genes are especially enhanced when combined with other epigenetic modifiers such as DNA methyltransferase inhibitors (5-azacytidine or decitabine) (19, 20). Clinical trials are already underway based on these preclinical data from several labs including our own. In a traditional phase II trial in women with refractory metastatic breast cancer, administration of oral vorinostat 200 mg bid for 14 days of each 21 day cycle was not associated with complete or partial responses in the 14 patients who enrolled in the first stage of the study, leading to trial closure. However, 4 patients (29%) had stable disease with times to progression of 4, 8, 9, and 14 months (21). Munster et al completed a phase II trial of vorinostat and tamoxifen in women with ER-positive metastatic breast cancer who have progressed on prior endocrine therapy. The investigators reported an objective response rate of 19% and a clinical benefit rate of 40%, including in patients who received prior tamoxifen (22). We used our preclinical and clinical results to initiate a study of daily vorinostat 400 mg and tamoxifen 20 mg for 14 days in the preoperative window setting. Two women with ER-positive breast cancer enrolled in the study and received the combination without significant side effects or surgical delays (data not shown) before difficulty in drug supply and poor accrual led to early trial closure. In an ongoing trial, we are evaluating the combination of entinostat and azacitidine in women with hormone resistant or triple negative breast cancer; women who develop progressive disease on this regimen may enroll in an optional continuation cohort, where either tamoxifen or an aromatase inhibitor is added to the epigenetic therapy to further test the possibility of epigenetic sensitization to other treatments (24).
In summary, short term administration of the oral HDAC inhibitor vorinostat to women with early stage breast cancer is well tolerated and associated with reduction in mRNA expression of proliferation index genes in primary breast cancer tissue. This observation in human tissues recapitulates results from extensive preclinical studies. This successful translation of preclinical data from lab to clinic supports ongoing efforts to move promising combination therapies such as HDAC inhibitor plus a demethylating agent or HDAC inhibitor plus chemotherapy agents into clinical investigation in the hope that such approaches may enhance cell kill, improve response rates, and improve long-term outcomes.
Supplementary Material
Statement of Translational Relevance.
Epigenetic alterations are common in multiple genes in breast cancer and may predict inferior prognosis and response to standard therapies. Agents that modulate epigenetic alterations including demethylating agents and histone deacetylase (HDAC) inhibitors are currently available. Preclinical studies in breast cancer model systems suggest that administration of HDAC inhibitors alone or combined with hormonal agents, cytotoxics, or other biologics is associated with significant anti-tumor activity. In human breast cancer cell lines, the HDAC inhibitor vorinostat induces growth arrest, resulting in differentiation or apoptosis. Here we demonstrate that short term oral vorinostat administered to women with primary breast cancer is associated with significant reduction in expression of proliferation-related genes. Our results confirm preclinical and clinical data suggesting that vorinostat may have single agent activity in breast cancer. Since these effects are modest and likely reversible upon drug discontinuation, agents that target epigenetic alterations should be studied in combination with other drugs.
Acknowledgments
We thank Drs. Pedram Argani, Ting Bao, Michael Carducci, Lorraine Tafra, and Stanley Watkins, Ms. Nina Kouprina, as well as Ms. Lillie Shockney and the staff of the Avon Breast Center for help in study design and conduct. We thank Dr. Steven Shak for collaboration and Roberto Bugarini for biostatistical support, and Diana Cherbavaz, PhD for scientific support in Genomic Health’s development laboratory. We thank the late Dr. Merrill Egorin for providing analytical support for vorinostat concentrations.
Financial Support: Support for this study was provided by the Cancer Therapy Evaluation Program (CTEP), National Institutes of Health (NIH) U01 CA 70095 (VS and MR) and SAIC (P6914 to VS), P30CA47904 (to M. Egorin) and U01-CA099168 (to M. Egorin), American Society of Clinical Oncology Advanced Clinical Research Award (to VS) and Young Investigator Award (to MH), and Specialized Program Of Research Excellence in Breast Cancer (P50 CA88843 to NED and SS) and by the Avon Foundation. Vorinostat for the vorinostat and tamoxifen study was supplied by Merck. The Oncotype DX® 21-gene assays were performed by Genomic Health, Inc.
Footnotes
Clinical Trial Registration: The description of the clinical study design can be found on http://www.clinicaltrials.gov (NCT00262834).
Conflicts of interest: VS received a research grant (investigator-initiated) from Merck. Calvin Chao and Carl Yoshizawa are employees of Genomic Health, Inc.
Bibliography
- 1.Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349(21):2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 2.Stearns V, Zhou Q, Davidson NE. Epigenetic regulation as a new target for breast cancer therapy. Cancer Invest. 2007;25(8):659–65. doi: 10.1080/07357900701719234. [DOI] [PubMed] [Google Scholar]
- 3.Kelly WK, Richon VM, O’Connor O, Curley T, MacGregor-Curtelli B, Tong W, et al. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res. 2003;9(10 Pt 1):3578–88. [PubMed] [Google Scholar]
- 4.Kelly WK, O’Connor OA, Krug LM, Chiao JH, Heaney M, Curley T, et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol. 2005;23(17):3923–31. doi: 10.1200/JCO.2005.14.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen LA, Marks PA, Rifkind RA, Amin S, Desai D, Pittman B, et al. Suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, suppresses the growth of carcinogen-induced mammary tumors. Anticancer Res. 2002;22(3):1497–504. [PubMed] [Google Scholar]
- 6.Huang L, Pardee AB. Suberoylanilide hydroxamic acid as a potential therapeutic agent for human breast cancer treatment. Mol Med. 2000;6(10):849–66. [PMC free article] [PubMed] [Google Scholar]
- 7.Munster PN, Troso-Sandoval T, Rosen N, Rifkind R, Marks PA, Richon VM. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 2001;61(23):8492–7. [PubMed] [Google Scholar]
- 8.Zhou Q, Chaerkady R, Shaw PG, Kensler TW, Pandey A, Davidson NE. Screening for therapeutic targets of vorinostat by SILAC-based proteomic analysis in human breast cancer cells. Proteomics. 2010;10(5):1029–39. doi: 10.1002/pmic.200900602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351(27):2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida M, Kijima M, Akita M, Beppu T. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265(28):17174–9. [PubMed] [Google Scholar]
- 11.Fackler MJ, McVeigh M, Mehrotra J, Blum MA, Lange J, Lapides A, et al. Quantitative multiplex methylation-specific PCR assay for the detection of promoter hypermethylation in multiple genes in breast cancer. Cancer Res. 2004;64(13):4442–52. doi: 10.1158/0008-5472.CAN-03-3341. [DOI] [PubMed] [Google Scholar]
- 12.Swift-Scanlan T, Blackford A, Argani P, Sukumar S, Fackler MJ. Two-color quantitative multiplex methylation-specific PCR. Biotechniques. 2006;40(2):210–9. doi: 10.2144/000112097. [DOI] [PubMed] [Google Scholar]
- 13.Parise RA, Holleran JL, Beumer JH, Ramalingam S, Egorin MJ. A liquid chromatography-electrospray ionization tandem mass spectrometric assay for quantitation of the histone deacetylase inhibitor, vorinostat (suberoylanilide hydroxamicacid, SAHA), and its metabolites in human serum. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;840(2):108–15. doi: 10.1016/j.jchromb.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 14.Ratain MJ. Bar the windows but open the door to randomization. J Clin Oncol. 2010;28(19):3104–6. doi: 10.1200/JCO.2010.29.3787. [DOI] [PubMed] [Google Scholar]
- 15.Cobleigh MA, Tabesh B, Bitterman P, Baker J, Cronin M, Liu ML, et al. Tumor gene expression and prognosis in breast cancer patients with 10 or more positive lymph nodes. Clin Cancer Res. 2005;11(24 Pt 1):8623–31. doi: 10.1158/1078-0432.CCR-05-0735. [DOI] [PubMed] [Google Scholar]
- 16.Sharma D, Saxena NK, Davidson NE, Vertino PM. Restoration of tamoxifen sensitivity in estrogen receptor-negative breast cancer cells: tamoxifen-bound reactivated ER recruits distinctive corepressor complexes. Cancer Res. 2006;66(12):6370–8. doi: 10.1158/0008-5472.CAN-06-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Connolly R, Stearns V. Epigenetics as a therapeutic target in breast cancer. J Mammary Gland Biol Neoplasia. 2012;17(3–4):191–204. doi: 10.1007/s10911-012-9263-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim MS, Blake M, Baek JH, Kohlhagen G, Pommier Y, Carrier F. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63(21):7291–300. [PubMed] [Google Scholar]
- 19.Zhu WG, Otterson GA. The interaction of histone deacetylase inhibitors and DNA methyltransferase inhibitors in the treatment of human cancer cells. Curr Med Chem Anticancer Agents. 2003;3(3):187–99. doi: 10.2174/1568011033482440. [DOI] [PubMed] [Google Scholar]
- 20.Yang X, Phillips DL, Ferguson AT, Nelson WG, Herman JG, Davidson NE. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2001;61(19):7025–9. [PubMed] [Google Scholar]
- 21.Luu TH, Morgan RJ, Leong L, Lim D, McNamara M, Portnow J, et al. A phase II trial of vorinostat (suberoylanilide hydroxamic acid) in metastatic breast cancer: a California Cancer Consortium study. Clin Cancer Res. 2008;14(21):7138–42. doi: 10.1158/1078-0432.CCR-08-0122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munster PN, Thurn KT, Thomas S, Raha P, Lacevic M, Miller A, et al. A phase II study of the histone deacetylase inhibitor vorinostat combined with tamoxifen for the treatment of patients with hormone therapy-resistant breast cancer. Br J Cancer. 2011;104(12):1828–35. doi: 10.1038/bjc.2011.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yardley DA, Ismail-Khan RPK. Results of ENCORE 301, a randomized, phase II, double-blind, placebo-controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor-positive (ER+) breast cancer progressing on a nonsteroidal aromatase inhibitor (AI) J Clin Oncol. 2011;29(suppl 27):abstr 268. doi: 10.1200/JCO.2012.43.7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Connolly RM, Jankowitz RC, Andreopoulou E, Allred JB, Jeter SC, Zorzi J, et al. A phase 2 study investigating the safety, efficacy and surrogate biomarkers of response of 5-azacitidine (5-AZA) and entinostat (MS-275) in patients with advanced breast cancer. Cancer Research. 2011;7(24) Supplement 3, OT-01-6: A. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.