

Abstract
Steric Bulk or Conformation Control? Optimization of P,N-bidentate ligands reveals the importance of conformation control in the development of highly efficient intermolecular trapping of reactive α-oxo gold carbene intermediates. While a pendant piperidine ring offers suitable steric bulk, fixing its conformation to provide better shielding to the highly electrophilic carbene center turned out to be crucial for the excellent reaction efficiency. A generally highly efficient and broadly applicable synthesis of carboxymethyl ketones from readily available carboxylic acids and terminal alkynes is developed under exceptionally mild reaction conditions.
Keywords: gold, oxidation, carbene, conformation, bidentate ligand
A few years ago we reported[1] that gold-catalyzed[2] intermolecular oxidation of alkynes offered an expedient access to synthetically highly versatile α-oxo gold carbene intermediates (Scheme 1A). This strategy circumvents the use of hazardous and potentially explosive α-diazo ketone precursors[3] and has led to the development of various efficient synthetic methods by us[1, 4] and others[5] based on their intramolecular trapping. The intermolecular counterpart, likely of exceptional synthetic utilities, however, proves to be very challenging due to the highly electrophilic nature of the carbene center, and is often plagued by over oxidation, reactions with solvents,[4b, 4g] and intractable side reactions. Earlier we reported for the first time that the reactivity of the gold carbene could be attenuated by using bidentate phosphine ligands so that it reacted with carboxamides efficiently, leading to a one-step synthesis of 2,4-disubstituted oxazoles.[6] Mor-DalPhos (see Scheme 1B),[7] a bulky P,N-bidentate ligand, was found as a uniquely effective ligand, and its role is proposed to enable the formation of a tris-coordinated gold carbene (e.g., A) instead of the typical bis-coordinated one (e.g., A’), leading to attenuated electrophilicity at the carbene center (Scheme 1B).
Scheme 1.

A) Trapping α-oxo gold carbenes generated via intermolecular alkyne oxidation by internal and external nucleophiles. B) Mor-DalPhos and the previously calculated relative energies of the corresponding gold carbene species, in kcal/mol, at PBE1PBE/6-311+G** level.[6]
To further develop alkynes as surrogates of hazardous α-diazo ketones in gold catalysis especially in the context of synthetically important intermolecular trapping, we embarked on expanding the scope of suitable external nucleophiles beyond carboxamide. Our first target was carboxylic acid (Scheme 1A), a weaker nucleophile in its neutral form than carboxamide. Notably, the reaction, if developed, would offer a novel and rapid access to synthetically versatile α-carboxymethyl ketones (i.e, 3, Scheme 1) from readily available terminal alkynes and carboxylic acids.
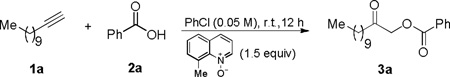
At the outset, we used 1-dodecyne and benzoic acid (1.2 equiv) as the reacting partners and 8-methylquinoline N-oxide[4d] as the oxidant, and the results of reaction optimization are shown in Table 1. Consistent with our previous study,[6] cationic gold complexes derived from typical ligands such as Ph3P, IPr, and BrettPhos were largely ineffective, resulting in complex mixtures with little desired product (entry 1). On the contrary, Mor-DalPhos[7] again proved to be an effective ligand, and the oxidative gold catalysis led to the desired α-benzoxymethyl ketone 3a in a fairly good yield (entry 3). An identical yield was observed with the ligand L1[7] (Figure 1A), which differs from Mor-DalPhos by having a piperidine ring instead of a morpholine ring (entry 4). However, the smaller Me-DalPhos (Figure 1A)[7] was much inferior (entry 2), suggesting that the steric size of the pendant secondary amine might be critical.
Table 1.
Optimization of the Reaction Conditions a
 | |||
|---|---|---|---|
| entry | 1a/2a | catalyst | yieldb |
| 1 | 1:1.2 | LAuCl (5 mol %)/NaBArF4 (10 mol %) | <7 c |
| 2 | 1:1.2 | Me-DalPhosAuCl (5 mol %)/NaBArF4(10 mol %) | 11% |
| 3 | 1:1.2 | Mor-DalPhosAuCl (5 mol %)/NaBArF4(10 mol %) | 68% |
| 4 | 1:1.2 | L1AuCl(5 mol %)/NaBArF4(10 mol %) | 68% |
| 5 | 1:1.2 | L2AuCl(5 mol %)/NaBArF4(10 mol %) | 68% |
| 6 | 1:1.2 | L3AuCl(5 mol %)/NaBArF4(10 mol %) | 18% |
| 7 | 1:1.2 | L4AuCl(5 mol %)/NaBArF4(10 mol %) | 75% |
| 8 | 1:1.2 | L5AuCl(5 mol %)/NaBArF4(10 mol %) | 84% |
| 9 | 1:1.2 | L6AuCl(5 mol %)/NaBArF4(10 mol %) | 30% |
| 10 | 1:1.2 | L7AuCl(5 mol %)/NaBArF4(10 mol %) | 79% |
| 11 | 1:1.2 | L8AuCl(5 mol %)/NaBArF4(10 mol %) | 63% |
| 12 | 1.3/1 | L7AuCl(5 mol %)/NaBArF4(10 mol %) | 86% |
| 13 | 1.3/1 | L5AuCl(5 mol %)/NaBArF4(10 mol %) | 98% d |
| 14 | 1.3/1 | L5AuCl(5 mol %)/NaBArF4(10 mol %)e | 95% |
| 15 | 1.3/1 | L5AuCl(5 mol %)/NaBArF4(10 mol %)f | 88% |
The reaction was run with everything except the oxidant in a vial capped with a septum, and the oxidant was introduced into the reaction mixture in 12 h using a syringe pump. Initially, [1a] = 0.1 M.
Measured by 1H NMR analysis using diethyl phthalate as the internal standard.
L = Ph3P, IPr, or BrettPhos; the crude 1H NMR spectra were mostly messy.
96% isolated yield.
DCE was used as the solvent.
toluene was used as the solvent.
Figure 1.

A) known ligands. B) Newly developed P,N-bidentate ligands
Consequently, we modified the piperidine ring of L1 with different substituents. While the installation of a methyl group at its 4-position was inconsequential (entry 5, see L2 in Figure 1B), a much bigger t-butyl was detrimental (entry 6, see L3 in Figure 1B). However, to our delight, a 3-methyl group, as in L4 (Figure 1B), led to a notable increase of the reaction yield (entry 7); moreover, the use of cis-3,5-dimethylpiperidine (as in L5) as the pendant amine group resulted in a higher 84% yield of 3a (entry 8). On the contrary, the ligand L6 with a trans-3,5-dimethylpiperidine ring led to a much less efficient reaction (entry 9), which could be attributed to deleterious steric congestion caused by an inevitable axially oriented methyl group. The piperidine ring in L5 could be replaced with a morpholine ring, as in L7, with only small, yet adverse impact on the reaction outcome (entry 10). Interestingly, replacing the methyl groups in L7 with bigger phenyl groups as in L8 resulted in a yield even lower than that by Mor-DalPhos (entry 11). This result, along with that by L3, suggesting that there is an optimal steric bulk for the pendant six-membered ring in this reaction. When benzoic acid was the limiting reagent, L5 was a significantly better ligand than L7 for the gold catalysis (comparing entries 12 and 13). Moreover, the reaction yield in the former case is nearly quantitative, which is impressive considering the difficulties previously encountered in trapping these reactive gold carbene species and really showcased the opportunities in method development based on ligand design/development. Other solvents such as DCE (entry 14) and toluene (entry 15) were suitable for this reaction albeit not nearly as good as PhCl. Although the oxidant, 8-methylquinoline N-oxide had to be introduced to the reaction slowly via a syringe pump in order to avoid over oxidation, the reaction proceeded smoothly at ambient temperature.
With the optimized reaction conditions revealed in Table 1, entry 13, the scope of the transformation was first examined with various carboxylic acids. As shown in Table 2, entries 1–6, various substituted benzoic acids were allowed, yielding the desired products in mostly excellent yields. Even a Bpin group was tolerated, and the relatively low isolated yield was due to the co-elution of the boronated product 3f with 8-methylquinoline (entry 5). The reaction also worked well with other conjugated acids such as thiophene-2-carboxylic acid (entry 7), trans-cinnamic acid (entry 8), trans-3-(2-furyl)acrylic acid (entry 9), and trans, trans-hexa-2,4-dienoic acid (entry 10), delivering the corresponding products in ≥90% yields. Acetic acids with the α-carbon functionalized by a 1-methylindol-3-yl (entry 11), a trimethylsilyl (entry 12), a chloro (entry 13), and a phenoxy (entry 14), were all suitable substrates, and functionalized α-carboxymethyl ketones were again isolated in good to excellent yields; the reaction with N-Boc protected proline, however, only resulted in a serviceable yield of the corresponding product (entry 15). Other carboxylic acids such as cyclopropanecarboxylic acid (entry 16) and adamantane-1-carboxyclic acid (entry 17) were also allowed. While these reactions were run in 0.2 mmol scale, a 3 mmol-scale reaction of entry 2 proceeded well even with only 2 mol % of the catalyst, affording 3c in a respectful 82% yield.
Table 2.
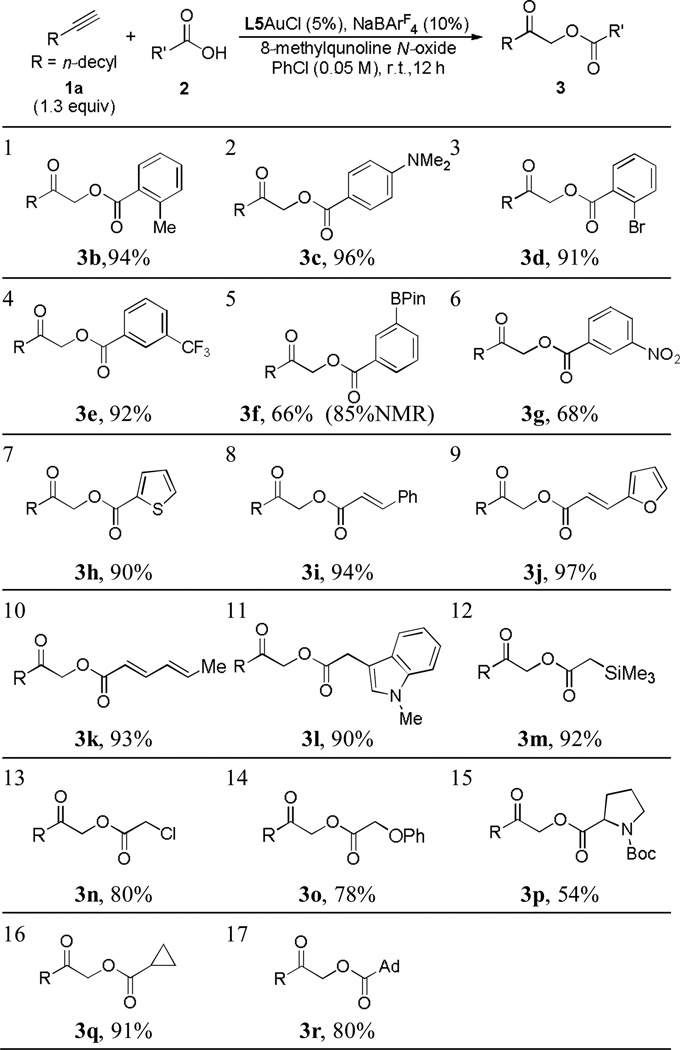 |
Initially [2] = 0.1 M; the oxidant was introduced to the reaction vial in 12 h using a syringe pump.
Isolated yields are shown.
The reaction also proceeded smoothly with various terminal alkynes. As shown in Table 3, phenylacetylene (entry 1) and 1-ethynylcyclohexene (entry 4) were excellent substrates, and so are the acetylenes substituted by cyclic alkyl groups (entries 2 and 3) or remotely functionalized linear ones (entries 5–7). The reaction yields were invariably excellent. Even with heteroatoms placed close to the C-C triple bond and hence to the in-situ formed electrophilic gold carbene center, this intramolecular reaction still led to a good (entry 8) or a serviceable yield (entry 9).
Table 3.
Reaction Scope with various alkynes[a]
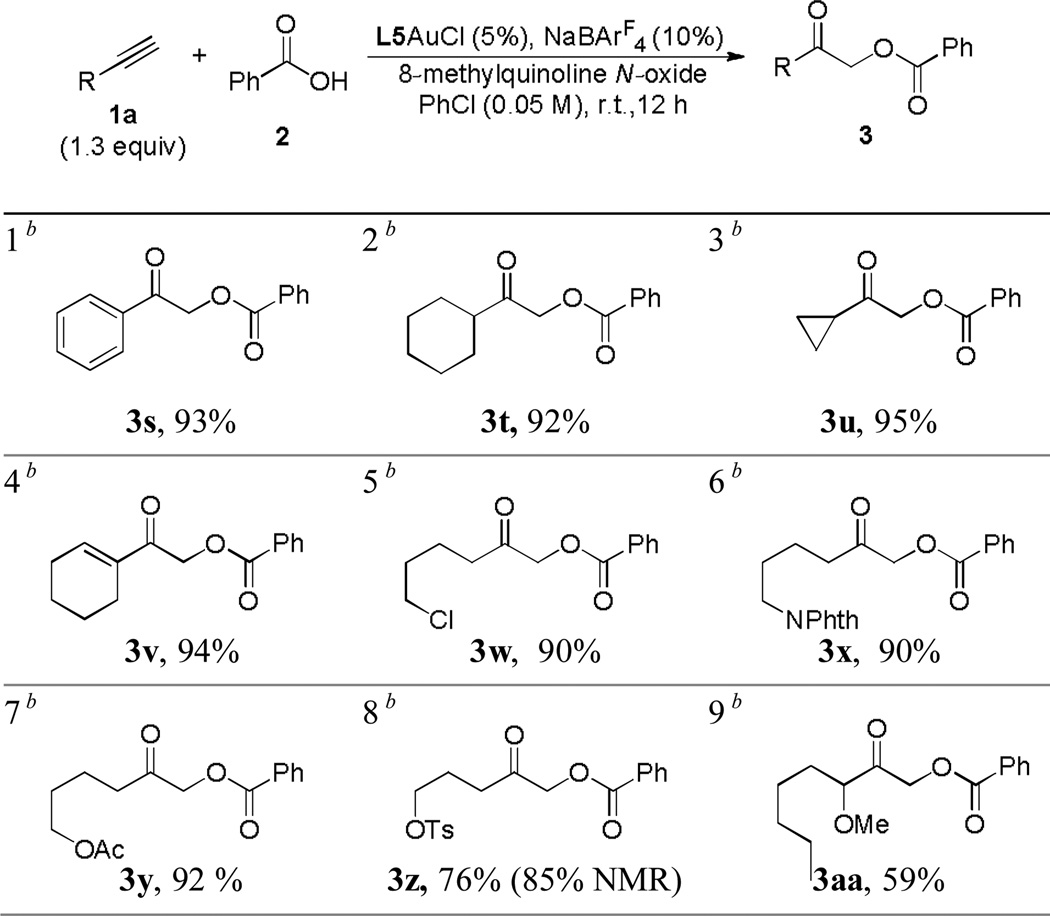 |
Initially [2] = 0.1 M; the oxidant was introduced to the reaction vial using a syringe pump.
Isolated yields are shown.
The ligand optimization via modification of the pendant piperidine/morpholine ring is worthy of commenting and further probing. In our previous carboxamide trapping chemistry,[6] the smaller Me-DalPhos was found as an equally effective bidentate ligand as Mor-DalPhos; On the contrary, in this chemistry, the steric size of the pendant amino group is apparently crucial for the reaction outcome (See Table 1). We attributed the difference to the decreased nucleophilicity of the carbonyl oxygen in carboxylic acid[8] in comparison to that in carboxamide. It is reasonable to suspect that steric shielding around the gold carbene center would facilitate, to a certain extent, slower reactions with small nucleophiles by minimizing sterically demanding side reactions. With regard to the ligand L1 and Mor-DalPhos, the enhanced efficiency with L5 could be attributed a priori to the bulkier piperidine ring; however, two cis methyl groups could at the same time impose conformation rigidity to the N-heterocycle. Scheme 2A outlines two chair conformers of the gold carbene intermediate with L1 as its ligand. It is noteworthy that the X-ray diffraction studies of Mor-DalPhos Pd complexes by Stradiotto[7a] reveal that the morpholine ring can adopt the chair conformation in B’ and twist boat conformations. The conformer B’, though understandably less stable and hence less populated, does not provide sufficient steric protection to the carbene center; consequently, the reaction could be improved if the conformers including B’ and other less shielding ones could be minimized. We surmise that the two cis methyl groups in L5 might play the role of locking the piperidine in a conformation identical to that in B. As such, the carbene center is constantly shielded and hence would react preferably with smaller nucleophiles such as carboxylic acids over bigger ones including the oxidant. X-ray diffraction study of L5AuCl confirmed the preferred shielding chair conformation for the cis-3,5-dimethylpiperidine ring (Scheme 2B). In the case of L8, the phenyl groups are likely too bulky to hinder the carbene formation and/or the productive approach by a carboxylic acid.
 |
(1) |
 |
Scheme 2.

A) Two chair conformers of the tris-coordinated gold carbene with L1 as the bidentate ligand. B) Ortep drawing of L5AuCl with the solvent molecules omitted. C) L9 with two chair conformers. D) The structure of L10.
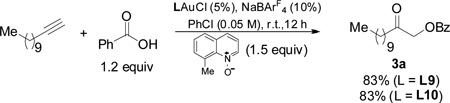
To offer more insight into the importance of conformation control, we prepared the ligand L9 (Scheme 2C), where an ortho methyl group on the benzene ring is installed to essentially prohibit the piperidine ring from adopting the chair conformation in L9-ax and likely minimizing the contribution of other non-chair conformers. Indeed, the gold catalyst afforded 3a in 83% yield (Eq. 1), much better than that with the ligand L1 (where the ortho-Me is absent) and virtually idential to that by L5 (see Table 1, entry 8). When the ligand L10 with an ortho-methyl group and a pendant cis-3,5-dimethylpiperidine was used for the gold catalysis, the reaction efficiency remained the same (Eq. 1), suggesting that the role of the piperidine methyl groups in the ligand L5 is fixing the desired chair conformation of the N-heterocycle instead of providing additional steric bulk.
 |
(4) |
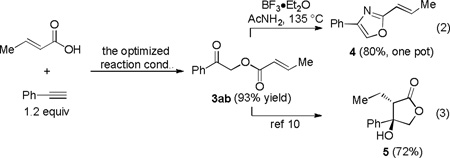
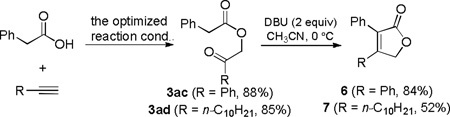
This one-step, generally applicable, and efficient synthesis of functionalized carboxylmethyl ketones permits rapid access to various useful cyclic structures. For example, 2-alkenyloxazole 4 was isolated in 80% yield by the combination of the gold catalysis and a subsequent BF3•Et2O-promoted cyclodehydration[9] in a one-pot process (Eq. 2); moreover, the phenone intermediate 3ab, isolated in 93% yield, has been previously converted into the γ-lactone 5 in 72% yield (Eq. 3).[10] Another approach to cyclized products are shown in Eq. 4, where DBU effectively promoted sequential intramolecular aldol reaction and dehydration of the ketones 3ac and 3ad, which in turn were obtained in excellent yields via the oxidative gold catalysis.
In conclusion, we have developed a generally highly efficient and broadly applicable synthesis of carboxymethyl ketones from readily available carboxylic acids and terminal alkynes under exceptionally mild conditions. In this oxidative gold catalysis, the highly electrophilic α-oxo gold carbene intermediate is most likely generated, and its challenging intermolecular trapping by weakly nucleophilic carboxylic acids is achieved upon extensive optimization of the P,N-bidentate ligands to the metal gold. While the steric bulk of the pendant amino group in these ligands is beneficial to a certain extent, controlling the conformation of the sterically suitable piperidine ring to provide better shielding to the carbene center appeared to be important to achieve the generally high efficiencies. Importantly, the reaction products can be rapidly converted into synthetically versatile cyclic structures. Further studies employing other types of nucleophiles including so-far unyielding enol ethers are currently underway.
Experimental Section
General procedure for gold-catalyzed synthesis of α-carboxymethyl ketones
To a 3 dram vial containing 2 mL of chlorobenzene were added sequentially a carboxylic acid (0.2 mmol), an alkyne (0.26 mmol), L5AuCl (0.01 mmol) and NaBArF4 (0.02 mmol). The resulting mixture was stirred at room temperature. To this vial a solution of N-oxide (47.7 mg, 0.3 mmol) in 4 mL of chlorobenzene was then added via a syringe pump in 12 h. Upon completion, the reaction mixture was concentrated under vacuum. The residue was purified by chromatography on silica gel (eluent: hexanes/ethyl acetate) to afford the desired product 3.
Supplementary Material
Acknowledgments
The authors thank NIGMS (R01 GM084254) for generous financial support.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Ye L, Cui L, Zhang G, Zhang L. J. Am. Chem. Soc. 2010;132:3258–3259. doi: 10.1021/ja100041e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected reviews, see: Hashmi ASK. Chem. Rev. 2007;107:3180–3211. doi: 10.1021/cr000436x. Fürstner A, Davies PW. Angew. Chem., Int. Ed. 2007;46:3410–3449. doi: 10.1002/anie.200604335. Zhang L, Sun J, Kozmin SA. Adv. Synth. Catal. 2006;348:2271–2296. Arcadi A. Chem. Rev. 2008;108:3266–3325. doi: 10.1021/cr068435d. Gorin DJ, Sherry BD, Toste FD. Chem. Rev. 2008;108:3351–3378. doi: 10.1021/cr068430g. Jiménez-Núñez Es, Echavarren AM. Chem. Rev. 2008;108:3326–3350. doi: 10.1021/cr0684319. Li Z, Brouwer C, He C. Chem. Rev. 2008;108:3239–3265. doi: 10.1021/cr068434l. Abu Sohel SM, Liu R-S. Chem. Soc. Rev. 2009;38:2269–2281. doi: 10.1039/b807499m. Wang S, Zhang G, Zhang L. Synlett. 2010:692–706. Zhong C, Shi X. Eur. J. Org. Chem. 2010:2999–3025. Patil NT, Yamamoto Y. Chem. Rev. 2008;108:3395–3442. doi: 10.1021/cr050041j. Liu L-P, Hammond GB. Chem. Asian J. 2009;4:1230–1236. doi: 10.1002/asia.200900091.
- 3.(a) Doyle MP, McKervey MA, Ye T. Modern catalytic methods for organic synthesis with diazo compounds: from cyclopropanes to ylides. Wiley; New York: 1998. [Google Scholar]; (b) Davies HML, Beckwith REJ. Chem. Rev. 2003;103:2861–2903. doi: 10.1021/cr0200217. [DOI] [PubMed] [Google Scholar]; (c) Taber DF. In: Carbon-Carbon σ-Bond Formation. Pattenden G, editor. Vol. 3. Oxford, England; New York: Pergamon Press; 1991. pp. 1045–1062. [Google Scholar]
- 4.(a) Ye L, He W, Zhang L. J. Am. Chem. Soc. 2010;132:8550–8551. doi: 10.1021/ja1033952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) He W, Li C, Zhang L. J. Am. Chem. Soc. 2011:8482–8485. doi: 10.1021/ja2029188. [DOI] [PubMed] [Google Scholar]; (c) Ye L, He W, Zhang L. Angew. Chem., Int. Ed. 2011;50:3236–3239. doi: 10.1002/anie.201007624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lu B, Li C, Zhang L. J. Am. Chem. Soc. 2010;132:14070–14072. doi: 10.1021/ja1072614. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Li C, Zhang L. Org. Lett. 2011;13:1738–1741. doi: 10.1021/ol2002607. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Wang Y, Ji K, Lan S, Zhang L. Angew. Chem., Int. Ed. 2012;51:1915–1918. doi: 10.1002/anie.201107561. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) He W, Xie L, Xu Y, Xiang J, Zhang L. Org. Biomol. Chem. 2012;10:3168–3171. doi: 10.1039/c2ob25235j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Li C-W, Pati K, Lin G-Y, Sohel SMA, Hung H-H, Liu R-S. Angew. Chem., Int. Ed. 2010;49:9891–9894. doi: 10.1002/anie.201004647. [DOI] [PubMed] [Google Scholar]; (b) Wang T, Zhang JL. Dalton Trans. 2010;39:4270–4273. doi: 10.1039/c0dt00024h. [DOI] [PubMed] [Google Scholar]; (c) Xu C-F, Xu M, Jia Y-X, Li C-Y. Org. Lett. 2011;13:1556–1559. doi: 10.1021/ol200270t. [DOI] [PubMed] [Google Scholar]; (d) Qian D, Zhang J. Chem. Commun. 2011;47:11152–11154. doi: 10.1039/c1cc14788a. [DOI] [PubMed] [Google Scholar]; (e) Vasu D, Hung H-H, Bhunia S, Gawade SA, Das A, Liu R-S. Angew. Chem., Int. Ed. 2011:6911–6914. doi: 10.1002/anie.201102581. [DOI] [PubMed] [Google Scholar]; (f) Bhunia S, Ghorpade S, Huple DB, Liu R-S. Angew. Chem., Int. Ed. 2012:2939–2942. doi: 10.1002/anie.201108027. [DOI] [PubMed] [Google Scholar]; (g) Yuan W, Dong X, Wei Y, Shi M. Chem. Eur. J. 2012;18:10501–10505. doi: 10.1002/chem.201201161. [DOI] [PubMed] [Google Scholar]; (h) Xu M, Ren T-T, Li C-Y. Org. Lett. 2012;14:4902–4905. doi: 10.1021/ol302238t. [DOI] [PubMed] [Google Scholar]; (i) Murai M, Kitabata S, Okamoto K, Ohe K. Chem. Commun. 2012;48:7622–7624. doi: 10.1039/c2cc32628k. [DOI] [PubMed] [Google Scholar]; (j) Dateer RB, Pati K, Liu R-S. Chem. Commun. 2012;48:7200–7202. doi: 10.1039/c2cc33030j. [DOI] [PubMed] [Google Scholar]; (k) Davies PW, Cremonesi A, Martin N. Chem. Commun. 2011;47:379–381. doi: 10.1039/c0cc02736g. [DOI] [PubMed] [Google Scholar]; (l) Qian D, Zhang J. Chem. Commun. 2012;48:7082–7084. doi: 10.1039/c2cc31972a. [DOI] [PubMed] [Google Scholar]; (m) Hashmi ASK, Wang T, Shi S, Rudolph M. J. Org. Chem. 2012 doi: 10.1021/jo301381z. [DOI] [PubMed] [Google Scholar]
- 6.Luo Y, Ji K, Li Y, Zhang L. J. Am. Chem. Soc. 2012;134:17412–17415. doi: 10.1021/ja307948m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Lundgren RJ, Peters BD, Alsabeh PG, Stradiotto M. Angew. Chem., Int. Ed. 2010;49:4071–4074. doi: 10.1002/anie.201000526. [DOI] [PubMed] [Google Scholar]; (b) Hesp KD, Stradiotto M. J. Am. Chem. Soc. 2010;132:18026–18029. doi: 10.1021/ja109192w. [DOI] [PubMed] [Google Scholar]
- 8.1H NMR studies of a mixture of benzoic acid and 8-methylquinoline in toluene-d8 confirmed that the benzoic acid is not deprotonated in the nonpolar solvent. As PhCl is also a nonpolar solvent, the acid should similarly stay in its neutral form in PhCl. Hence, it is most likely that the gold carbene intermediate are trapped by the neutral carboxylic acid in this reaction.
- 9.Huang W, Pei J, Chen B, Pei W, Ye X. Tetrahedron. 1996;52:10131–10136. [Google Scholar]
- 10.Lam HW, Joensuu PM. Org. Lett. 2005;7:4225–4228. doi: 10.1021/ol051649h. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.