

Abstract
The histone methyltransferase Mixed Lineage Leukemia (MLL) is essential to maintain hematopoietic stem cells and is a leukemia protooncogene. Although clustered homeobox genes are well-characterized targets of MLL and MLL fusion oncoproteins, the range of Mll-regulated genes in normal hematopoietic cells remains unknown. Here, we identify and characterize part of the Mll-dependent transcriptional network in hematopoietic stem cells with an integrated approach by using conditional loss-of-function models, genomewide expression analyses, chromatin immunoprecipitation, and functional rescue assays. The Mll-dependent transcriptional network extends well beyond the previously appreciated Hox targets, is comprised of many characterized regulators of self-renewal, and contains target genes that are both dependent and independent of the MLL cofactor, Menin. Interestingly, PR-domain containing 16 emerged as a target gene that is uniquely effective at partially rescuing Mll-deficient hematopoietic stem and progenitor cells. This work highlights the tissue-specific nature of regulatory networks under the control of MLL/Trithorax family members and provides insight into the distinctions between the participation of MLL in normal hematopoiesis and in leukemia.
Keywords: proliferation, HSC, epigenetics
Epigenetic regulation is an important mechanism by which gene expression fidelity is maintained during development. The trithorax-group (trx-G) and Polycomb-group (Pc-G) genes encode epigenetic factors that act as opposing regulators of clustered homeobox (Hox) gene expression and of axial patterning in most metazoa (1, 2). In addition, numerous studies implicate Pc-G and trx-G homologs in mammals in the maintenance of broader gene expression programs in embryonic and tissue stem cells and in cancer (1, 2). Because of the reversible nature of epigenetic lesions in cancer, targeting oncogenes and tumor supressors that use epigenetic mechanisms is a promising an approach for targeted therapy (3).
The human protooncogene Mixed Lineage Leukemia (MLL) was the first mammalian trx homolog identified because of its characteristic rearrangement in ∼70% of infant leukemia. Rearrangement of the human MLL gene by chromosomal translocation also occurs at a lower frequency in childhood acute lymphoblastic leukemia (ALL), acute myelogenous leukemia (AML), and treatment-related and de novo AML in adults (4, 5). Most translocations produce MLL fusion oncoproteins that retain the chromatin-targeting N terminus and acquire a transcriptional effector domain from the C-terminal partner. Partner proteins frequently recruit protein complexes that result in increased histone H3 lysine 79 dimethylation at MLL-fusion targets, overexpression of these target genes, and leukemic transformation (6). Because many of the chromatin-targeting motifs are shared between oncogenic MLL fusions and wild-type MLL, targeting of MLL-fusion oncoproteins will also require a thorough understanding of normal MLL-dependent regulatory pathways.
Wild-type MLL exists in cells as part of a large multiprotein, chromatin-associated complex that contains chromatin remodeling and histone acetylation/methylation activities (7, 8). MLL itself is thought to regulate genes in part through a highly conserved histone methyltransferase motif, the Su(var)3-9, Enhancer of Zeste, and Trithorax (SET) domain. MLL, like Trithorax, maintains precise domains of Hox gene expression during embryo development (9, 10). In addition, MLL has been shown to regulate other tissue-specific processes in immune, hematopoietic, vascular, and neural cell types (11–14). Germ-line disruption of Mll is generally embryonic lethal with multiple developmental defects (9, 15–17); however, conditional deletion of Mll in specific cell types revealed unique functions. For example, hematopoietic-specific deletion of Mll demonstrated that it is essential for maintaining hematopoietic stem and progenitor cells (HSPCs), but dispensable for lineage-committed precursors (13, 18, 19). The breadth of target genes regulated by MLL in specific tissues is largely unknown, although Hox genes are consistently down-regulated in many Mll-deficient cell types (9, 13, 14).
In this study, we investigate the molecular circuitry underlying the critical role of Mll in maintaining hematopoiesis as a means to understand trx-G function in normal and pathologic gene regulation. We used inducible loss-of-function models to identify hematopoietic stem cells (HSC)-specific MLL-regulated genes and delineated a network of transcriptional regulators that are direct transcriptional targets of MLL. We then tested reexpression of a subset of these genes in Mll-deficient hematopoietic cells to determine the epistatic relationships among transcriptional targets, to identify cross-regulatory relationships, and assess their individual ability to restore function in Mll-deficient cells. These studies reveal a coherent MLL pathway that coordinates self-renewal, proliferation, and lineage-specific gene expression fidelity in HSCs. Furthermore, this work distinguishes the MLL-dependent transcriptional network from that controlled by MLL fusion oncoproteins in leukemia.
Results
Short-Term Consequences of Mll Deletion in HSCs.
To identify Mll-dependent genes involved in maintaining HSCs, we analyzed differentially expressed transcripts after Mll deletion. Lineage-negative, stem cell antigen-1 (Sca-1)+, c-Kit+, CD48− (LSK/CD48neg) HSC-enriched cells from the bone marrow (BM) of polyinosinic:polycytidylic acid (pI:pC)-injected control MllF/F or Mx1-cre;MllF/F animals were purified 6 d after the first pI:pC injection, the optimal timing for Mll deletion, cell yield, and down-regulation of homeobox protein a9 (Hoxa9), a bona fide Mll target gene (13). Assessment of normalized gene expression differences between control and Mll-deficient LSK/CD48neg cells revealed 1,935 differentially expressed genes using Significance Analysis of Microarrays (which does not impose a fold cutoff; Fig. 1) (20). Functional classification of genes differentially expressed in Mll-deficient HSCs compared with controls resulted in three global observations: (i) more genes were up-regulated than down-regulated, (ii) a subset of erythroid-specific genes were up-regulated, and (iii) the largest category of annotated down-regulated genes was comprised of transcriptional regulators.
Fig. 1.

Identification of Mll-regulated genes in HSCs. General overview of genes up-regulated (A) or down-regulated (B) in Mll-deficient LSK/CD48neg cells compared with controls. Cells were sorted from pI:pC-injected control MllF/F or Mx1-cre;MllF/F animals at day six. Gene Ontology assignments were based on the criteria in Datasets S1 and S2. (C) The top down-regulated transcription factors in Mll-deficient LSK/CD48neg cells listed by fold reduction (see also Dataset S2). (D) RT-qPCR validating down-regulated genes in independent control MllF/F (blue) or Mll-deficient (red) LSK/CD48neg cells, n = 8 animals per genotype; ND, not detected. (E) RT-qPCR validation of transcripts in LSK cells sorted from control ER-cre;MllF/+ (blue) or ER-cre;MllF/F animals (red) cultured for 72 h after initiating Mll deletion. Relative expression levels were determined by normalizing to Gapdh, n = 4 animals per genotype. Error bars represent 95% confidence interval (CI). *P ≤ 0.07, **P ≤ 0.05. ER-cre, estrogen receptorT2 mutant fused to cre recombinase.
Among the up-regulated genes, the largest group corresponds to HSC proliferation and ribosome or mitochondrial biogenesis (Fig. 1A and Dataset S1). Up-regulation of genes involved in ribosome biogenesis reflected the greater proportion of cycling Mll-deficient LSK/CD48neg cells (45% G0 in Mll-deleted cells versus 75% G0 in controls; ref. 13). Ten percent in this category and 17% in the mitochondrial group were also identified in proliferating HSCs (21), (Dataset S1). Thus, many of the up-regulated genes reflect the expected changes based on the proliferation state of Mll-deficient LSK/CD48neg cells. Unexpectedly, 5% of the genes that were up-regulated in Mll-deficient LSK/CD48neg cells encode erythroid-specific proteins including transcriptional regulators such as GATA binding protein 1 (Gata1) and Kruppel-like factor 1 (Klf1), as well as spectrin, Kell protein (Kel), Erythropoietin receptor (EpoR), and hemoglobin biosynthesis genes (Dataset S1). Gene set enrichment analysis (GSEA) also identified a GATA1-induced gene signature and a tendency toward erythroid identity (Fig. S1 A and B). The up-regulation of erythroid genes was validated by using an independent in vitro Mll deletion system, illustrating that the scale of gene up-regulation was consistent with derepression rather than full induction of erythroid genes (Fig. S1 C and D). Furthermore, this derepression was not sufficient to impart erythroid fate as demonstrated by colony assay (Fig. S1E). Derepression of erythroid genes likely occurs through an indirect mechanism, thus we focused on the down-regulated genes as potential MLL effectors in the maintenance of HSCs.
Identifying an Mll-Dependent Transcriptional Network.
Transcriptional regulators comprised the largest single annotated category of down-regulated genes in Mll-deleted LSK/CD48neg cells (Fig. 1B and Dataset S2). Because many of these regulators are highly expressed in HSCs relative to more differentiated cell types (22), we asked whether Mll-deficient HSCs exhibit a global shift in cell fate by assessing the relatedness of our gene expression data to other hematopoietic populations (23, 24). This analysis showed an enrichment of erythroid identity as described earlier, but did not suggest that HSCs were generally differentiated, because HSC and multipotent progenitor signatures were equivalently enriched by GSEA (Fig. S1F). Mll itself (Fig. S1G) and well-characterized MLL targets such as Hoxa9 were down-regulated although the majority of the genes in this category were not previously known to be Mll targets (Fig. 1C). We confirmed the Mll dependence for all annotated transcription factors >2.5-fold down-regulated by quantitative RT-PCR (RT-qPCR) using independently sorted samples from Mx1-cre;MllF/F animals (Fig. 1D), as well as cells in which Mll was deleted in vitro by using 4-hydroxytamoxifen (4-OHT; Fig. 1E). Each inducible knockout model has its characteristic limitations, so to discover genes that were truly Mll-dependent, we only pursued genes down-regulated in both Mx1-cre and ER-cre systems. Of the annotated transcription factors down-regulated >2.5-fold (Fig. 1C), MDS and Evi1 complex locus (Mecom), Prdm16, Pre-B cell leukemia homeobox protein 1 (Pbx1), Eyes absent homolog 1 (Eya1) and Hoxa9 were consistently Mll-dependent (Fig. 1E). Tripartate motif-containing 30b (Trim30b) is not characterized, so we focused on the other five genes for the following studies.
Several of the transcriptional regulators identified above individually play critical roles in HSC homeostasis. For example, the proteins encoded by the Pbx1, Prdm16, and Mecom genes act to restrain HSC proliferation and/or promote self-renewal (25–29), as has been demonstrated for Mll (13, 18). Interestingly, Mecom and Prdm16 were not Mll-dependent in fibroblasts or in Mll knockout embryos overall, despite coexpression of Mll and these genes (Fig. S2).
MLL Binds Directly to the Promoter Regions of a Subset of Mll-Dependent Genes.
Mll and its homolog Trithorax typically act to maintain expression of their direct target genes (30), thus we evaluated the down-regulated transcription factors as potential direct MLL targets. To assess whether MLL acts directly to promote expression of the identified transcriptional regulators, we used a mini-ChIP procedure optimized for 5 × 104 BM cells (31). Based on previous results demonstrating MLL binding near transcription start sites (TSS) in cell lines (32, 33), we assessed MLL binding within 2 kb of the TSS by using 3–5 amplicons per gene. Mll-dependence was similarly observed in the BM lineage-negative (linneg) population and LSK cells (Fig. S3A). Control ChIP experiments demonstrated MLL binding to the Hoxa9 but not Gapdh TSS regions (Fig. S3B). Using linneg BM cells, we observed specific MLL binding around each TSS of the Mecom locus [both Myelodysplastic syndrome 1 (Mds1) and Ecotropic virus integration site 1 (Evi1) promoter regions], as well as the Prdm16, Pbx1, and Eya1 genes (Fig. 2 and Fig. S3 C–G). Interestingly, we did not observe MLL binding to the Early growth response 1 (Egr1) promoter (Fig. 2B and Fig. S3H), consistent with the observation that this gene was not Mll-dependent in both model systems (Fig. 1E). Therefore, we conclude that like Hoxa9, the expression of Mecom, Prdm16, Pbx1, and Eya1 is maintained directly by MLL in normal linneg BM cells.
Fig. 2.

MLL binds directly to the promoter regions of a subset of genes identified by expression array. ChIP results demonstrating specific enrichment at the Mecom locus (Mds1 and Evi1 start sites) and the Prdm16, Pbx1, and Eya1 promoter regions. Anti-MLL C-terminal (black) or control (anti-GAL4, gray) antibodies were used for ChIP, and enrichment was determined by using quantitative PCR assays. Amplicon position is indicated relative to the TSS for each gene. Results using additional primers surrounding the TSS are shown in Fig. S4. Data represents averages ± SEM for two to four PCR replicates and are representative of at least four independent experiments.
Only a Subset of Mll-Dependent Genes Are Affected by Men1 Deletion.
MLL itself does not harbor sequence-specific DNA binding motifs. One important chromatin-targeting mechanism occurs through an N-terminal interaction with Menin and p75/lens epithelium-derived growth factor (LEDGF), thought to be essential for targeting wild-type MLL to promoter regions based on studies using MLL fusion oncoproteins (34). To understand how the MLL complex localizes to its targets in HSCs, we assessed the Menin dependence of Egr1, Hoxa9, Prdm16, Mecom, Pbx1, and Eya1. Consistent with a previous study (35), we found that Hoxa9 expression was reduced in Menin (Men) 1-deficient LSK cells. Interestingly, Mecom and Eya1 were slightly reduced, but the latter was not statistically significant (Fig. 3A). Despite efficient excision of Men1 (Fig. 3B), Prdm16 and Pbx1 levels were not affected (Fig. 3A), suggesting that a subset of HSC-specific Mll-dependent genes do not require Menin. These data demonstrate that the MLL complex differentially requires the Menin chromatin-targeting cofactor to regulate distinct classes of target genes.
Fig. 3.
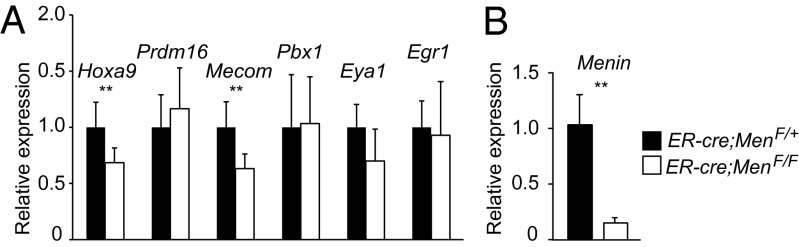
Menin loss affects some but not all MLL targets in LSK cells. (A) RT-qPCR of Mll-regulated genes in LSK cells sorted from control ER-cre;Men1F/+ (black) or ER-cre;Men1F/F cells (white) cultured for 72 h after initiating Menin deletion. Expression levels were normalized to rRNA. (B) Menin transcript levels in LSK cells treated as in A. Error bars represent 95% CI; n = 4–8 animals per genotype. **P ≤ 0.05.
Structure of the Mll-Dependent Transcriptional Network.
We considered that some of the Mll-dependent transcriptional regulators act in interconnected pathways to modulate HSC function. For example, it has been reported that overexpression of Evi1 up-regulates Pbx1 in c-Kit–enriched BM cells (36). To identify potential expression interrelationships and determine whether the identified genes represent a linear or branched pathway downstream of MLL, we overexpressed Hoxa9, Prdm16, Eya1, Pbx1, or Mecom isoforms (Mds1-Evi1 and Evi1) in wild-type or Mll-deficient LSK cells and assessed the effect on other genes in this network 48 h later. Focusing first on the effects of overexpression in wild-type cells, we found that Hoxa9 could increase levels of Prdm16, Evi1 could increase both Prdm16 and Hoxa9, and Prdm16 could increase Hoxa9 levels. For Mll-deficient LSK cells infected with the empty retrovirus, we observed reduced expression of Hoxa9, Prdm16, Mecom, Pbx1, and Eya1 (Fig. 4, empty) as observed in unmanipulated Mll-deficient LSK cells (Fig. 1). However, reexpression of Hoxa9, Prdm16, Eya1, or Pbx1 did not restore expression of the other tested genes to wild-type levels in Mll-deficient LSK cells (Fig. 4). In contrast, expression of either of the Mecom isoforms altered the expression of other genes in this network in Mll-deficient LSK cells. Evi1 expression increased Prdm16 and Hoxa9 transcripts in Mll-deficient LSK cells back to the wild-type levels (Fig. 4 A and B). Mds1-Evi1 suppressed Prdm16, Hoxa9, Pbx1, and Eya1 expression in wild-type cells to the low levels observed in Mll-deficient LSK cells (Fig. 4 A, B, D, and E), consistent with previous observations that Mds1-Evi1 and Evi1 have opposing activities on hematopoietic differentiation and cytokine-stimulated growth (37, 38). These data illustrate that overexpression of individual transcription factors can influence the expression levels of other regulators in this network primarily in wild-type LSK cells, yet in most cases cannot restore normal levels of any of the network genes in Mll-deficient cells. The exception is Evi1, which is capable of restoring the expression of two of the five genes in this network in Mll-deficient LSK cells. Taken together, these data exclude that these transcriptional regulators are organized in a linear pathway downstream of MLL and, instead, suggest that they each perform independent functions as downstream effectors of MLL.
Fig. 4.

Effect of reexpression of individual Mll targets on others in the network. RT-qPCR of genes in LSK cells reexpressing the cDNA indicated below each set of bars. Cells were produced in vivo by pI:pC injection, sorted 6 d later, then infected with a retrovirus without an added cDNA (empty) or cDNA as indicated. Two days later, retrovirally infected cells were sorted and RT-qPCR assays were performed. (A) Prdm16 expression levels in control MllF/F (blue) or Mll-deficient (red) LSK cells infected with the retrovirus indicated below each set of bars. Expression levels were normalized to the average expression level empty retrovirus-infected MllF/F cells and to Gapdh in each sample. Expression of Hoxa9 (B), Mecom transcripts (C), Pbx1 (D), and Eya1 (E) were analyzed and normalized as in A. Dashed lines indicate the average expression level in wild-type or Mll-deficient, empty retrovirus infected cells; four to five animals per genotype were used for each experiment, and error bars represent 95% CI. P values are shown for the comparison between pairs of empty vector and Evi1-expressing cells, calculated with the paired Student t test. ND, not detected.
Prdm16 Exhibits a Unique Capacity to Partially Rescue Mll-Deficient Cells.
One to two weeks after inducing cre, the attrition of BM cells in Mx1-cre;MllF/F animals results in animal death accompanied by multiple defects in HSPCs (13). To evaluate the relative functional importance of the identified Mll targets, we assessed whether reexpression of individual genes could rescue Mll-deficient cell attrition from BM chimeras. To this end, the Mll target genes identified above were overexpressed individually in sorted LSK cells from uninduced control MllF/F or Mx1-cre;MllF/F mice, then engrafted into lethally irradiated recipients together with uninfected wild-type BM cells. After stable engraftment, Mll excision was induced by pI:pC injection and the persistence of Mll-deficient BM cells expressing the reintroduced gene was determined 2 wk later (Fig. 5A). Thus, in this assay, “rescue” is defined as the selective persistence of retrovirus-infected cells within the population of Mll-deleted cells (Fig. S4A). The use of Mll itself as a positive control was precluded by the large size of the Mll transcript (>11 kb), because it could not be packaged into a retrovirus.
Fig. 5.

Reexpression of Prdm16 partially rescues Mll deficiency. (A) Experimental scheme to determine effects of reexpression of Mll-dependent genes. LSK cells were sorted from control MllF/F or Mx1-cre;MllF/F donor animals then infected with the indicated retrovirus. The entire pool of infected and uninfected cells was transplanted into irradiated recipients, which were analyzed 6 wk later. (B) Results of reexpression of each individual gene in control MllF/F (blue) or Mll-deficient LSK cells (red); each point represents an individual recipient animal, n = 3–10 recipients per condition. The percentage of donor-type (CD45.1+) BM cells that are GFP+ or hCD4+ 2 wk after Mll deletion is shown. Data are representative of three independent experiments. *P ≤ 0.05 was calculated by using the Wilcoxon rank-sum test.
Upon Mll deletion, uninfected or empty retrovirus-expressing donor cells were lost rapidly from chimeric animals as expected (Fig. S4 B and C). Hoxa9 overexpression resulted in the expansion of donor-derived cells in chimeras (Hoxa9 versus empty) but also Hoxa9 expressing Mll-deficient cells were protected from attrition, as evidenced by their overrepresentation in the Mll-deficient population (Fig. 5B, red versus blue). Surprisingly, Prdm16 reexpression resulted in the most significant rescue of Mll-deficient cells. Despite its greater ability to influence other network genes, reexpression of Evi1 only marginally protected Mll-deficient cells from attrition, and Mds1-Evi1, Pbx1, and Eya1 had no specific activity in this assay (Fig. 5B). Because of the low contribution of Evi1-expressing cells in chimeras, we considered in this case that overexpression may suppress hematopoiesis overall, but we found that a retrovirus producing ∼10-fold less Evi1 produced similar results (Fig. S4 E–H). Complete Mll deletion in the persisting cells of chimeras was confirmed by a quantitative genomic PCR assay (Fig. S4D). We found that retroviral overexpression of the individual genes resulted in a similar contribution to lymphoid and myeloid lineages, with the exception being the suppression of B-lymphopoiesis by Prdm16 (Fig. S4I) as has been noted (26). Taken together, these data suggest that in addition to Hoxa9, Prdm16 is an important direct target of MLL in HSCs and is capable of partially rescuing Mll-deficient hematopoietic cells from attrition in BM chimeras without restoring the entire transcriptional network.
Prdm16 Can Correct the Intrinsic Proliferation Defect of Mll-Deficient HSCs.
To determine the mechanism by which Prdm16 partially rescued Mll-deleted BM cells, we examined the consequences of Prdm16 reexpression on LSK cell proliferation. We demonstrated that more Mll-deleted LSK cells are in S phase compared with wild-type, and that the CD48neg subset of these cells were largely in G1/S rather than G0 (13). Thus, we first assessed whether we could recapitulate any aspects of the hyperproliferative phenotype in vitro, then assessed the impact of Prdm16 in this setting.
To directly assess proliferation kinetics in vitro, wild-type (MllF/F) or Mll-deleted (Mx1-cre;MllF/F) LSK/CD48neg cells were sorted from pI:pC-injected animals, deposited into wells as single cells and cultured in serum-free medium containing cytokines to maintain HSC identity and function (39) (Fig. 6A). Importantly, the percentage of surviving clones was similar between wild-type and Mll-deleted cells (Fig. S5A), confirming previous observations that apoptosis is not induced in Mll-deleted HSPCs (13). Integrating individual observations for 158 wild-type and 240 Mll-deleted LSK/CD48neg cells, we found that the proliferation kinetics of the latter were consistently more advanced than wild type (Fig. 6E). After 48 h, the mode (greatest number of cells) of Mll-deleted LSK/CD48neg clones had progressed approximately one-half a division further than the wild-type clones (Fig. 6C), and by 72 h, the mode was one full cell division ahead (Fig. 6D). To address the possibility that Mll-deficient LSK/CD48neg cells exhibit earlier cell division because more are initially in G1/S compared with wild type, we performed higher resolution studies examining the initial three cell divisions (Fig. 6E). We found that Mll-deficient LSK/CD48neg cells enter the cell cycle earlier at all cell divisions; in fact, Mll-deficient cells had a shorter cell cycle (∼1 h) than wild-type cells (Fig. S5B). Therefore, Mll-deficiency results in a cell-intrinsic increase in proliferation that is recapitulated in vitro in conditions that maintain HSC identity. This system likely models the increased proportion of LSK cells in S phase we observed in vivo but does not represent the defect in maintaining G0 (13).
Fig. 6.
The intrinsic proliferation defect of Mll-deficient HSCs is corrected by reexpression of Prdm16. (A) Scheme to determine proliferation kinetics of individual LSK/CD48neg cells. Mll deletion was performed in vivo, and double-sorted LSK/CD48neg cells were deposited at 1 cell per well. Cell divisions were scored every 24 h. (B–D) Cumulative proliferation data from individual control MllF/F (blue) or Mll-deficient LSK/CD48neg cells (red). Data represent 158 control MllF/F and 240 Mll-deficient cells; n = 3–5 animals per genotype. The difference between modes of each line is indicated by gray fill. The Pearson’s χ2 test was performed to determine statistical significance, shown on B–D. (E) Higher-resolution proliferation kinetics of control MllF/F (blue) or Mll-deficient LSK/CD48neg cells (red). Cells were prepared as in A, n = 2–3 animals per genotype, 93 control MllF/F and 38 Mll-deficient cells. The percentage of cells past the first, second, and third divisions are graphed separately (1°, 2°, 3°). (F) Scheme to determine the impact of Prdm16 reexpression in Mll-deficient LSK cells. (G) Accumulation of LSK cells expressing an empty (solid) or hCD4-Prdm16 retrovirus (dashed). LSK cells were sorted from control ER-cre;MllF/+ (blue) or ER-cre;MllF/F animals (red), cultured in 4-OHT during the retroviral infection to induce Mll deletion then enumerated every 24 h for 3 d. Data represent averages ± 95% CI, n = 4 animals per genotype, 3 replicates per time point.
To investigate whether Prdm16 reexpression influenced the proliferation phenotype observed in Mll-deficient cells, we sorted LSK cells from control ER-cre;MllF/+and ER-cre;MllF/F mice, retrovirally introduced Prdm16, and concurrently incubated with 4-OHT to induce Mll deletion (Fig. 6F). ER-cre;MllF/F cells infected with an empty control retrovirus displayed greater cell accumulation than the ER-cre;MllF/+ control cells, consistent with the single cell observations. However, Prdm16 reexpression restored the growth of Mll-deficient LSK cells to within the normal range of the control LSK cells (Fig. 6G). Together, these data suggest that the mechanism by which Prdm16 can correct Mll deficiency is, in part, by restraining proliferation within HSPCs.
Discussion
Using two complementary conditional knockout models (Mx1-cre and ER-cre), we have identified genes that are consistently Mll dependent in HSC-enriched cell populations. The acute nature of Mll deletion and the use of highly purified cells resulted in the identification of a succinct list of transcriptional regulators with a high level of reproducibility and enrichment for genes that control self-renewal and proliferation specifically in HSCs. Thus, we refer to this set of genes as core components of the MLL HSC-specific transcriptional network. Among the down-regulated genes, Prdm16, Mecom, Pbx1, Eya1, and Hoxa9 emerged as a series of interconnected Mll-regulated transcriptional nodes, with Prdm16 exhibiting the greatest activity to replace Mll function in HSCs. We tested these genes individually by overexpression to uncover dominant nodes downstream of Mll, but our data are consistent with the concept that this network functions coordinately to sustain HSC homeostasis through diverse functions, hence the inability of any individual gene to completely replace Mll in the gene expression or functional assays. In fact, each of these genes has distinct targets and loss-of-function phenotypes (25, 27–29, 40). Ultimately, identification of the minimal network of genes sufficient to replace Mll function will require simultaneous expression of physiologic levels of multiple genes.
Given the mechanisms by which MLL family members regulate gene expression, one surprising finding was the large number of up-regulated genes in Mll-deficient HSCs. However, the majority of these genes reflect the enhanced proliferation that we observe in Mll-deficient HSC-enriched populations in vivo, a finding that we also observe at single-cell resolution in the current study. The direct connection between Mll and enhanced proliferation in HSCs could be explained by three mechanistically distinct hypotheses. First, Pbx1, Mecom, and Prdm16 have all been suggested to suppress HSC proliferation, based on the analysis of hematopoietic populations in the corresponding knockout animals (25, 27, 29). Thus, the reduction in these three factors would be predicted to result in unrestrained proliferation, specifically in HSCs. Interestingly, responsiveness to TGFβ signaling is attenuated in hematopoietic cells from each of these knockouts (25, 29, 41), suggesting that the overall effect may have a significant impact on TGFβ signaling (Fig. S5 C and D). Alternatively, a distinct mechanism has been proposed to link Mll to proliferation in the setting of DNA damage. In this case, DNA damage-induced delay in origin of replication activation is enforced by wild-type MLL (42). In our conditional knockout system, it is possible that the loss of MLL (even in the absence of overt DNA damage) also results in unrestrained origin activation, a more rapid S phase, and shorter overall cell cycle duration. Finally, a recent demonstration that Mds1-Evi1 and Prdm16 are H3K9 monomethylases (43) suggests that global derepression of heterochromatinized genes could potentially have a broad impact on the suppression of proliferation or erythropoiesis in Mll-deficient HSCs.
By identifying this transcriptional network, we discovered three important features of this HSC-specific Mll pathway. First, some (e.g., Hoxa9, Mecom), but not all (e.g., Pbx1, Prdm16), of the direct Mll target genes also require the cofactor Menin. This finding illustrates that MLL uses distinct chromatin-targeting motifs for distinct categories of its direct target genes. Second, the genes identified here as Mll dependent in HSCs are not universally regulated by Mll in other tissues, with the exception of Hoxa9. This observation suggests that tissue-specific targeting and restriction mechanisms are behind the tissue-specific activity of MLL family members. Third, we note that not all of the HSC-specific, Mll target genes are up-regulated in leukemia, possibly reflecting the distinction between the chromatin targeting/activation mechanisms used by fusion oncoproteins in contrast to those used by wild-type MLL. For example, it is clear that Hoxa9 is consistently overexpressed in MLL translocation leukemia, whether T-cell ALL (T-ALL), B-cell ALL (B-ALL), or AML (44–46). Evi1 and Eya1 have recently been implicated as targets of MLL fusion oncoproteins in some leukemia subsets (33, 47), but they are not consistently up-regulated in either ALL or AML harboring an MLL rearrangement. Prdm16 is not up-regulated in MLL-translocation leukemia yet can be activated by retroviral insertion in leukemia by translocation in other contexts, therefore has leukemogenic potential (48). Thus, our data begin to delineate a normal and reversible HSC-specific maintenance pathway, of which a selective portion is subverted to result in leukemia. Interestingly, Hoxa9, Mecom, and possibly Eya1 are the Mll-dependent genes we found to be affected by Menin loss, providing an intriguing connection between chromatin-targeting mechanism and leukemogenic versus normal HSC regulatory networks. The selective dependence on particular protein–protein interactions may render leukemia-specific gene programs driven by Mll-fusion oncogenes more sensitive to inhibitors than normal HSCs, as suggested by the study of compounds that disrupt the Menin–MLL interaction (49). Our work illustrates that MLL family members control exquisitely tissue-specific gene programs despite their ubiquitous expression patterns, underscoring the complexity of mechanisms that must be used to regulate diverse gene expression programs in vivo.
Materials and Methods
Mice and in Vivo Induction of cre Recombinase.
Mx1-cre;MllF/F animals and cre induction have been described (13). Men1F/F mice (kind gift of Matthew L. Meyerson, Harvard Medical School, Boston, MA) were back-crossed by using the DartMouse speed congenic facility then crossed to the ER-cre strain.
Flow Cytometry, Cell Sorting, and Culture.
Flow cytometry and cell sorting were performed on a FACSCalibur and FACSAria, respectively (BD Biosciences). Data were analyzed by using FlowJo software (Tree Star). Fluorochrome-labeled antibodies and procedures are detailed in SI Materials and Methods.
Plasmids, Retroviral Infection, Cell Culture, and Transplantation.
Murine stem cell virus (MSCV)-based retroviral expression plasmids were constructed by using cDNAs obtained or cloned as described in SI Materials and Methods. Viral supernatants were prepared by cotransfection, and sorted LSK cells were infected by using retronectin (Takara). Retrovirally infected cells were cotransplanted into lethally irradiated (950 Rads, split dose) C57BL/6J female mice. For proliferation assays, LSK and LSK/CD48neg cells were cultured in HSC expansion medium [StemSpan Serum Free Expansion Medium (SFEM); 300 ng/mL recombinant murine (rm) SCF, 20 ng/mL rmIL-11, and 4 ng/mL rmFlt3L; StemCell Techologies and R&D Systems]. To induce deletion using the ER-cre strain, HSC expansion medium was supplemented with 300–400 nM 4-OHT (Sigma).
ChIP.
Rabbit polyclonal anti-MLL C terminus (50) or anti-Gal4 (Santa Cruz; SC-577) antibodies were used for ChIP by using linneg or LSK cells (31) with refinements as indicated in SI Materials and Methods. Primer sequences and genomic positions are described in Dataset S3.
Microarray Sample Preparation and Data Analyses.
Affymetrix microarray analyses were performed by using sorted LSK/CD48neg cells from five MllF/F or Mx1-cre; MllF/F mice 6 d after cre induction. Detailed methods and bioinformatic analyses are found in SI Materials and Methods and Dataset S4.
Statistical Analyses.
Unless indicated otherwise, the unpaired Student t test was used to determine significance, and error bars represent 95% CI. Statistical analyses were performed by using Excel (Microsoft) or Prism (GraphPad) Software.
Supplementary Material
Acknowledgments
We thank R. Mako Saito, Chris Vakoc, Steve Smale, Hanna Mikkola, Emmanuelle Passegué, and Adolfo Ferrando for critical comments; Thomas Milne and Joanna Attema for advice on ChIP; and Drs. Perkins, Morishita, and Spiegelman for providing plasmids. E.L.A. and B.P.M. were partially supported by the Lady Tata Memorial Trust. E.K.Y.C. is a Japan Society for the Promotion of Science Foreign Postdoctoral Fellow, and A.W.M. was supported by a Japan Society for the Promotion of Science Grants-in-Aid Young Scientist (B) and a RIKEN Brain Sciences Institute core grant. This work was supported in part by National Institutes of Health Grants HL090036 and RR16437, American Cancer Society Grant RSG-10-242-LIB, and funds from the Gabrielle's Angel Foundation for Cancer Research and Lauri Strauss Leukemia Foundation.
Footnotes
Conflict of interest statement: P.E. is a shareholder of Amgen stock.
This article is a PNAS Direct Submission.
Data deposition: The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE47205).
See Commentary on page 11670.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1301278110/-/DCSupplemental.
References
- 1.Schuettengruber B, Chourrout D, Vervoort M, Leblanc B, Cavalli G. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–745. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 2.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6(11):846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 3.Deshpande AJ, Bradner J, Armstrong SA. Chromatin modifications as therapeutic targets in MLL-rearranged leukemia. Trends Immunol. 2012;33(11):563–570. doi: 10.1016/j.it.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daser A, Rabbitts TH. Extending the repertoire of the mixed-lineage leukemia gene MLL in leukemogenesis. Genes Dev. 2004;18(9):965–974. doi: 10.1101/gad.1195504. [DOI] [PubMed] [Google Scholar]
- 5.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7(11):823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 6.Marschalek R. Mechanisms of leukemogenesis by MLL fusion proteins. Br J Haematol. 2011;152(2):141–154. doi: 10.1111/j.1365-2141.2010.08459.x. [DOI] [PubMed] [Google Scholar]
- 7.Nakamura T, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10(5):1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 8.Yokoyama A, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24(13):5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378(6556):505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- 10.Ingham PW. A clonal analysis of the requirement for the trithorax gene in the diversification of segments in Drosophila. J Embryol Exp Morphol. 1985;89:349–365. [PubMed] [Google Scholar]
- 11.Lim DA, et al. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature. 2009;458(7237):529–533. doi: 10.1038/nature07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamashita M, et al. Crucial role of MLL for the maintenance of memory T helper type 2 cell responses. Immunity. 2006;24(5):611–622. doi: 10.1016/j.immuni.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 13.Jude CD, et al. Unique and independent roles for MLL in adult hematopoietic stem cells and progenitors. Cell Stem Cell. 2007;1(3):324–337. doi: 10.1016/j.stem.2007.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diehl F, Rössig L, Zeiher AM, Dimmeler S, Urbich C. The histone methyltransferase MLL is an upstream regulator of endothelial-cell sprout formation. Blood. 2007;109(4):1472–1478. doi: 10.1182/blood-2006-08-039651. [DOI] [PubMed] [Google Scholar]
- 15.Yagi H, et al. Growth disturbance in fetal liver hematopoiesis of Mll-mutant mice. Blood. 1998;92(1):108–117. [PubMed] [Google Scholar]
- 16.Ayton P, et al. Truncation of the Mll gene in exon 5 by gene targeting leads to early preimplantation lethality of homozygous embryos. Genesis. 2001;30(4):201–212. doi: 10.1002/gene.1066. [DOI] [PubMed] [Google Scholar]
- 17.Yokoyama A, et al. Proteolytically cleaved MLL subunits are susceptible to distinct degradation pathways. J Cell Sci. 2011;124(Pt 13):2208–2219. doi: 10.1242/jcs.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon KA, et al. Mll has a critical role in fetal and adult hematopoietic stem cell self-renewal. Cell Stem Cell. 2007;1(3):338–345. doi: 10.1016/j.stem.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 19.Gan T, Jude CD, Zaffuto K, Ernst P. Developmentally induced Mll1 loss reveals defects in postnatal haematopoiesis. Leukemia. 2010;24(10):1732–1741. doi: 10.1038/leu.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA. 2001;98(9):5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venezia TA, et al. Molecular signatures of proliferation and quiescence in hematopoietic stem cells. PLoS Biol. 2004;2(10):e301. doi: 10.1371/journal.pbio.0020301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seita J, et al. Gene Expression Commons: An open platform for absolute gene expression profiling. PLoS ONE. 2012;7(7):e40321. doi: 10.1371/journal.pone.0040321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novershtern N, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He S, Kim I, Lim MS, Morrison SJ. Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev. 2011;25(15):1613–1627. doi: 10.1101/gad.2052911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ficara F, Murphy MJ, Lin M, Cleary ML. Pbx1 regulates self-renewal of long-term hematopoietic stem cells by maintaining their quiescence. Cell Stem Cell. 2008;2(5):484–496. doi: 10.1016/j.stem.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deneault E, et al. A functional screen to identify novel effectors of hematopoietic stem cell activity. Cell. 2009;137(2):369–379. doi: 10.1016/j.cell.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aguilo F, et al. Prdm16 is a physiologic regulator of hematopoietic stem cells. Blood. 2011;117(19):5057–5066. doi: 10.1182/blood-2010-08-300145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goyama S, et al. Evi-1 is a critical regulator for hematopoietic stem cells and transformed leukemic cells. Cell Stem Cell. 2008;3(2):207–220. doi: 10.1016/j.stem.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, et al. PR-domain-containing Mds1-Evi1 is critical for long-term hematopoietic stem cell function. Blood. 2011;118(14):3853–3861. doi: 10.1182/blood-2011-02-334680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schuettengruber B, Martinez AM, Iovino N, Cavalli G. Trithorax group proteins: Switching genes on and keeping them active. Nat Rev Mol Cell Biol. 2011;12(12):799–814. doi: 10.1038/nrm3230. [DOI] [PubMed] [Google Scholar]
- 31.Weishaupt H, Attema JL. A method to study the epigenetic chromatin states of rare hematopoietic stem and progenitor cells; MiniChIP-Chip. Biol Proced Online. 2010;12(1):1–17. doi: 10.1007/s12575-010-9031-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guenther MG, et al. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci USA. 2005;102(24):8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang QF, et al. MLL fusion proteins preferentially regulate a subset of wild-type MLL target genes in the leukemic genome. Blood. 2011;117(25):6895–6905. doi: 10.1182/blood-2010-12-324699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yokoyama A, et al. The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell. 2005;123(2):207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- 35.Maillard I, et al. Menin regulates the function of hematopoietic stem cells and lymphoid progenitors. Blood. 2009;113(8):1661–1669. doi: 10.1182/blood-2009-01-135012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shimabe M, et al. Pbx1 is a downstream target of Evi-1 in hematopoietic stem/progenitors and leukemic cells. Oncogene. 2009;28(49):4364–4374. doi: 10.1038/onc.2009.288. [DOI] [PubMed] [Google Scholar]
- 37.Sitailo S, Sood R, Barton K, Nucifora G. Forced expression of the leukemia-associated gene EVI1 in ES cells: A model for myeloid leukemia with 3q26 rearrangements. Leukemia. 1999;13(11):1639–1645. doi: 10.1038/sj.leu.2401585. [DOI] [PubMed] [Google Scholar]
- 38.Sood R, Talwar-Trikha A, Chakrabarti SR, Nucifora G. MDS1/EVI1 enhances TGF-beta1 signaling and strengthens its growth-inhibitory effect but the leukemia-associated fusion protein AML1/MDS1/EVI1, product of the t(3;21), abrogates growth-inhibition in response to TGF-beta1. Leukemia. 1999;13(3):348–357. doi: 10.1038/sj.leu.2401360. [DOI] [PubMed] [Google Scholar]
- 39.Uchida N, Dykstra B, Lyons KJ, Leung FY, Eaves CJ. Different in vivo repopulating activities of purified hematopoietic stem cells before and after being stimulated to divide in vitro with the same kinetics. Exp Hematol. 2003;31(12):1338–1347. doi: 10.1016/j.exphem.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 40.Chuikov S, Levi BP, Smith ML, Morrison SJ. Prdm16 promotes stem cell maintenance in multiple tissues, partly by regulating oxidative stress. Nat Cell Biol. 2010;12(10):999–1006. doi: 10.1038/ncb2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Avagyan S, Aguilo F, Kamezaki K, Snoeck HW. Quantitative trait mapping reveals a regulatory axis involving peroxisome proliferator-activated receptors, PRDM16, transforming growth factor-β2 and FLT3 in hematopoiesis. Blood. 2011;118(23):6078–6086. doi: 10.1182/blood-2011-07-365080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu H, et al. Phosphorylation of MLL by ATR is required for execution of mammalian S-phase checkpoint. Nature. 2010;467(7313):343–346. doi: 10.1038/nature09350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinheiro I, et al. Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell. 2012;150(5):948–960. doi: 10.1016/j.cell.2012.06.048. [DOI] [PubMed] [Google Scholar]
- 44.Armstrong SA, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30(1):41–47. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 45.Ferrando AA, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: Dominance of HOX dysregulation. Blood. 2003;102(1):262–268. doi: 10.1182/blood-2002-10-3221. [DOI] [PubMed] [Google Scholar]
- 46.Ross ME, et al. Gene expression profiling of pediatric acute myelogenous leukemia. Blood. 2004;104(12):3679–3687. doi: 10.1182/blood-2004-03-1154. [DOI] [PubMed] [Google Scholar]
- 47.Arai S, et al. Evi-1 is a transcriptional target of mixed-lineage leukemia oncoproteins in hematopoietic stem cells. Blood. 2011;117(23):6304–6314. doi: 10.1182/blood-2009-07-234310. [DOI] [PubMed] [Google Scholar]
- 48.Morishita K. Leukemogenesis of the EVI1/MEL1 gene family. Int J Hematol. 2007;85(4):279–286. doi: 10.1532/IJH97.06174. [DOI] [PubMed] [Google Scholar]
- 49.Grembecka J, et al. Menin-MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol. 2012;8(3):277–284. doi: 10.1038/nchembio.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ. Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol. 2003;23(1):186–194. doi: 10.1128/MCB.23.1.186-194.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.