

Abstract
Studies in humans and in mice have highlighted the importance of short telomeres and impaired mitochondrial function in driving age-related functional decline in the heart. Although telomere and mitochondrial dysfunction have been viewed mainly in isolation, recent studies in telomerase-deficient mice have provided evidence for an intimate link between these two processes. Telomere dysfunction induces a profound p53-dependent repression of the master regulators of mitochondrial biogenesis and function, peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α and PGC-1β in the heart, which leads to bioenergetic compromise due to impaired oxidative phosphorylation and ATP generation. This telomere-p53-PGC mitochondrial/metabolic axis integrates many factors linked to heart aging including increased DNA damage, p53 activation, mitochondrial, and metabolic dysfunction and provides a molecular basis of how dysfunctional telomeres can compromise cardiomyocytes and stem cell compartments in the heart to precipitate cardiac aging.
Keywords: heart aging, telomere, mitochondria, metabolism, PGC-1α, myocardial regeneration, telomerase, transcription factors, transcriptional coactivator
Societies around the world are experiencing a dramatic increase in the elderly population (people 65 years and older). It is predicted that by the year 2030, 1 billion individuals (1 in 8 individuals) will be 65 years or older worldwide. In the United States, the percentage of the elderly population is expected to increase from 12.9% to 19.7% (40.2 to 88.5 million people total) from 2010 to 2050. Within the elderly, the oldest old (85 years and older) is the fastest growing section and is projected to grow from 1.8% to 4.2% (5.7 to 19.0 million) in the same time period.1 This rise of an aging population is accompanied by a sharp increase in the prevalence of age-related chronic diseases ranging from cardiovascular disease (CVD) and cancer to metabolic syndrome and Alzheimer’s disease.
CVD includes coronary artery disease (CAD), hypertension, and chronic heart failure and is a major cause of chronic disability and the leading cause of death in the United States. The prevalence of CVD for men and women combined jumps from 12% in individuals younger than 40% to 38% in the 40 to 60 group to a staggering 83% in those older than 80 years of age. CAD is the most common CVD and manifests as angina pectoris and myocardial infarction, both of which are significantly more common in the elderly. Eighty-two percent of patients who die of CAD are 65 years or older. Similarly, aging is associated with a dramatic increase in the prevalence of hypertension and chronic heart failure.2 Although substantial progress in the prevention and treatment of CVD has led to a significant decline in death rates over the last 2 decades, morbidity and mortality rates associated with CVD continue to be high and remain a tremendous burden for the national health care system. In 2010 alone, CVD-related costs were estimated to be $444 billion.3
A long-term solution to this social and health care crisis will require a better understanding of how aging per se drives cardiovascular decline. This would facilitate the development of effective preventive and therapeutic strategies. Telomeres, repetitive TTAGGG sequences at the ends of chromosomes, have been strongly implicated in aging. Cumulative studies in humans with telomere maintenance disorders and telomerase knock-out mice have demonstrated that short telomeres precipitate functional decline in different tissues, including the cardiovascular system.4 Mechanistically, telomere dysfunction–driven tissue compromise is thought to be secondary to the activation of DNA damage signaling pathways that converge on p53, a central executor of the DNA damage response pathway.5 p53 activation induces senescent and apoptosis pathways, particularly in stem cell and progenitor compartments of highly regenerative organs. The elimination of stem and progenitor cells is thought to be the driving force in the development of tissue defects. Telomeres are also linked to aging and disease development in more quiescent tissues such as the heart, and recent studies have uncovered a different mechanism that could explain how telomere dysfunction affects not only proliferative but also quiescent tissues that rely less on stem cell–driven regeneration. These studies have demonstrated that telomere dysfunction–activated p53 directly leads to mitochondrial and metabolic compromise through the repression of the master regulators of mitochondrial biogenesis and function, peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α and PGC-1β.6 PGC repression is associated with a profound compromise in mitochondrial biogenesis and function and subsequent decline in ATP generation, indicating that a fundamental problem of energy maintenance drives the aging process.
Independent studies in the aging heart have noted a significant a decline of mitochondrial function, including compromised oxidative phosphorylation, ATP generation capacity, and β-oxidation. This has lead to the hypothesis that human heart failure is a consequence of mitochondrial dysfunction and energy starvation.7,8 In the present report, we review the role of these two drivers of cardiac aging—the mitochondrial/energy maintenance and the telomere hypotheses—in CVD, with the main focus on heart disease, and discuss how telomere dysfunction converges on mitochondria and energy maintenance pathways to drive cardiac aging and failure.
Mitochondrial Alterations in the Aging Heart
The heart undergoes a number of functional and structural changes during aging, even when no contributing cardiac risk factors such as hypertension or diabetes exist (Table). These changes include a 35% loss of cardiomyocytes and a 20% decrease of sinoatrial nodal pacemaker cells due to apoptosis, necrosis, and potentially autophagy.9,10 The aging heart also contains more senescent cardiomyocytes as defined by the expression of senescence markers p53, p21, and p16 and the presence of shorter telomeres.11 This continued loss of functional resident cardiac cells is paralleled by a decline in regenerative activity from 1% per year at age 20 to 0.4% at the age of 75.12 Age-related changes often include left ventricular hypertrophy and diastolic dysfunction, which lead to increased end-diastolic pressure.13 The cumulative effect of all these changes together with the coexistence of other diseases predisposes older individuals to develop typical aged-associated heart disease: diastolic heart failure (defined as presence of heart failure symptoms in patients who have preserved left ventricular function but impaired diastolic function), atrial fibrillation, and cardiac ischemia.14,15
Table.
Age-Associated Changes in the Heart
|
The heart contains a very high density of mitochondria due to its extraordinary demand for ATP, which is mainly provided by oxidative phosphorylation (OXPHOS). Other biochemical processes involved in energy production such as fatty acid oxidation (FAO), Krebs cycle, and tricarboxylic acid cycle also take place in mitochondria. Except for 13 subunits of the oxidative phosphorylation chain that are encoded by the mitochondrial genome, all mitochondrial proteins are encoded within the nucleus. Mitochondrial biogenesis is regulated on the transcriptional level by a set of nuclear encoded coactivators and transcription factors. The first coactivator demonstrated to regulate mitochondrial biogenesis and function was identified as a coactivator of nuclear receptor peroxisome proliferator-activated receptor (PPAR)γ in brown adipose tissue and named PGC-1α.16 Subsequently, 2 more related coactivators were identified: PGC-1β and PGC-related coactivator (PRC).17,18 Of these 3 PGC members, PGC-1α and PGC-1β have been well studied and demonstrated to coordinate mitochondrial biogenesis and function through association with different transcription factors, including nuclear respiratory factors 1 and 2 (NRF-1 and NRF-2), estrogen-related receptor α (ERRα), and PPAR, to drive the expression of mitochondrial genes important for mitochondrial energy transduction, ATP generation, and mitochondrial biogenesis.19
Age-related mitochondrial changes include increased reactive oxygen species (ROS), impaired OXPHOS, reduced ATP generation, impaired FAO, and increased mtDNA mutations.20-23 The pathophysiological relevance of alterations in each of these processes for cardiac disease has been documented in patients with genetic disorders.24 Patients with loss-of-function mutations in genes essential for mtDNA integrity, OXPHOS, or β-oxidation develop significant cardiac compromise and cardiomyopathy.24-26 Studies in genetically modified mice have supported these clinical observations. Mice carrying mutations in genes resulting in compromised fatty acid transport and oxidation, ATP generation, protection from ROS, and mtDNA proofreading capacity all develop cardiac dysfunction. Although the study of single gene mutations has highlighted the importance of specific processes for the maintenance of cardiac function, it has been noted that there is often compromise of multiple mitochondrial processes in aged heart tissues.27,28 According to the mitochondrial variant of the free radical theory of aging, this is due to increasing levels of mitochondrial ROS, which leads to a progressive forward-feedback spiral of increasing damage to mtDNA and other mitochondrial components. This in turn induces more ROS through damage to the electron transport chain, furthering ROS production and more mtDNA damage and ultimately leading to bioenergetic failure.27,28 Support for an important role of ROS in driving cardiac aging comes from findings in mice overexpressing catalase (an antioxidant enzyme) selectively in heart mitochondria.29 Mitochondrial catalase overexpression reduces ROS-mediated damage to proteins and mtDNA, prevents functional decline in the heart and increases median lifespan.29 Interestingly, catalase overexpression also improves the cardiomyopathy observed in mice expressing a proofreading-impaired variant of mitochondrial polymerase γ, which is involved in mtDNA replication and repair.30 The expression of this variant is associated with a substantial increase in mtDNA point mutations and deletions and an accelerated aging phenotype including cardiomyopathy (so-called mutator mouse).31,32 The beneficial effect of catalase overexpression on cardiac function in the mutator mice is surprising, given the absence of increased ROS levels in these mice. However, it can be hypothesized that the neutralization of even very mildly increased (and undetected) ROS levels are beneficial or that alternatively, in the setting of increased mtDNA damage, even normal levels of ROS might be deleterious.
Although the excessive levels of mtDNA damage observed in mutator mice are beyond the levels seen during natural aging in human and rodents, the development of cardiomyopathy in these mice highlights the importance of mtDNA integrity for normal cardiac function.33 Increased mtDNA mutations and deletions have been documented in aged heart tissues of humans and rodents, and this might be facilitated by the lack of mtDNA histones and the limited mtDNA repair capacity. Mechanistically, the increase in mtDNA damage is thought to compromise the electron transport chain through dysregulated transcription of mtDNA encoded respiratory chain components. This view is supported by the findings in the mutator mouse, which have depressed complex I and IV activities, reduced ATP generation, and subsequently develop dilated cardiomyopathy.32 The decline in electron transport chain activity has been also noted in naturally aged hearts in humans and rodents but might affect only a subset of mitochondria.27,34 The decline in electron chain activity is often associated with impaired ATP generation.8,34 Other age-dependent changes in mitochondrial energy pathways have been noted and are thought to contribute to ATP depletion phenotype, including decreased FAO rates and an increase in glycolysis in animal models of pressure overload–induced cardiac hypertrophy and heart failure.8
In humans, there is growing evidence that cardiac hypertrophy and heart failure have also altered energy pathways, although the changes are not uniform. For example, whereas hypertension-induced cardiac hypertrophy and heart failure is characterized by impaired FAO and a shift to glucose utilization resembling the fetal situation with reliance of glycolysis for ATP generation,35 the heart of diabetic patients utilizes primarily fatty acids.36-38 Furthermore, the degree of cardiac dysfunction appears to be an important determinant for fuel preference based on studies demonstrating that FAO is unchanged or increased in early heart failure but significantly decreased in later stages. Similarly, glucose utilization is increased early in heart failure but drops significantly in advanced heart failure.8 The progression to heart failure in humans is associated with a decline of ATP content and reduction in the ATP transfer capacity.8 PCr/ATP ratios are reduced with heart failure severity and are good predictors of cardiovascular mortality.39
The Role of PGCs in Mitochondrial and Heart Decline
Multiple signaling pathways converge to compromise mitochondrial function during aging in the heart (Figure 1). Among these, PGC family members have gained particular interest because of their capacity to drive virtually all aspects of mitochondrial biogenesis in the heart, including mitochondrial number, mitochondrial respiration, expression of OXPHOS and FAO genes, and ROS levels.19,40 Gain-of-function and loss-of-function studies highlight the critical role of PGC family members in the heart. One study showed that transgenic PGC-1α overexpression in mice leads to significant increase in mitochondrial number in the heart, leading to lethal cardiomyopathy.40 Mice lacking either PGC-1α or PGC-1β have no overt cardiac phenotype.41-43 However, more detailed analysis has uncovered a compromise in bioenergetics (reduced FAO, ATP synthesis, and myocardial efficiency) and defects in cardiac function (isolated hearts showed decreased cardiac work and output) at baseline.41,43 Under conditions of increased hemodynamic stress and increased bioenergetic demands, PGC-1α– and PGC-1β–deficient mice develop overt cardiac failure.42,44 Decreased PGC-1α expression has been also linked to the development of heart failure in other mouse models, including mice overexpressing cyclin-dependent kinase 9 (Cdk9).45 Cardiac failure is even more pronounced in mice lacking both coactivators, indicating a functional overlap of these coactivators. In contrast to mice deficient for PGC-1α or PGC-1β, double knock-out mice die shortly after birth, with severe cardiac and mitochondrial defects.46 The relevance of reduced activity of the PGC regulatory network in cardiac aging is further supported by the development of mitochondrial dysfunction and cardiomyopathy in mice lacking PGC targets such as ERRα and Tfam.47,48
Figure 1. Pathways implicated in contributing to mitochondrial dysfunction in the heart with aging.
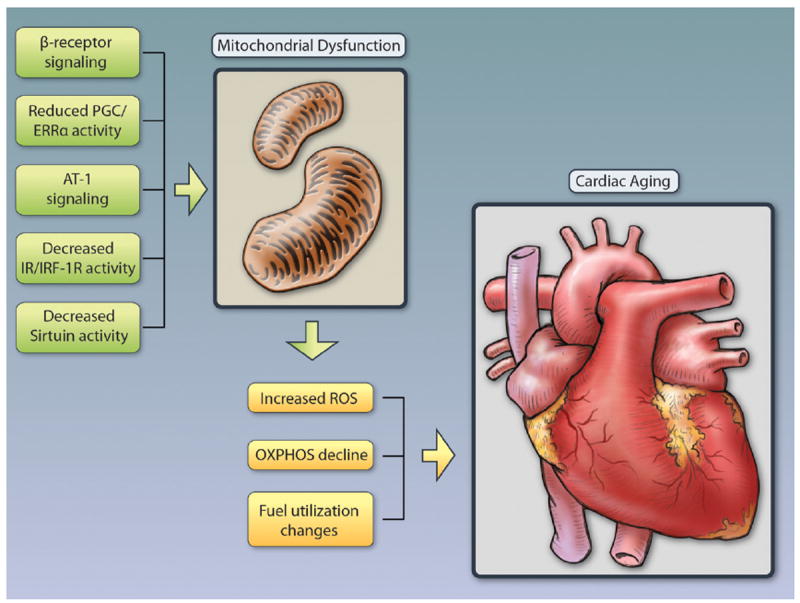
Multiple signaling pathways have been implicated in driving mitochondrial decline with age. Besides reduced activity of the PGC/ERRα network (see text) Angiotensin II has been suggested to play a role as activation of the Angiotensin receptor type I (AT-1) impairs mitochondrial biogenesis and mitochondrial respiratory chain activity and deletion of the AT-1 in mice is associated with increased number of mitochondria, decreased ROS induced oxidative damage, and improved cardiac function.117 Elevated norepinephrine and activation of β-adrenergic receptors in the heart increases ROS, impairs mitochondrial function and deletion of AC5 (the predominant isoform of adenyl cyclase in the heart), and protects from oxidative stress through activation of the RAF/MEK/ERK signaling pathway.118 Similarly, β-receptor blockade with carvedilol reduces oxidative stress levels in the heart.119,120 The insulin/IGF-1 pathway has been suggested to play a protective role, based on findings in mice deficient for either the insulin (CIRKO), the insulin-like growth factor 1 (IGF-1), or both receptors (MI2RKO).121,122 These mice have varying degrees of reduced oxidative phosphorylation with decreased ATP generation, increased ROS, reduced tricarboxylic acid cycle and fatty acid oxidation genes, and impaired cardiac function.123 Of the 7 mammalian sirtuins, Sirtuin 1 and Sirtuin 3 have been particularly implicated in delaying cardiac aging and protecting from oxidative stress. Illustration credit: Cosmocyte/Ben Smith.
Studies in patients with cardiac hypertrophy and heart failure have suggested that alterations in the PGC network underlie cardiac hypertrophy and failure.49-51 However, these studies also highlight the complexity of PGC biology. For example, in one study, PGC-1α targets such as ERRα, PPARδ, as well as OXPHOS genes were downregulated whereas PGC-1α expression was increased.52 One important point to consider in understanding these discrepancies is the complex posttranslational control of PGC activity, which may not correlate with expression levels. The functional redundancy of the different PGC members and their dependence on downstream transcription factors further complicate the analysis of the PGC network.
Together, these studies indicate that mitochondrial decline is a feature of the aging heart and that dysregulation at the transcriptional level underlies some of the changes. What causes PGC repression is not entirely clear, but there is evidence that increased DNA damage due to shortened telomeres and activation of p53 play a role. This issue will be discussed next.
Telomere Structure
Telomeres consist of several kilobases of tandem-repeat double-stranded TTAGGG sequences that are followed by a single-stranded, few hundred nucleotide–long 3′-overhang that loops back into telomeres to form a so-called t-loop. The essential function of the t-loop is to prevent telomere ends from being recognized as break points by the DNA damage repair machinery. Telomeres associate with a number of proteins, 6 of which are highly telomere specific (TRF1, TRF2, POT1, TIN2, TPP1, and Rap1) and form the shelterin complex. The shelterin complex is essential for the formation of the t-loop, maintenance of telomere length, and the suppression of the DNA damage response (DDR; Figure 2). Other non–telomeric-specific proteins contribute to telomere maintenance and include DNA repair (XPF/ERCC1, Rad51D, Ku70/80, Apollo, Mre11), DNA replication (RecQ helicase and ORC), and DNA damage signaling proteins (Mre11 and 9–1–1 complex).53 Although it was widely believed that the only role of telomeres is to provide structural stability to chromosomal ends, recent studies have demonstrated that mammalian telomeres are transcribed into telomeric repeat-containing RNA (TERRA).54 Akin to previous work in flies, trypanosomes, and birds, TERRA transcription in mammalian cells starts at subtelomeric regions and moves outward to generate TERRA of various lengths which localize at telomeres.55-58 Functionally, TERRA has been shown to regulate telomerase activity, telomere length, heterochromatin formation, and the DDR.58,59 Thus, telomere length and integrity are regulated through a complex interplay of proteins (shelterin and nonshelterin proteins), TERRA, and telomerase. Even in the absence of significant telomere length attrition, changes in the t-loop structure, composition of the shelterin complex, and telomeric proteins can induce telomere dysfunction and activate the DDR. Telomere length is regulated in a species-, tissue- and cell type–specific manner. There is a growing appreciation that telomere length is not fixed and can be modulated by exercise or psychological factors.60-64 Telomeres are maintained by the enzyme telomerase. Telomerase is composed of a catalytic active component TERT and the telomerase RNA component (TERC), which serves as a template during telomere biosynthesis (Figure 2). In the absence of telomerase, cells are not capable of solving the end-replication problem and experience telomere shortening with each successive round of DNA replication.65,66 In humans, with the exception of progenitor and stem cell compartments and some other notable exceptions such as activated lymphocytes, most tissues have no or very little telomerase activity primarily due to transcriptional repression of TERT. However, even in compartments with documented telomerase activity, such as CD34 positive hematopoietic stem cells, telomere length is not maintained as a function of age in humans, pointing to other levels of regulation in the maintenance of telomeres.4
Figure 2. Telomere structure and telomerase.
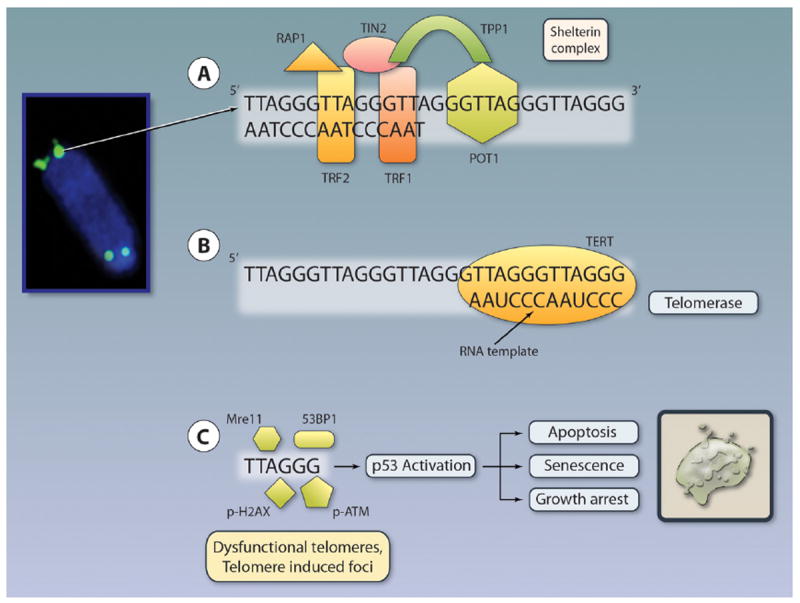
Telomeres (shown in green by fluorescence in situ hybridization) are repetitive TTAGGG sequences at the ends of chromosomes. The 3′ end contains a G-rich, single-stranded overhang that loops and invades the double-stranded telomeric region to form a so-called t-loop (not depicted). Telomeres exist in association with many different proteins that contribute to the overall stability. Six telomere-binding proteins (POT1, TRF1, TRF2, TIN2, TPP1, and Rap13) are telomere-specific and form the shelterin complex. Three, POT1, TRF1, and TRF2 directly bind to telomeres, whereas TIN2, TPP1, and Rap13 connect them (A). Telomeres are maintained by the enzyme telomerase, which consists of a RNA subunit (AAUCCC) and the reverse transcriptase, TERT, which are illustrated. The catalytic component TERT uses the RNA component to synthesize new telomeres (B). Progressive telomere shortening leads to telomere shortening and activation of the DNA damage machinery (53BP1, Mre11, and (phosphorylated) p-H2AX, p-ATM, and others) and subsequent activation of p53 and induction of apoptosis, growth arrest, and senescence (C). Telomere length is measured traditionally by Terminal Restriction Fragment (TRF) length analysis by Southern blot. TRF assesses the average telomere length within a cell population with up to 1-kb resolution. Telomere length can be alternatively measured by fluorescence in situ hybridization (FISH) with greater sensitivity (resolution, 0.3 kb) on metaphase or interphase chromosomes using directly fluorescently labeled (CCCTAA)3 peptide nucleic acid (PNA) probes. This method can be combined with immunohistochemical staining to detect TIFs. A variant of FISH has been adopted for flow (Flow-FISH). Newer methods use PCR methods with greater sensitivity (Single Telomere Length Analysis or STELA, Q-PCR, and monochrome multiplex Q-PCR). Illustration credit: Cosmocyte/Ben Smith.
Telomeres and Telomerase in Aging
The importance of adequate telomerase activity and maintenance of telomere length for both replicative potential in cultured cells and organismal aging was initially inferred from a series of seminal studies using cultured human fibroblasts. These in vitro experiments demonstrated that continuous passage of fibroblasts resulted in telomere shortening and eventual cellular senescence after a finite number of cell divisions, a barrier known as the Hayflick limit.67 The critical role of adequate telomere length in this senescence process was demonstrated by the capacity of enforced TERT expression to stabilize telomere length and enable unlimited replicative potential.68 These paradigmatic studies strongly implicated telomeres in processes of cellular senescence or “aging,” which were further substantiated in humans and in telomerase deficient mice.
Patients with autosomal dominant dyskeratosis congenita carry mutations in TERC or TERT and have shortened telomeres, signs of accelerated aging, and a reduced lifespan.69 TERC or TERT mutations have also been linked to more organ-restricted disease manifestations such as idiopathic pulmonary fibrosis (a fatal lung disease with excessive fibrosis) and bone marrow failure syndromes.70 Interestingly, telomerase mutations, although not widely recognized to confer predisposition to develop cardiac disease, have been recently linked to the development of cardiac disease such as cardiac fibrosis and cardiomyopathy in few reports (see below). The factors that determine disease localization and extent of clinical manifestations in patients with telomerase mutations remain unknown, although the degree of telomere dysfunction plays an important role as severity and onset of disease manifestation correlate with telomere length.70
Similarly, Werner syndrome patients with mutations in the WRN gene, which encodes a RecQ helicase involved in DNA repair, DNA recombination, and telomere maintenance, manifest clinically with multiple premature aging pathologies after puberty, including senile cataracts, osteoporosis, hair graying, myocardial infarction, and cancer.71 A potential role of telomeres in human disease pathogenesis was inferred by the capacity of enforced TERT expression to rescue the limited replicative potential of Werner syndrome fibroblasts with shortened telomeres.72-74 Taken together, studies of inherited human degenerative conditions support the view that telomere dysfunction is a rate-limiting pathogenic element underlying degenerative phenotypes and shortened lifespan.
The understanding of the in vivo relevance and specific roles of telomeres in aging and degenerative diseases was initially provided by the generation of mice null for either the Tert or Terc component of telomerase.4,75 Deletion of either component was found to be fully compatible with normal embryonic and postnatal development, indicating that telomerase per se is dispensable for life.75 However, the limited impact of telomerase deficiency was found to be related to the long starting telomeres in inbred laboratory mouse strains. Generation of mice with increasingly shorter telomeres through continuous interbreeding (G1×G1>G2, G2×G2>G3, etc) revealed that onset of telomere dysfunction dramatically diminished organismal fitness, promoted age-associated diseases, and reduced lifespan.76 On the tissue level, “late-generation” Terc−/− mice with short dysfunctional telomeres show widespread tissue atrophy with impaired organ function.76 Whereas proliferative tissues such as the hematopoietic system (anemia and lymphopenia), intestine (atrophy), skin (alopecia, hair graying, decreased wound healing), reproductive system (infertility), and skeletal system (kyphosis and osteoporosis) are prominently affected, there is also significant tissue pathology in more quiescent tissues such as the heart (cardiomyopathy, see also below) as well as generalized metabolic aberrations including insulin resistance. The severity of these conditions is dependent on the degree of telomere dysfunction, as measured by the number of chromosomal fusions, anaphase bridges, and number of DNA damage foci at telomeres (telomere-induced-foci [TIFs]).77 Studies in the mouse have further established that telomere dysfunction in a few chromosomes is sufficient to drive degenerative changes as observed in “classic” late-generation TERC−/− mice.78 The relevance of limiting telomeres to the aging process is further substantiated through intercrossing telomerase deficient mice with mice with classic human degenerative-disease mutations such as WRN or ATM.79,80 Strikingly, these mutant mice fail to recapitulate the classic human phenotypes unless telomeres are reduced to a more human-like length, thus supporting the view that telomere dysfunction plays a critical role in the pathogenesis of these conditions.
The Relevance of Telomeres in CVD
Studies in Tert and Terc knock-out mice have advanced our understanding of the relevance of critical short telomeres for cardiovascular aging and CVD. Late-generation Terc and Tert knock-out mice develop hallmark features of cardiomyopathy with severe systolic and diastolic dysfunction. This cardiomyopathy is linked to increased p53 levels and cardiomyocyte apoptosis.81 Cardiomyocyte specific Tert overexpression maintains telomere length in the adult mouse heart, inhibits apoptosis and extends the capacity of cardiomyocytes to proliferate from 8 to 12 weeks.82 Intriguingly, continued expression of Tert induces cardiac hypertrophy without fibrosis or impaired myocardial function, indicating a role for telomerase beyond modulation of proliferation and apoptosis in the heart.82
A causal role for telomere dysfunction in the pathogenesis of human CVD is supported by 3 lines of evidence. First, case reports indicate that CVD might be more common in patients with telomerase germline mutations than previously appreciated. Two members of a family with autosomal dominant mutations in TERT developed cardiac disease at an early age. One member developed dilated cardiomyopathy, whereas a second one had severe cardiac fibrosis.83 In a different study, myocardial infarction at a young age was documented in a patient carrying a mutation in telomerase.84 Accordingly, other telomere maintenance disorders such as Werner syndrome are associated with premature CVD including atherosclerosis, aortic stenosis, valvular disease, mitral regurgitation, and calcification.85 Although a mouse model of Werner syndrome develops cardiac fibrosis, only 1 instance of florid cardiac fibrosis in a 17-year-old with Werner syndrome has been reported.86,87 Although these reports are intriguing, systematic studies in asymptomatic patients are needed to more precisely quantify the prevalence and extent of cardiac compromise in patients with telomere maintenance disorders. Second, human cardiomyocytes and cardiac stem cells display an age-dependent decline in telomere length and express senescence markers such as p53, p21, and p16. These changes correlate with a decreased proliferative and regenerative capacity of aged cardiomyocytes and cardiac stem cells.11,88 Notably, failing human hearts with decreased expression of telomere repeat binding-factor 2 (TRF2) contain 25% shorter telomeres than age-matched healthy control subjects. Consistent with this documented telomere attrition, there is robust activation of DNA damage kinase, checkpoint kinase 2 (Chk2) and increased cardiomyocyte apoptosis.89 Third, the relevance of critical short telomeres for human heart aging and disease is inferred from a large number of epidemiological studies demonstrating associations between shortened telomeres, aging, and CVD. In these studies, leukocyte telomere length correlates inversely with multiple cardiovascular risk factors, including cigarette smoking, obesity, diabetes, systemic inflammation, and physical inactivity.90-92 In case-control studies, decreased leukocyte telomere length is associated with CAD and premature myocardial infarction as well as intermediate phenotypes such as carotid media thickness and coronary calcification.93,94 Prospective cohort studies show that decreased leukocyte telomere length independently predicts mortality and cardiovascular events in the general population, patients with stable CAD, and patients with severe congestive heart failure.95,96,97 These studies are complemented by a nested case-control study, carried out as part of a large, randomized trial involving statin treatment. In this study, individuals with shorter leukocyte telomere length had a significantly higher risk for developing coronary heart disease over 5 years.98 In contrast to the above studies, longer telomeres have been associated with increased left ventricular mass in 2 independent studies, but the relationship between these 2 observations and the consistent association of short telomeres with adverse outcomes requires further study.99,100
The interpretation of the above studies remains complicated, given that most studies are cross-sectional and capture telomere length only at a single time point. As such, leukocyte telomere length may be a consequence of coronary heart disease rather than causative. Furthermore, the extent to which telomere length in circulating leukocytes reflects telomere dynamics in tissues such as the heart remains largely unclear, although a close correlation between leukocyte and aortic wall tissue telomere length has been reported.101
Cellular and Molecular Mechanisms of Telomere-Driven Aging in the Heart
The primary mechanism of telomere-driven aging rests on the principle that cellular turnover results in progressive loss of telomere reserves, which ultimately generates short dysfunctional telomeres. These dysfunctional telomeres were shown to activate p53-dependent DNA damage checkpoint mechanisms such as proliferative arrest, apoptosis, and senescence. This telomere-driven process was particularly evident in highly proliferative tissues with high renewal requirements, which aligns with the depletion of stem and progenitor cell reserves in these tissues.102,103
In the heart, cardiac stem and progenitor cells have been reported to undergo telomere attrition during aging despite telomerase activity, similar to what has been described in other stem cells.104 The documented telomere shortening in aged cardiac stem/progenitor cells could impair cardiomyocyte renewal.104 However, whereas the decline in cardiac stem/progenitor cell–dependent regeneration might contribute to cardiac aging, the overall renewal capacity, even of young hearts, is low and inadequate to replace ongoing age-associated cardiomyocyte loss.105
The impact of dysfunctional telomeres on mature cardiomyocytes is evident on 2 levels. First, telomere attrition has been demonstrated to modestly induce cardiomyocyte apoptosis, and, conversely, cardiac specific telomerase overexpression is associated with decreased apoptosis in vivo and in cultured cardiomyocytes.82 Second, telomere dysfunction might impair the regenerative capacity of cardiomyocytes or the ability of a subset of cardiomyocytes to regenerate, similar to what has been shown in the liver, where terminally differentiated hepatocytes can reenter cell cycle but are impaired in the setting of telomere dysfunction.106 Thus, the combined negative impact of dysfunctional telomeres on cardiac stem/progenitor cells and mature cardiomyocytes might explain some aspects of the failing heart in the aged.
More significantly, recent genetic studies in the mouse have established that telomere dysfunction not only induces the cellular phenotypes of proliferative arrest, apoptosis, and senescence but compromises cardiomyocyte function through repression of PGC-1α and PGC-1β.6 The combined PGC-1α and PGC-1β repression is associated with impaired mitochondrial biogenesis and function in the heart and other tissues.46 The decrease in mitochondrial mass and impairment in the electron transport chain negatively affects ATP synthesis capacity and leads to a decrease in cardiac ATP levels. Furthermore, metabolic processes such as β-oxidation and fatty acid synthesis are affected. Importantly, the mitochondrial and metabolic changes are reversible as PGC-1α overexpression in telomere dysfunctional mice rescues the oxidative electron chain defect.6 Interestingly, cardiomyocyte-specific telomerase overexpression prevents the decline in telomere length during postnatal heart maturation and induces hypertrophy.82 The basis for the hypertrophy was not identified in this study. Given the recent findings that telomere dysfunction leads to widespread metabolic changes, it is tempting to speculate that telomerase reactivation can reverse a more global metabolic decline. In this regard, the fundamental demonstration that aged tissues in telomere dysfunctional mice can be rejuvenated by endogenous telomerase reactivation allows investigation into what extent this rejuvenation process is dependent on metabolic changes in postmitotic cells and tissues.107
These two arms—increased apoptosis/senescence and impaired mitochondrial and metabolic compromise—do not have to be mutually exclusive (Figure 3). Such a view is supported by the finding that older telomere dysfunctional mice have increased apoptosis in addition to mitochondrial dysfunction whereas young telomere dysfunctional mice have compromised mitochondrial function without any evidence of significant apoptosis. These findings also indicate that mitochondrial dysfunction is the primary event. The degree of telomere dysfunction and the associated DDR response probably will dictate whether a cell will be only metabolically compromised or undergo apoptosis or senescence.
Figure 3. Model of how telomere dysfunction and other pathways cause cardiac aging through either cellular or metabolic compromise.
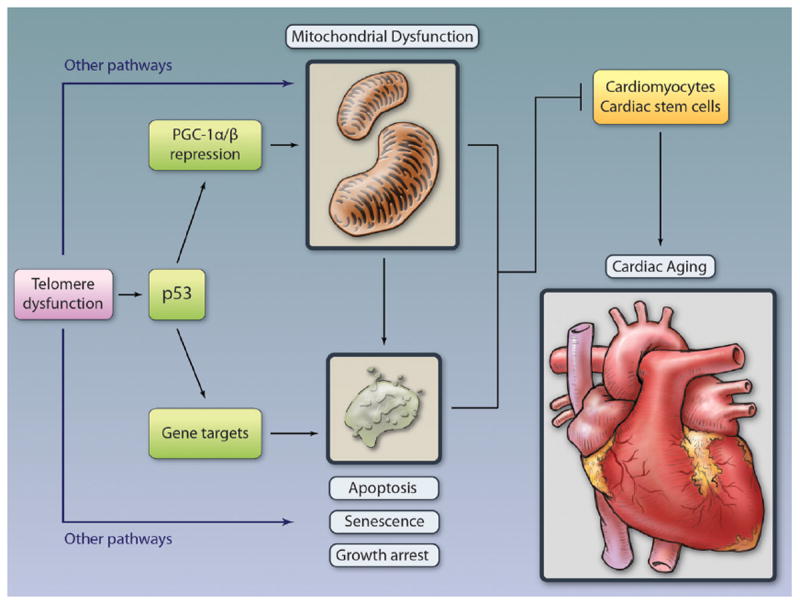
Telomere dysfunction–induced p53 plays an pivotal role in inducing age-associated functional decline.5 The pathways engaged by p53 are mutually nonexclusive and involve repression of PGC and consequent mitochondrial dysfunction, as well as induction of classic cellular outcomes of apoptosis, senescence, and growth arrest.6,5 Mitochondrial dysfunction per se can lead to growth arrest, senescence, or apoptosis, depending on extent and duration as well as other cellular events. Telomere dysfunction activates other p53-independent pathways to mediate mitochondrial dysfunction and cellular phenotypes. Illustration credit: Cosmocyte/Ben Smith.
Because most studies have assessed the metabolic and mitochondrial changes in whole heart, it is unclear whether these mitochondrial and metabolic changes also occur in human or mouse cardiac stem/progenitor cells during aging. Previously, it could be demonstrated that telomere dysfunction represses PGCs also in hematopoietic stem cells, and this is associated with their severe functional compromise. Along these lines, it is worth noting that hematopoietic stem cells (HSCs) appear to be exquisitely sensitive to metabolic perturbation as shown, for example, in LKB1-deficient HSCs, which display a profound self-renewal defect along with metabolic and mitochondrial compromise.108 The importance of PGC for stem cell maintenance is also suggested by studies in drosophila. Here, overexpression of the Drosophila PGC-1 homolog (dPGC-1/spargel) and increasing mitochondrial activity in stem and progenitor cells of the digestive tract is sufficient to extend the lifespan.109 That said, further study is needed to firmly establish the relevance of these mitochondrial and metabolic changes in stem cell function, particularly given recent studies demonstrating that HSCs are more dependent on glycolysis than oxidative phosphorylation.110 Whether this is true for cardiac stem cells is unclear.
Although the molecular pathways that sense telomere dysfunction and activate cellular checkpoint responses as well as PGC repression have yet to be established, p53 has been shown to be the central effector of telomere dysfunction (Figure 3).5 The importance of p53 activation for the development of degenerative disorders was demonstrated by the rescue of most degenerative phenotypes in telomerase knock-out mice also null for p53.5 This widespread rescue by p53 deficiency was mainly attributed to the rescue of proliferative arrest, apoptosis, and senescence. In more recent studies, however, the role of p53 has been expanded by the discovery that activated p53 can directly repress PGC-1α and PGC-1β gene express on the transcriptional level in a manner dependent on the binding of p53 to PGC-1α and PGC-1β promoters. Accordingly, p53 deficiency leads to increased PGC expression and partially rescues mtDNA copy number in heart and liver tissues in telomere dysfunctional mice. On a functional level, p53 deficiency improves low-dosage, doxorubicin-induced cardiomyopathy in telomere dysfunctional mice.6 The prominent role of p53 in driving cardiac failure/degeneration is further supported by the increase in p53 in aging hearts and in heart failure.81,111,112 Genetic evidence for an essential role of p53 in the development of cardiac failure derives further support from cardiac-specific deletion of Mdm4, a negative regulator of p53. These mice develop dilated cardiomyopathy through increased apoptosis and senescence.113 Furthermore, chronic p53 elevation inhibits hypoxia-inducible factor-1 (Hif-1)-dependent induction of angiogenic factors and induces the transition from hypertrophy to heart failure in a mouse model of cardiac hypertrophy.112 Similarly, hyperactivation of p53 in the setting of cardiac specific deletion of Sirt1 (which inhibits p53 through deacetylation) predisposes cardiomyocytes to cell death, and, conversely, modest Sirt1 overexpression in cardiomyocytes attenuates age-dependent cardiomyopathy.107,114,115 Given that Sirt1 is an important activator of PGC, cardiac specific increased PGC expression could be a major contributor to the attenuated cardiomyopathy phenotype in this model.
Conclusion and Perspective
The study of human telomere maintenance disorders and genetic model systems has advanced our understanding of the role of telomeres in the aging process. These studies have pointed to the importance of telomere integrity in maintaining tissue homeostasis and regulating lifespan. Whereas the role of telomere dysfunction in driving tissue atrophy in high turnover tissues is well established, there is also growing recognition that tissues with less proliferative profiles are also affected. Increasing evidence indicates that critical short telomeres play an important role in cardiac aging and disease. Some of the conflicting results relate to the differences in assessing telomere status and the reliance on measurements of telomere length in peripheral leukocytes. For future human studies, it would be desirable to standardize protocols for analyzing the degree of telomere dysfunction (ie, TIFs) rather than telomere length in cardiomyocytes, where possible. It will be also important to identify new markers that might allow more straightforward and technically less challenging assessment of telomere dysfunction. How telomere dysfunction develops in cells with a low degree of cell turnover such as cardiomyocytes is largely unclear and remains an important are of investigation. The relevance of telomeres for CVD might find further clarification from much-needed prospective studies in patients with telomere maintenance syndromes. Systematic analysis of cardiovascular function in these patients will establish the prevalence of cardiac compromise, which might be underreported due to the absence of symptoms.
On a mechanistic level, recent reports linking telomere dysfunction to metabolic and mitochondrial compromise provide a novel mechanism as to how dysfunctional telomeres can compromise cardiac function. This telomere-p53-PGC-mitochondrial axis aligns with many changes seen in aged hearts: impaired OXPHOS, decreased ATP generation, and increased ROS levels. Other metabolic changes, including compromised FAO and a shift to glucose utilization, can be expected to be altered with telomere dysfunction, based on the downregulation of the PGC-PPARα axis in mice with critical short telomeres. It will be important to establish whether similar mitochondrial/metabolic decline occurs in hearts and other tissues of patients with telomerase mutations and short telomeres.
Besides p53, other genes and pathways appear to be involved in compromising mitochondrial function in the setting of telomere dysfunction, as p53 deficiency only partially rescues PGC expression and mitochondrial dysfunction. The identification of these other pathways will expand our understanding of how critical short telomeres compromise cellular function and precipitate aging. The telomere-p53-PGC link might be useful in both identifying aging biomarkers as well as illuminating targeted therapeutic avenues in the prevention and reversal of age-associated cardiovascular decline. In keeping with this, therapeutic strategies to stabilize DNA integrity, attenuate DDR signaling, or enhance mitochondrial function through pharmacological and nonpharmacological means such as continuous exercise—which is demonstrated to reverse the premature aging phenotype of the mutator mouse—are possible avenues to pursue.116 This telomere-mitochondrial link further adds to the notion that compromised energy maintenance pathways are central to heart aging and failure. Accordingly, metabolic pathways can be considered a promising target to delay and treat heart failure.
Acknowledgments
We thank Drs Ramin Farzeneh-Far, Rajan Sah, and Marc Liesa Roig for critical review of the manuscript. We apologize to all those investigators whose work could not be cited.
Sources of Funding
J.M. is supported by a National Institutes of Health training grant for cardiovascular research (K08HL097031) and the Watkins Cardiovascular Discovery Award (from Brigham and Women’s Hospital). E.S. was supported by the Deutsche Forschungsgemeinschaft, and R.A.D. is supported by R01CA84628 and 1U01CA141508–01 grants from the NIH National Cancer Institute and the Robert A. and Renee E. Belfer Foundation. R.A.D. was supported by an Ellison Foundation for Medical Research Senior Scholar and an American Cancer Society Research Professor Award.
Non-standard Abbreviations and Acronyms
- CAD
coronary artery disease
- CVD
cardiovascular disease
- DDR
DNA damage response
- FAO
fatty acid oxidation
- HSC
hematopoietic stem cell
- mtDNA
mitochondrial DNA
- OXPHOS
oxidative phosphorylation
- PGC
peroxisome proliferator-activated receptor gamma coactivator
- PPAR
peroxisome proliferator-activated receptor
- PRC
PGC-related coactivator
- ROS
reactive oxygen species
- TERC
telomerase RNA component
- TERT
telomerase reverse transcriptase
- TERRA
telomeric repeat-containing RNA
- TIF
telomere-induced foci
Footnotes
Disclosures
None.
Contributor Information
Javid Moslehi, Division of Cardiovascular Medicine, the Department of Medicine, Brigham and Women’s Hospital, and the Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA.
Ronald A. DePinho, Department of Cancer Biology, University of Texas MD Anderson Cancer Center, Houston, TX
Ergün Sahin, Huffington Center on Aging and the Department of Molecular Physiology and Biophysics, Baylor College of Medicine, Houston, TX.
References
- 1. [April 2, 2012];National Population Projections. http://www.census.gov/population/www/projections/natproj.html.
- 2.Roger VL, Go AS, Lloyd-Jones DM, et al. Heart disease and stroke statistics: 2011 update: a report from the American Heart Association. Circulation. 2011;123:e18–e209. doi: 10.1161/CIR.0b013e3182009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. [April 2, 2012];Heart Disease and Stroke Prevention. http://www.cdc.gov/chronicdisease/resources/publications/aag/pdf/2011/Heart-Disease-and-Stroke-AAG-2011.pdf.
- 4.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chin L, Artandi SE, Shen Q, Tam A, Lee SL, Gottlieb GJ, Greider CW, DePinho RA. P53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 6.Sahin E, Colla S, Liesa M, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. 2007;12:331–343. doi: 10.1007/s10741-007-9034-1. [DOI] [PubMed] [Google Scholar]
- 8.Neubauer S. The failing heart: an engine out of fuel. N Engl J Med. 2007;356:1140–1151. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]
- 9.Olivetti G, Melissari M, Capasso JM, Anversa P. Cardiomyopathy of the aging human heart: myocyte loss and reactive cellular hypertrophy. Circ Res. 1991;68:1560–1568. doi: 10.1161/01.res.68.6.1560. [DOI] [PubMed] [Google Scholar]
- 10.Chien KR, Karsenty G. Longevity and lineages: toward the integrative biology of degenerative diseases in heart, muscle, and bone. Cell. 2005;120:533–544. doi: 10.1016/j.cell.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal-Ginard B, Kajstura J, Anversa P, Leri A. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res. 2004;94:514–524. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 12.Bergmann O, Bhardwaj RD, Bernard S, Zdunek S, Barnabe-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA, Druid H, Jovinge S, Frisen J. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises, part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. [DOI] [PubMed] [Google Scholar]
- 14.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises, part I: aging arteries: a “set up” for vascular disease. Circulation. 2003;107:139–146. doi: 10.1161/01.cir.0000048892.83521.58. [DOI] [PubMed] [Google Scholar]
- 15.Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises, part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003;107:490–497. doi: 10.1161/01.cir.0000048894.99865.02. [DOI] [PubMed] [Google Scholar]
- 16.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 17.Andersson U, Scarpulla RC. Pgc-1-related coactivator, a novel, serum-inducible coactivator of nuclear respiratory factor 1-dependent transcription in mammalian cells. Mol Cell Biol. 2001;21:3738–3749. doi: 10.1128/MCB.21.11.3738-3749.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin J, Puigserver P, Donovan J, Tarr P, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1beta (pgc-1beta), a novel pgc-1-related transcription coactivator associated with host cell factor. J Biol Chem. 2002;277:1645–1648. doi: 10.1074/jbc.C100631200. [DOI] [PubMed] [Google Scholar]
- 19.Leone TC, Kelly DP. Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harbor Symp Quantitative Biol. 2011;LXXVI:1–7. doi: 10.1101/sqb.2011.76.011965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zahn JM, Poosala S, Owen AB, et al. Agemap: a gene expression database for aging in mice. PLoS Genet. 2007;3:e201. doi: 10.1371/journal.pgen.0030201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brink TC, Demetrius L, Lehrach H, Adjaye J. Age-related transcriptional changes in gene expression in different organs of mice support the metabolic stability theory of aging. Biogerontology. 2009;10:549–564. doi: 10.1007/s10522-008-9197-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marin-Garcia J, Pi Y, Goldenthal MJ. Mitochondrial-nuclear cross-talk in the aging and failing heart. Cardiovasc Drugs Ther. 2006;20:477–491. doi: 10.1007/s10557-006-0584-6. [DOI] [PubMed] [Google Scholar]
- 23.Asakura M, Kitakaze M. Global gene expression profiling in the failing myocardium. Circ J. 2009;73:1568–1576. doi: 10.1253/circj.cj-09-0465. [DOI] [PubMed] [Google Scholar]
- 24.Wallace DC. Mitochondrial defects in cardiomyopathy and neuro-muscular disease. Am Heart J. 2000;139:S70–S85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 25.DiMauro S, Schon EA. Mitochondrial respiratory-chain diseases. N Engl J Med. 2003;348:2656–2668. doi: 10.1056/NEJMra022567. [DOI] [PubMed] [Google Scholar]
- 26.Kelly DP, Strauss AW. Inherited cardiomyopathies. N Engl J Med. 1994;330:913–919. doi: 10.1056/NEJM199403313301308. [DOI] [PubMed] [Google Scholar]
- 27.Marin-Garcia J, Goldenthal MJ. Mitochondrial centrality in heart failure. Heart Fail Rev. 2008;13:137–150. doi: 10.1007/s10741-007-9079-1. [DOI] [PubMed] [Google Scholar]
- 28.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 29.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kujoth GC, Hiona A, Pugh TD, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 32.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 33.Kujoth GC, Bradshaw PC, Haroon S, Prolla TA. The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 2007;3:e24. doi: 10.1371/journal.pgen.0030024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatarkova Z, Kuka S, Racay P, Lehotsky J, Dobrota D, Mistuna D, Kaplan P. Effects of aging on activities of mitochondrial electron transport chain complexes and oxidative damage in rat heart. Physiol Res. 2011;60:281–289. doi: 10.33549/physiolres.932019. [DOI] [PubMed] [Google Scholar]
- 35.van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res. 2004;61:218–226. doi: 10.1016/j.cardiores.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 36.Carley AN, Severson DL. Fatty acid metabolism is enhanced in type 2 diabetic hearts. Biochim Biophys Acta. 2005;1734:112–126. doi: 10.1016/j.bbalip.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 37.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–E1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 38.Paulson DJ, Crass MF., III Endogenous triacylglycerol metabolism in diabetic heart. Am J Physiol. 1982;242:H1084–H1094. doi: 10.1152/ajpheart.1982.242.6.H1084. [DOI] [PubMed] [Google Scholar]
- 39.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 40.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arany Z, He H, Lin J, et al. Transcriptional coactivator pgc-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Riehle C, Wende AR, Zaha VG, et al. Pgc-1β deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res. 2011;109:783–793. doi: 10.1161/CIRCRESAHA.111.243964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehman JJ, Boudina S, Banke NH, Sambandam N, Han X, Young DM, Leone TC, Gross RW, Lewandowski ED, Abel ED, Kelly DP. The transcriptional coactivator pgc-1alpha is essential for maximal and efficient cardiac mitochondrial fatty acid oxidation and lipid homeostasis. Am J Physiol Heart Circ Physiol. 2008;295:H185–H196. doi: 10.1152/ajpheart.00081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sano M, Wang SC, Shirai M, Scaglia F, Xie M, Sakai S, Tanaka T, Kulkarni PA, Barger PM, Youker KA, Taffet GE, Hamamori Y, Michael LH, Craigen WJ, Schneider MD. Activation of cardiac cdk9 represses pgc-1 and confers a predisposition to heart failure. EMBO J. 2004;23:3559–3569. doi: 10.1038/sj.emboj.7600351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai L, Leone TC, Zechner C, Schaeffer PJ, Kelly SM, Flanagan DP, Medeiros DM, Kovacs A, Kelly DP. Transcriptional coactivators PGC-1alpha and PGC-lbeta control overlapping programs required for perinatal maturation of the heart. Genes Dev. 2008;22:1948–1961. doi: 10.1101/gad.1661708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, Giguere V, Murphy E, Kelly DP. The nuclear receptor ERRalpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. 2007;6:25–37. doi: 10.1016/j.cmet.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 48.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 49.Garnier A, Zoll J, Fortin D, N′Guessan B, Lefebvre F, Geny B, Mettauer B, Veksler V, Ventura-Clapier R. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circ Heart Fail. 2009;2:342–350. doi: 10.1161/CIRCHEARTFAILURE.108.812099. [DOI] [PubMed] [Google Scholar]
- 50.Rowe GC, Jiang A, Arany Z. Pgc-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–838. doi: 10.1161/CIRCRESAHA.110.223818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sebastiani M, Giordano C, Nediani C, Travaglini C, Borchi E, Zani M, Feccia M, Mancini M, Petrozza V, Cossarizza A, Gallo P, Taylor RW, d’Amati G. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. 2007;50:1362–1369. doi: 10.1016/j.jacc.2007.06.035. [DOI] [PubMed] [Google Scholar]
- 52.Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. Pgc-1alpha and ERRalpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. 2009;46:201–212. doi: 10.1016/j.yjmcc.2008.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 54.Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- 55.Morcillo G, Barettino D, Carmona MJ, Carretero MT, Diez JL. Telomeric DNA sequences differentially activated by heat shock in two chironomus subspecies. Chromosoma. 1988;96:139–144. doi: 10.1007/BF00331046. [DOI] [PubMed] [Google Scholar]
- 56.Rudenko G, Van der Ploeg LH. Transcription of telomere repeats in protozoa. EMBO J. 1989;8:2633–2638. doi: 10.1002/j.1460-2075.1989.tb08403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Solovei I, Gaginskaya ER, Macgregor HC. The arrangement and transcription of telomere DNA sequences at the ends of lampbrush chromosomes of birds. Chromosome Res. 1994;2:460–470. doi: 10.1007/BF01552869. [DOI] [PubMed] [Google Scholar]
- 58.Luke B, Lingner J. Terra: telomeric repeat-containing RNA. EMBO J. 2009;28:2503–2510. doi: 10.1038/emboj.2009.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feuerhahn S, Iglesias N, Panza A, Porro A, Lingner J. Terra biogenesis, turnover and implications for function. FEBS Lett. 2010;584:3812–3818. doi: 10.1016/j.febslet.2010.07.032. [DOI] [PubMed] [Google Scholar]
- 60.Epel ES, Blackburn EH, Lin J, Dhabhar FS, Adler NE, Morrow JD, Cawthon RM. Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101:17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Epel ES, Lin J, Wilhelm FH, Wolkowitz OM, Cawthon R, Adler NE, Dolbier C, Mendes WB, Blackburn EH. Cell aging in relation to stress arousal and cardiovascular disease risk factors. Psychoneuroendocrinology. 2006;31:277–287. doi: 10.1016/j.psyneuen.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Kotrschal A, Ilmonen P, Penn DJ. Stress impacts telomere dynamics. Biol Lett. 2007;3:128–130. doi: 10.1098/rsbl.2006.0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Puterman E, Lin J, Blackburn E, O’Donovan A, Adler N, Epel E. The power of exercise: buffering the effect of chronic stress on telomere length. PLoS One. 2010;5:e10837. doi: 10.1371/journal.pone.0010837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wolf SA, Melnik A, Kempermann G. Physical exercise increases adult neurogenesis and telomerase activity, and improves behavioral deficits in a mouse model of schizophrenia. Brain Behav Immun. 2011;25:971–980. doi: 10.1016/j.bbi.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 65.McEachern MJ, Krauskopf A, Blackburn EH. Telomeres and their control. Annu Rev Genet. 2000;34:331–358. doi: 10.1146/annurev.genet.34.1.331. [DOI] [PubMed] [Google Scholar]
- 66.Cech TR. Beginning to understand the end of the chromosome. Cell. 2004;116:273–279. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 67.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 68.Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 69.Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009;1792:371–379. doi: 10.1016/j.bbadis.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Multani AS, Chang S. Wrn at telomeres: implications for aging and cancer. J Cell Sci. 2007;120:713–721. doi: 10.1242/jcs.03397. [DOI] [PubMed] [Google Scholar]
- 72.Wyllie FS, Jones CJ, Skinner JW, Haughton MF, Wallis C, Wynford-Thomas D, Faragher RG, Kipling D. Telomerase prevents the accelerated cell ageing of Werner syndrome fibroblasts. Nat Genet. 2000;24:16–17. doi: 10.1038/71630. [DOI] [PubMed] [Google Scholar]
- 73.Choi D, Whittier PS, Oshima J, Funk WD. Telomerase expression prevents replicative senescence but does not fully reset mRNA expression patterns in Werner syndrome cell strains. FASEB J. 2001;15:1014–1020. doi: 10.1096/fj.00-0104com. [DOI] [PubMed] [Google Scholar]
- 74.Crabbe L, Verdun RE, Haggblom CI, Karlseder J. Defective telomere lagging strand synthesis in cells lacking Wrn helicase activity. Science. 2004;306:1951–1953. doi: 10.1126/science.1103619. [DOI] [PubMed] [Google Scholar]
- 75.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 76.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 77.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 78.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 79.Chang S, Multani AS, Cabrera NG, Naylor ML, Laud P, Lombard D, Pathak S, Guarente L, DePinho RA. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nat Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 80.Wong KK, Maser RS, Bachoo RM, Menon J, Carrasco DR, Gu Y, Alt FW, DePinho RA. Telomere dysfunction and atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 81.Leri A, Franco S, Zacheo A, Barlucchi L, Chimenti S, Limana F, Nadal-Ginard B, Kajstura J, Anversa P, Blasco MA. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003;22:131–139. doi: 10.1093/emboj/cdg013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oh H, Taffet GE, Youker KA, Entman ML, Overbeek PA, Michael LH, Schneider MD. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci U S A. 2001;98:10308–10313. doi: 10.1073/pnas.191169098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Basel-Vanagaite L, Dokal I, Tamary H, Avigdor A, Garty BZ, Volkov A, Vulliamy T. Expanding the clinical phenotype of autosomal dominant dyskeratosis congenita caused by tert mutations. Haematologica. 2008;93:943–944. doi: 10.3324/haematol.12317. [DOI] [PubMed] [Google Scholar]
- 84.Vulliamy T, Marrone A, Szydlo R, Walne A, Mason PJ, Dokal I. Disease anticipation is associated with progressive telomere shortening in families with dyskeratosis congenita due to mutations in terc. Nat Genet. 2004;36:447–449. doi: 10.1038/ng1346. [DOI] [PubMed] [Google Scholar]
- 85.Capell BC, Collins FS, Nabel EG. Mechanisms of cardiovascular disease in accelerated aging syndromes. Circ Res. 2007;101:13–26. doi: 10.1161/CIRCRESAHA.107.153692. [DOI] [PubMed] [Google Scholar]
- 86.Massip L, Garand C, Turaga RV, Deschenes F, Thorin E, Lebel M. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the Werner syndrome gene homologue. Exp Gerontol. 2006;41:157–168. doi: 10.1016/j.exger.2005.10.011. [DOI] [PubMed] [Google Scholar]
- 87.Tri TB, Combs DT. Congestive cardiomyopathy in Werner’s syndrome. Lancet. 1978;1:1052–1053. doi: 10.1016/s0140-6736(78)90786-9. [DOI] [PubMed] [Google Scholar]
- 88.Cesselli D, Beltrami AP, D’Aurizio F, et al. Effects of age and heart failure on human cardiac stem cell function. Am J Pathol. 2011;179:349–366. doi: 10.1016/j.ajpath.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Oh H, Wang SC, Prahash A, Sano M, Moravec CS, Taffet GE, Michael LH, Youker KA, Entman ML, Schneider MD. Telomere attrition and chk2 activation in human heart failure. Proc Natl Acad Sci U S A. 2003;100:5378–5383. doi: 10.1073/pnas.0836098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366:662–664. doi: 10.1016/S0140-6736(05)66630-5. [DOI] [PubMed] [Google Scholar]
- 91.Gardner JP, Li S, Srinivasan SR, Chen W, Kimura M, Lu X, Berenson GS, Aviv A. Rise in insulin resistance is associated with escalated telomere attrition. Circulation. 2005;111:2171–2177. doi: 10.1161/01.CIR.0000163550.70487.0B. [DOI] [PubMed] [Google Scholar]
- 92.Cherkas LF, Hunkin JL, Kato BS, Richards JB, Gardner JP, Surdulescu GL, Kimura M, Lu X, Spector TD, Aviv A. The association between physical activity in leisure time and leukocyte telomere length. Arch Intern Med. 2008;168:154–158. doi: 10.1001/archinternmed.2007.39. [DOI] [PubMed] [Google Scholar]
- 93.Samani NJ, Boultby R, Butler R, Thompson JR, Goodall AH. Telomere shortening in atherosclerosis. Lancet. 2001;358:472–473. doi: 10.1016/S0140-6736(01)05633-1. [DOI] [PubMed] [Google Scholar]
- 94.Mainous AG, III, Codd V, Diaz VA, Schoepf UJ, Everett CJ, Player MS, Samani NJ. Leukocyte telomere length and coronary artery calcification. Atherosclerosis. 2010;210:262–267. doi: 10.1016/j.atherosclerosis.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 95.Farzaneh-Far R, Cawthon RM, Na B, Browner WS, Schiller NB, Whooley MA. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: data from the heart and soul study. Arterioscler Thromb Vasc Biol. 2008;28:1379–1384. doi: 10.1161/ATVBAHA.108.167049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cawthon RM, Smith KR, O’Brien E, Sivatchenko A, Kerber RA. Association between telomere length in blood and mortality in people aged 60 years or older. Lancet. 2003;361:393–395. doi: 10.1016/S0140-6736(03)12384-7. [DOI] [PubMed] [Google Scholar]
- 97.van der Harst P, de Boer RA, Samani NJ, Wong LS, Huzen J, Codd V, Hillege HL, Voors AA, van Gilst WH, Jaarsma T, van Veldhuisen DJ. Telomere length and outcome in heart failure. Ann Med. 2010;42:36–44. doi: 10.3109/07853890903321567. [DOI] [PubMed] [Google Scholar]
- 98.Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, Packard CJ, Samani NJ. Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007;369:107–114. doi: 10.1016/S0140-6736(07)60071-3. [DOI] [PubMed] [Google Scholar]
- 99.Kuznetsova T, Codd V, Brouilette S, Thijs L, Gonzalez A, Jin Y, Richart T, van der Harst P, Diez J, Staessen JA, Samani NJ. Association between left ventricular mass and telomere length in a population study. Am J Epidemiol. 2010;172:440–450. doi: 10.1093/aje/kwq142. [DOI] [PubMed] [Google Scholar]
- 100.Vasan RS, Demissie S, Kimura M, Cupples LA, White C, Gardner JP, Cao X, Levy D, Benjamin EJ, Aviv A. Association of leukocyte telomere length with echocardiographic left ventricular mass: the Framingham Heart Study. Circulation. 2009;120:1195–1202. doi: 10.1161/CIRCULATIONAHA.109.853895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wilson WR, Herbert KE, Mistry Y, Stevens SE, Patel HR, Hastings RA, Thompson MM, Williams B. Blood leucocyte telomere DNA content predicts vascular telomere DNA content in humans with and without vascular disease. Eur Heart J. 2008;29:2689–2694. doi: 10.1093/eurheartj/ehn386. [DOI] [PubMed] [Google Scholar]
- 102.Sharpless NE, DePinho RA. How stem cells age and why this makes us grow old. Nat Rev Mol Cell Biol. 2007;8:703–713. doi: 10.1038/nrm2241. [DOI] [PubMed] [Google Scholar]
- 103.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, II, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 104.Kajstura J, Rota M, Urbanek K, Hosoda T, Bearzi C, Anversa P, Bolli R, Leri A. The telomere-telomerase axis and the heart. Antioxid Redox Signal. 2006;8:2125–2141. doi: 10.1089/ars.2006.8.2125. [DOI] [PubMed] [Google Scholar]
- 105.Murry CE, Lee RT. Development biology: turnover after the fallout. Science. 2009;324:47–48. doi: 10.1126/science.1172255. [DOI] [PubMed] [Google Scholar]
- 106.Rudolph KL, Chang S, Millard M, Schreiber-Agus N, DePinho RA. Inhibition of experimental liver cirrhosis in mice by telomerase gene delivery. Science. 2000;287:1253–1258. doi: 10.1126/science.287.5456.1253. [DOI] [PubMed] [Google Scholar]
- 107.Jaskelioff M, Muller FL, Paik JH, Thomas E, Jiang S, Adams AC, Sahin E, Kost-Alimova M, Protopopov A, Cadinanos J, Horner JW, Maratos-Flier E, Depinho RA. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, Depinho RA. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468:701–704. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Rera M, Bahadorani S, Cho J, Koehler CL, Ulgherait M, Hur JH, Ansari WS, Lo T, Jr, Jones DL, Walker DW. Modulation of longevity and tissue homeostasis by the drosophila pgc-1 homolog. Cell Metab. 2011;14:623–634. doi: 10.1016/j.cmet.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, Sadek HA. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7:380–390. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leri A, Liu Y, Malhotra A, Li Q, Stiegler P, Claudio PP, Giordano A, Kajstura J, Hintze TH, Anversa P. Pacing-induced heart failure in dogs enhances the expression of p53 and p53-dependent genes in ventricular myocytes. Circulation. 1998;97:194–203. doi: 10.1161/01.cir.97.2.194. [DOI] [PubMed] [Google Scholar]
- 112.Sano M, Minamino T, Toko H, et al. P53-induced inhibition of hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- 113.Xiong S, Van Pelt CS, Elizondo-Fraire AC, Fernandez-Garcia B, Lozano G. Loss of mdm4 results in p53-dependent dilated cardiomyopathy. Circulation. 2007;115:2925–2930. doi: 10.1161/CIRCULATIONAHA.107.689901. [DOI] [PubMed] [Google Scholar]
- 114.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 115.Hsu CP, Zhai P, Yamamoto T, Maejima Y, Matsushima S, Hariharan N, Shao D, Takagi H, Oka S, Sadoshima J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation. 2010;122:2170–2182. doi: 10.1161/CIRCULATIONAHA.110.958033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 117.Benigni A, Corna D, Zoja C, Sonzogni A, Latini R, Salio M, Conti S, Rottoli D, Longaretti L, Cassis P, Morigi M, Coffman TM, Remuzzi G. Disruption of the Ang II type 1 receptor promotes longevity in mice. J Clin Invest. 2009;119:524–530. doi: 10.1172/JCI36703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 119.Piotrkowski B, Koch OR, De Cavanagh EM, Fraga CG. Cardiac mitochondrial function and tissue remodelling are improved by a non-antihypertensive dose of enalapril in spontaneously hypertensive rats. Free Radic Res. 2009;43:390–399. doi: 10.1080/10715760902801517. [DOI] [PubMed] [Google Scholar]
- 120.Nakamura K, Kusano K, Nakamura Y, Kakishita M, Ohta K, Nagase S, Yamamoto M, Miyaji K, Saito H, Morita H, Emori T, Matsubara H, Toyokuni S, Ohe T. Carvedilol decreases elevated oxidative stress in human failing myocardium. Circulation. 2002;105:2867–2871. doi: 10.1161/01.cir.0000018605.14470.dd. [DOI] [PubMed] [Google Scholar]
- 121.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–639. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Laustsen PG, Russell SJ, Cui L, Entingh-Pearsall A, Holzenberger M, Liao R, Kahn CR. Essential role of insulin and insulin-like growth factor 1 receptor signaling in cardiac development and function. Mol Cell Biol. 2007;27:1649–1664. doi: 10.1128/MCB.01110-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Boudina S, Bugger H, Sena S, et al. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–1283. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]