

Abstract
Progressive DNA damage and mitochondrial decline are both considered to be prime instigators of natural ageing. Traditionally, these two pathways have been viewed largely in isolation. However, recent studies have revealed a molecular circuit that directly links DNA damage to compromised mitochondrial biogenesis and function via p53. This axis of ageing may account for both organ decline and disease development associated with advanced age and could illuminate a path for the development of relevant therapeutics.
Ageing and its inevitable companions — disease and death — have fascinated mankind for millennia and have spurred the search for eternal youth. Long believed to be unchangeable, life expectancy and ‘healthspan’ have increased dramatically over the past two centuries. This has been fuelled by advances in medical science, improved hygiene and nutrition, and significant declines in mortality rates among the young1.
Now, the question is: can lifespan be further increased, or have we reached a biologically determined maximum lifespan? Scientists have long tried to identify pathways that are relevant for ageing, but it was not until the 1990s that the first genetic foothold was established in ageing: a loss-of-function mutation in daf-2 (which encodes an insulin/insulin-like growth factor 1 (IGF1)-like receptor) doubled the lifespan of worms2. Since then, the interest in the molecular pathways that control ageing has exploded, and many more mutations in metabolic pathways have been shown to affect lifespan in different model systems, ranging from yeast to mice3. Studies suggest that these pathways are also relevant for human lifespan, but how they might extend lifespan is not entirely clear4,5. However, among others, maintenance of mitochondrial function has been suggested to be an important mechanism of extending lifespan, as decreased mitochondrial function, impaired ATP generation and increased reactive oxygen species (ROS) levels have been implicated in driving the ageing process6.
In addition to the role of metabolic pathways, telomere maintenance has been shown to be linked to ageing7. Cultured human fibroblasts divide a finite number of times before entering a non-dividing state called senescence8. Subsequent work established that the ends of chromosomes, or telomeres, became shorter with each round of replication. These observations fuelled speculation that the loss of telomeres represents a type of molecular clock that drives ageing9-12. These studies underscored the general importance of DNA integrity, as dysfunctional telomeres are recognized as DNA damage and activate the DNA damage response pathway, which leads to the activation of p53 (REFS 13,14). p53, in turn, induces growth arrest, apoptosis and senescence in stem and progenitor cells15-18.
“a model of how different pathways … intersect and converge on mitochondria”
Recent studies have uncovered additional mechanistic insights into telomere-mediated ageing, in which telomere shortening and associated DNA damage responses promote mitochondrial dysfunction, diminished oxidative defence and compromised energy-generating processes. This general decline in energy maintenance could account for the decline seen in stem and progenitor cells as well as in post-mitotic tissues19.
In this Opinion article we present a model of how different pathways — DNA damage and metabolic pathways — intersect and converge on mitochondria to compromise energy maintenance and drive ageing. This integrated view provides a better understanding of the mechanisms that control the fundamental process of ageing and may open new avenues for therapeutic interventions for ageing and age-associated diseases.
Metabolic pathways in ageing
Several metabolic pathways and molecules regulate lifespan in response to nutrient availability and balance energy expenditure with mitochondrial function (BOX 1; FIG. 1). The insulin and IGF1 pathway was the first evolutionarily conserved pathway shown to regulate lifespan. Mammalian target of rapamycin (mTOR) signalling has also been implicated3,20 (BOX 1). There is increasing recognition that these metabolic pathways are intimately interconnected. For instance, AMP-activated protein kinase (AMPK), which is a central sensor of energy homeostasis that modulates mTOR signalling, activates forkhead box O (FOXO) transcription factors, which are targets of insulin and IGF1 signalling. This increases the expression of genes that are involved in stress resistance and energy balance21. AMPK also activates PPARγ co-activator 1α (PGC1α), which is a central regulator of mitochondrial biogenesis and function. Similarly, AMPK induces sirtuin 1 (SIRT1), which activates PGC1α and FOXO transcription factors22. There is also a close relationship between FOXO proteins and PGC1α, as FOXO1 and FOXO3 have been shown to increase PGC1α activity, and PGC1α itself can augment the transcriptional activity of FOXO3 (REFS 23-25). SIRT1 also inactivates the ‘guardian of the genome’, p53 (REFS 26,27). p53 itself intersects with several different longevity pathways, including insulin and IGF1, mTOR and AMPK28. The activation of AMPK and the repression of the insulin and IGF1 pathway and the mTOR pathway by p53 demonstrate the ability of p53 to positively regulate pathways that are essential for cell integrity and longevity28.
Box 1. Metabolic pathways and genes implicated in lifespan regulation.
Evolutionary conserved insulin and IGF1 signalling pathway
The insulin and insulin-like growth factor 1 (IGF1) pathway was the first evolutionarily conserved pathway shown to regulate lifespan in Caenorhabditis elegans. Components of this pathway include phosphoinositide 3-kinase (PI3K; AGE-1 in C. elegans), 3′-phosphoinositide-dependent kinase 1 (PDK-1), AKT (also known as PKB) and forkhead box O (FOXO) family transcription factors (DAF-16 in C. elegans). In response to insulin and IGF-1, PI3K activates PDK-1, which in turn activates AKT. AKT phosphorylates multiple downstream targets to promote cell survival and cell growth (see the figure, part a). Decreased activity of the insulin and IGF-1 pathway owing to loss-of-function mutations in genes that encode key components of this pathway or proteins that regulate its activity, such as growth hormones, extends lifespan in many species. The lifespan extension effect in these mutants is mediated by several transcription factors and their transcriptional targets, including DAF-16, heat shock factor 1 (HSF-1) and SKN-1 (a transcription factor involved in the oxidative stress response), which stimulate the expression of genes that increase stress resistance, oxidative defence and mitochondrial function3.
mTOR signaling
Mammalian target of rapamycin (mTOR) is an evolutionarily conserved protein kinase that exists in two complexes, mTORC1 (see the figure, part b) and mTORC2 (not shown). mTORC2 is involved in cytoskeletal remodelling, whereas mTORC1 is a potent regulator of cellular growth and lifespan. mTORC1 activates the ribosomal protein S6 kinase (S6K) and inhibits eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1; a negative regulator of translation) to stimulate translation and cell growth137. Inhibition of mTOR through caloric restriction, rapamycin treatment or genetic means extends lifespan in species ranging from yeast to mice29. Similarly, inhibiting S6K increases lifespan in worms and flies, and female mice lacking S6K have prolonged lifespan and are protected from age-related pathologies such as insulin resistance, immunological decline and motor dysfunction3,29. Lifespan extension in these S6K-deficient mice seems to be mediated by AMP-activated protein kinase (AMPK), which is a central sensor of energy homeostasis36. AMPK is potently activated by an increased AMP/ATP ratio, turns off anabolic pathways through indirect inhibition of mTORC1 and switches on catabolic pathways to generate ATP under energy stress. AMPK increases ATP levels by stimulating mitochondrial biogenesis and function, as well as fatty acid oxidation, among others138. Activation of AMPK in mice with metformin (an antidiabetic drug) enhances lifespan, and deletion of the AMPK orthologue aak-2 in C. elegans abrogates the lifespan extension induced in daf-2 mutants, which supports the notion that the regulation of energy pathways and preservation of mitochondrial function is integral to longevity3,29.
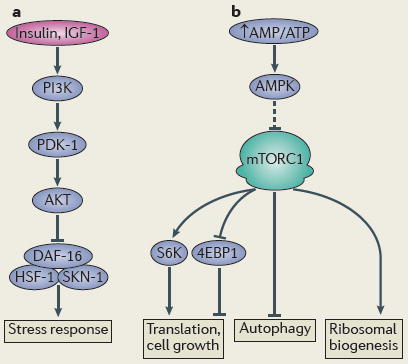
Figure 1. Proposed causes of ageing.
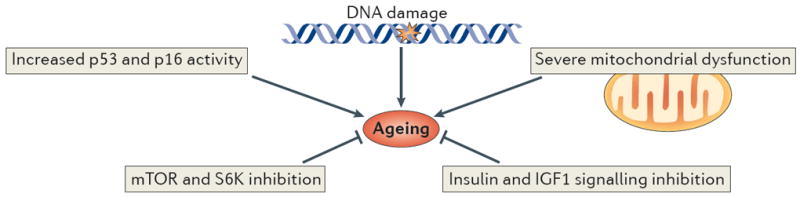
Major cellular pathways are implicated in the ageing process. Increased DNA damage, p53 and p16 activity and mitochondrial dysfunction have been shown to promote functional decline and ageing. By contrast, decreased activity in the mammalian target of rapamycin (mTOR), S6 kinase (S6K) and the insulin and insulin-like growth factor 1 (IGF1) pathways increase lifespan in different organisms.
How these pathways, alone or in combination, regulate lifespan is being extensively investigated, and many different mechanisms seem to be involved3,29. Among these, mitochondrial function and ROS defence are considered to be important modulators of lifespan. Along these lines, decreased activity of the insulin and IGF1 pathway is associated with improved mitochondrial function, as demonstrated in long-lived Ames mice (which have very low levels of IGF1) and in mice with decreased levels of insulin receptor substrate 2 (IRS2)30,31. Reduced insulin and IGF1 signalling results in the activation of FOXO transcription factors, which induce the expression of antioxidants, such as manganese superoxide dismutase (MnSOD) and catalase; accordingly, FOXO-deficient mice display increased levels of ROS and stem cell depletion32-34. Decreased mTOR activity during dietary restriction is also associated with improved mitochondrial function, and lifespan extension in this model depends on increased levels of respiration35. Furthermore, mice lacking S6 kinase (S6K), which is a downstream component of mTOR signalling, are long lived and show increased oxidative phosphorylation (OXPHOS) and oxygen consumption36. AMPK enhances SIRT1 activity (through increased NAD+ levels) and this, in turn, stimulates PGC1α and subsequently increases levels of OXPHOS and mitochondrial biogenesis37.
Further evidence for an essential and direct role of the maintenance of mitochondrial function in lifespan comes from mice expressing a proofreading-deficient mitochondrial DNA polymerase-γ (Polγ) variant that causes a premature ageing syndrome with shortened lifespan (the ‘mutator mice’)38,39. Overexpression of the antioxidant enzyme catalase specifically in mitochondria reduces ROS-induced damage and significantly improves age-related cardiac decline in both wild-type and mutator mice40,41. However, many other ROS-related studies have yielded conflicting results and question the role of increased ROS levels for the ageing process42,43. Subjecting mutator mice to continuous exercise potently rescues the premature ageing phenotype, which indicates that mitochondrial biogenesis and turnover (both of which are quality control mechanisms) are important for slowing down the ageing process in these mice44.
Although these studies indicate that mitochondrial decline drives ageing, other studies show a more complex picture, as mild impairment of mitochondrial function can extend lifespan in yeast, worms and mice3,45-49. This dichotomous role of mitochondria in lifespan has also been noted with other molecules that are involved in ageing, including p53 and AMPK50-52. Thus, a more detailed view of how these key molecules are tightly regulated in the context of mitochondrial biology is required to optimally control the molecular circuitry of ageing.
Telomeres and ageing
A separate line of extensive research has implicated telomere integrity as a major regulator of longevity53,54. Telomeres are repetitive TTAGGG sequences that cap chromosomes and prevent the ends from being recognized as DNA damage55. Most human cells lack adequate levels of telomerase to maintain telomeres, and this results in telomere shortening with each round of replication9,11,56. The importance of telomere length in ageing was initially inferred from seminal studies carried out in primary human fibroblasts in the early 1960s by Hayflick and Moorhead8. These cells divide a finite number of times in vitro and undergo telomere shortening with continuous passaging and, eventually, senescence8. Telomerase reactivation elongates telomeres and allows fibroblasts to bypass senescence and grow indefinitely, which demonstrates the causal role of shortened telomeres in cellular ageing57.
Indeed, telomere length has been shown to gradually decline with age in many human tissues, including proliferative compartments and more quiescent tissues11,58-60. Interestingly, even cells that express telomerase undergo telomere shortening over time, which points towards a complex regulation of telomere length61. Moreover, many studies have found a positive correlation between telomere shortening in human peripheral leukocytes and the risk of typical age-associated diseases62.
Further support for the role of telomeres in ageing comes from patients with loss-of-function mutations in genes that are crucial for telomere length maintenance, as these mutations predispose individuals to accelerated ageing. Mutations in TERC (the RNA component of telomerase) and TERT (the catalytic component of telomerase) are found in patients with the premature ageing syndrome dyskeratosis congenita63. Mutations in the genes encoding Werner syndrome ATP-dependent helicase (WRN) and ataxia telangiectasia mutated (ATM) cause Werner syndrome and the neurodegenerative disorder ataxia telangiectasia, respectively64. In addition to these multisystem disorders, TERC and TERT loss-of-function mutations are associated with the development of more organ-restricted diseases such as liver fibrosis, idiopathic pulmonary fibrosis and bone marrow failure syndromes63. The manifestation of degenerative phenotypes in telomere maintenance conditions depends on the degree of telomere dysfunction, as evidenced by the earlier and more severe occurrence of pathologies in subsequent generations of patients with dyskeratosis congenita, who have shorter telomeres (known as the anticipation effect)65. Although these studies of telomere maintenance disorders have provided evidence for the importance of telomeres for organ integrity and lifespan regulation, some pathologies seen in these patients are not typically observed during normal ageing and caution against a simple extrapolation of these findings to normal ageing. The exacerbated phenotypes in these patients might relate to excessive telomere shortening beyond what is seen in many proliferative and more static tissues during normal ageing in humans and may also be driven by environmental factors.
A link between telomeres and ageing has also been obtained from studies in mice. There is increasing recognition that telomere length and integrity are compromised during ageing in wild-type mice66,67. Interestingly, the presence of one or a few dysfunctional telomeres in cells seems to be sufficient to trigger a DNA damage response, and this could underline the development of pathologies68,69. Accordingly, overexpression of telomerase can delay some age-associated changes in mice that have been engineered to be resistant to cancer70. Furthermore, the role of telomeres for organismal fitness and lifespan has been substantiated in mice lacking telomerase activity, which develop numerous age-associated degenerative phenotypes once their telomeres become short and die prematurely71,72. Moreover, mouse models of Werner syndrome and ataxia telangiectasia develop classical human-like pathologies only when their telomeres are short, which demonstrates the essential role of critically short telomeres in disease manifestation16,73,74. Finally, telomerase overexpression can reverse age-associated decline in multiple tissues in mice with established degenerative phenotypes75.
These studies indicate that telomere dysfunction can drive the functional decline of tissues, promote ageing and shorten lifespan and, importantly, that the ageing process can be prevented or even be reversed by telomerase reactivation. However, it is clear that currently there is only an elemental understanding of the precise role of telomeres in natural ageing and how telomeres might influence age-associated pathologies.
Telomere–mitochondrion connection
How do waning telomeres precipitate such widespread degeneration? One clue comes from the observation that patients with dyskeratosis congenita, Werner syndrome and ataxia telangiectasia, and mice with dysfunctional telomeres, develop organ failure particularly in highly proliferative organs such as the intestines, skin and bone marrow76-78. These organs rely on continuous regeneration, which is mediated by resident stem and progenitor cells. This observation has led to the hypothesis that telomere-based ageing is primarily a stem cell defect caused by the activation of p53 and the induction of growth arrest, senescence and apoptosis in these cellular compartments79. Indeed, telomere shortening is accompanied by increased p53 activity in these cells and, consequently, high levels of apoptosis15,18. Mice lacking p53 or its downstream targets show functional rescue of stem and progenitor cells in the haematopoietic system, skin and gastrointestinal tract, as well as concomitant rescue of tissue pathologies15,18,80. Although the stem cell theory of ageing helps to rationalize the failure of highly regenerative tissues, it does not readily explain age-dependent changes in more quiescent tissues that dependent less on stem and progenitor cell activity for tissue homeostasis.
Indeed, general metabolic disorders and functional decline in mostly post-mitotic tissues such as the heart, liver and pancreas are well-recognized features in the aged, in patients with telomere maintenance disorders and in telomere-dysfunctional mice65,81. For instance, individuals with dyskeratosis congenita, Werner syndrome and ataxia telangiectasia are prone to develop insulin resistance and diabetes65,82,83. Moreover, cardiomyopathy has been recognized in patients with dyskeratosis congenita, in telomere-dysfunctional mice and in mouse models of ataxia telangiectasia81,84,85. Liver fibrosis and pulmonary fibrosis represent other pathophysiological manifestations in patients with dyskeratosis congenita. Hepatic toxicity is a major side effect in patients with dyskeratosis congenita, who receive cytotoxic chemotherapy for aplastic anaemia-related bone marrow transplantation65. Quiescent tissues such as the heart and liver have also been reported to undergo age-dependent telomere shortening, the basis for which is unclear58,60. Together, these observations suggest additional mechanisms of telomere-induced ageing beyond traditional p53-dependent checkpoint responses of apoptosis and senescence.
Some clues for additional mechanisms have surfaced from recent work in TERT-deficient mice with telomere dysfunction. This study reported a marked compromise in mitochondrial biogenesis and function in diverse tissues, including liver, heart and haematopoietic stem cells, which raises the possibility that a fundamental problem in energy maintenance might contribute to the premature ageing phenotypes in these mice19. These marked mitochondrial changes seem to be caused by the combined suppression of the transcriptional co-activators PGC1α and PGC1β and their downstream targets (FIG. 2). This is mediated by direct binding of p53 to the promoters of PGC1α and PGC1β; accordingly, telomere-dysfunctional mice lacking p53 have normal PGC expression, increased mitochondrial DNA (mtDNA) content, improved gluconeogenesis and blunted doxorubicin-induced cardiomyopathy. In line with the important role of PGCs in regulating diverse processes, TERT-deficient mice show reduced expression of genes that are essential for gluconeogenesis, β-oxidation and ROS defence, and they have greatly compromised OXPHOS, with reduced ATP generation, impaired gluconeogenic capacity and age-dependent cardiomyopathy. Of note, these changes are more pronounced with increasing telomere dysfunction. Importantly, overexpression of TERT or PGC1α in mice with telomere dysfunction improves mitochondrial respiration and gluconeogenesis, which confirms that the phenotype of TERT-deficient mice is caused by PGC suppression19. This telomere–mitochondrion link is also suggested by other studies, including those demonstrating increased ROS levels and mitochondrial dysfunction in cultured human fibroblasts, in fibroblasts overexpressing a mutant form of TERT and in heart tissues of TERT-deficient mice86-88. The basis for these defects has been suggested to be secondary to the activation of the p21–transforming growth factor-β (TGFβ)–p53 pathways and increased mtDNA damage86-88. Moreover, a recent study found reduced mitochondrial membrane hyperpolarization and impaired Ca2+ influx in telomere-dysfunctional mice, which leads to reduced insulin release in β-cells89.
Figure 2. Telomere–p53–PGC pathway.
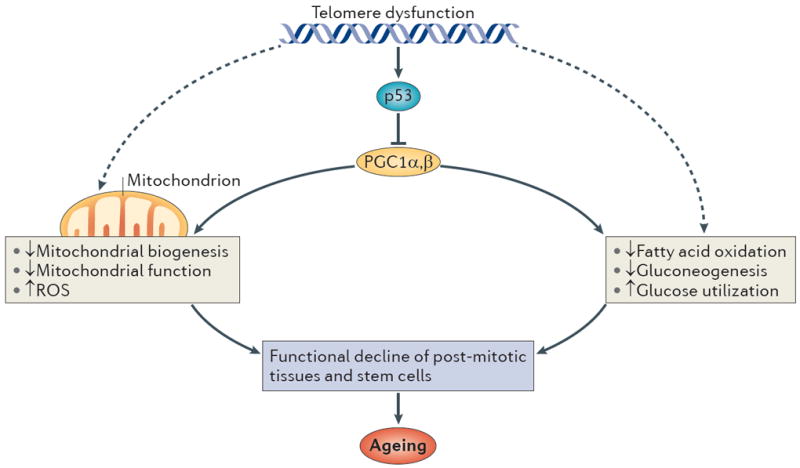
p53 induced by telomere dysfunction binds to the promoters of PPARγ co-activator 1α (PGC1α) and PGC1β and represses the expression of PGC1A and PGC1B. The repression of both co-activators impairs overall mitochondrial biogenesis and function and leads to defective ATP generation and increased levels of reactive oxygen species (ROS). PGCs are also involved in energy metabolism by regulating different biochemical pathways such as fatty acid oxidation, gluconeogenesis, glucose uptake and oxidation. The compromise in mitochondrial function and other biochemical pathways might equally lead to functional decline in tissue stem cells and post-mitotic tissues and drive ageing. Telomerase reactivation or PGC overexpression can reverse PGC-associated metabolic and mitochondrial changes in mice with established telomere dysfunction. Telomere dysfunction may also lead to compromised mitochondrial function and energy metabolism through other pathways (dashed arrows).
Previous studies have suggested that TERT has telomere elongation-independent functions90-94. However, TERC-deficient mice, which lack telomerase activity but have intact TERT expression, showed similar robust changes centred on PGC and mitochondrial suppression, which indicates that telomere dysfunction is the determining factor driving these changes19. Moreover, Tert- and Terc-knockout mice have indistinguishable phenotypes and similar transcriptomic profiles95. That said, these genetic studies were not designed to rigorously exclude more subtle telomere-independent roles of TERT. Indeed, continued study is warranted, as TERT has been shown to localize to mitochondria, retain reverse transcriptase activity, carry out different mitochondrial functions (such as modulating mtDNA integrity, improving respiratory chain function and affecting ROS production) and to potentially activate other pathways such as the WNT pathway (although this has been questioned recently) 88,96-100.
Furthermore, aged tissues often demonstrate concomitant telomere dysfunction, increased DNA damage and p53 activity as well as reduced PGC levels and mitochondrial function7. Importantly, studies in cells derived from patients and mouse models corroborate this link between telomeres and mitochondria101-103. For example, cells derived from patients with Werner syndrome have compromised mitochondrial function and increased ROS levels103. Although these observations support the importance of the telomere–p53–mitochondrion axis of ageing, more work is required to assess whether mitochondrial biogenesis and function are consistently impaired in human telomere-shortening conditions.
An integrated view of ageing
The connection between telomeres and mitochondria supports a model for ageing whereby DNA damage-induced p53 activation leads to mitochondrial dysfunction through the suppression of the master regulators of mitochondrial biogenesis and function, PGC1α and PGC1β.
The telomere–p53–mitochondrion model of ageing integrates many factors that have been shown to be important in the ageing process. On the genomic level, it accounts for ageing driven by DNA damage. This damage can stem from telomere shortening or from a decrease in the expression of genes that mediate DNA stability and DNA repair. Second, the model accounts for ageing syndromes documented in mice with hyperactive Tp53 alleles and mouse models that show increased DNA damage owing to mutations in Terc, Tert, the DNA repair genes Ku80 (also known as Xrcc5) and breast cancer 1 (Brca1), and Zmpste24, which encodes a metalloproteinase involved in lamin A maturation (lamin A is a crucial component of the nuclear envelope, and mutations of this metalloproteinase lead to premature ageing)104,105. Finally, the model accounts for ageing phenotypes that stem from mitochondrial dysfunction, as mice lacking PGC1α, PGC1β, BMI (a negative regulator of p16 that is upregulated in many tissues) or FOXO develop accelerated tissue degeneration and mitochondrial dysfunction106-109.
This model could also explain both the slow but progressive physiological decline and the precipitous nature of the ageing process (FIG. 3). Ensuing mitochondrial dysfunction sustains a feed-forward cycle of DNA damage and further mitochondrial dysfunction through the generation of DNA-damaging ROS and possibly other mitochondrion-derived factors, such as iron–sulphur (Fe–S) clusters and NADH/NAD. This boost in ROS production fuels a detrimental cycle of increased genotoxic damage, particularly to the G-rich sequences of telomeres, followed by sustained activation of p53, further mitochondrial decline, more ROS generation, and so on110,111. Increased ROS levels also damage other cellular components, including mtDNA, which further sustains this feed-forward spiral of damage by suppressing the expression of mtDNA-encoded genes for OXPHOS. Under conditions of severe nuclear or mtDNA damage, however, this cycle could be bypassed, and the premature ageing phenotype could be driven by increased apoptosis across different tissues, as reported in mutator mice (which carry a mutant form of Polγ; see above)38,39.
Figure 3. A unified theory of ageing.
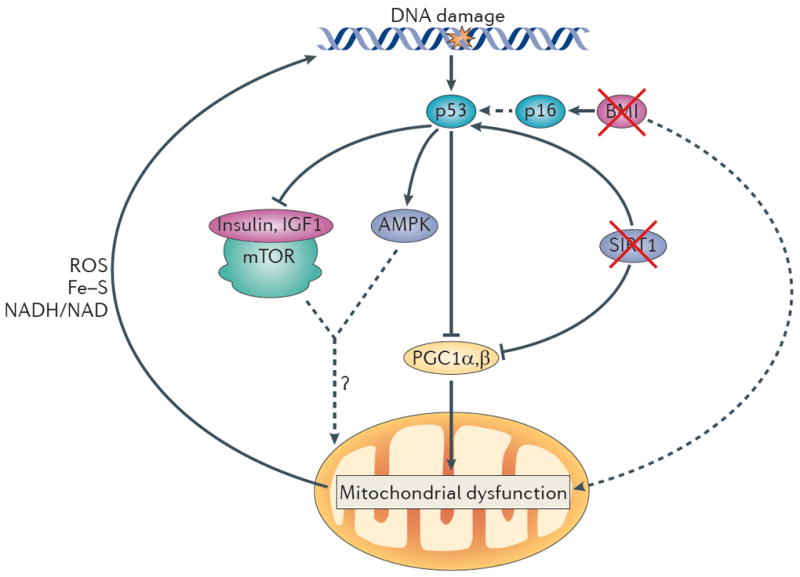
In this model, increased DNA damage (for example, owing to telomere attrition, impaired DNA repair and increased reactive oxygen species (ROS) levels) activates p53, and increasing levels of p53 ultimately lead to compromised mitochondrial function through the repression of PPARγ co-activator 1α (PGC1α) and PGC1β (which promote mitochondrial biogenesis). This p53-mediated mitochondrial dysfunction triggers a cycle of DNA damage (by affecting the production of ROS, iron–sulphur (Fe–S) clusters and NADH/NAD), which in turn leads to further p53 activation and mitochondrial compromise. This feed-forward loop could also account for the divergent and opposite effects of many players in the ageing process. Under mild stress conditions, several components depicted here (p53, mitochondria and AMP-activated protein kinase (AMPK)) have been shown to preserve cellular function but to promote cellular ageing under more severe stress conditions (see text). The interplay between p53 and other pathways that have been implicated in ageing is also indicated. p53 represses the activity of the insulin and insulin-like growth factor 1 (IGF1) pathway and the mammalian target of rapamycin (mTOR) pathway and activates AMPK. How the altered activity of these pathways modifies mitochondrial function and the ageing process in the setting of increased DNA damage is not clear. Other p53-dependent and p53-independent pathways might cooperate in inducing mitochondrial dysfunction. For example, BMI1 indirectly inhibits p53 activation, and BMI1 loss upregulates p16 expression, which increases p53 activity indirectly (dashed arrow) by interacting with MDM2, the negative regulator of p53 (not shown). BMI1 has also been shown to induce mitochondrial dysfunction (probably indirectly). In addition, loss of sirtuins may contribute to mitochondrial dysfunction, as active sirtuin 1 (SIRT1) decreases p53 activity, and loss of SIRT1 promotes p53 activation and its downstream functions. SIRT1 also activates PGC1α and thereby boosts mitochondrial biogenesis. The consequences of mitochondrial dysfunction are, when mild, functional impairment (for example, decreased ATP generation and β-oxidation) without cell loss. However, under increased stress conditions, mitochondrial dysfunction leads to functional impairment and concomitant loss of parenchymal mass owing to increased apoptosis and senescence.
The continuous, escalating nature of this cycle could explain the differential effect of p53 on ageing in response to varying degrees of DNA damage. Under low levels of genotoxic stress, p53 induces the expression of antioxidants, thereby favouring cell survival. By contrast, increasing levels of DNA damage promote the expression of pro-oxidants, which further promote cell damage112. Similarly, p53 has been shown to promote mitochondrial function and biogenesis in wild-type mice or in cells with low levels of p53, but under conditions of genotoxic stress p53 is associated with impaired mitochondrial function19,113-115. There is also evidence that p53 either does not change lifespan (as demonstrated in mice with a hypomorphic Mdm2 allele and in transgenic mice carrying an extra copy of the wild-type Tp53 locus) or delays ageing (which was shown in mice carrying one extra copy of Tp53 and increased copies of the tumour suppressor Arf (also known as Cdkn2a))116,117. Furthermore, studies in worms have demonstrated that the p53 orthologue CEP-1 can extend or decrease lifespan depending on the degree of mitochondrial impairment, which indicates that the dichotomous role of p53 might be preserved during evolution118.
In the presented model, mild DNA damage and mild or moderate levels of p53 activation would allow repair and maintenance of cellular function, whereas excessive DNA damage and p53 activation would eliminate cells that are not fit to survive or carry too much damage. These genetic findings also highlight the differential effects of mitochondrial function on lifespan observed in worms, flies and mice and indicate that the functional consequences of mitochondrial impairment could also depend on the activity levels of p53. The mild inhibition of mitochondrial respiration could activate longevity pathways (including those governed by p53), whereas more pronounced impairment of mitochondrial respiration with additional defects in biochemical processes, such as β-oxidation, could trigger a lifespan-shortening programme.
How p53 switches from a pro-survival to a pro-ageing protein is largely unknown, but the intersection of p53 with the insulin and IGF1, mTOR and AMPK pathways might provide some clues28. p53 suppresses the insulin and IGF1 pathway and the mTOR pathway through direct transcriptional upregulation of the negative regulators phosphatase and tensin homologue (PTEN), IGF1-binding protein 3 (IGF1BP3) and tuberous sclerosis protein 2 (TSC2; also known as tuberin), and p53 activates AMPK through direct transcriptional upregulation of its β-subunit28. Interestingly, prematurely aged mice with increased levels of DNA damage or with p53 hyperactivation show suppression of the insulin and IGF1 pathway and the mTOR pathway119,120. These paradoxical findings can be reconciled by the view that the activation of these pathways represents a compensatory mechanism to maintain and prolong lifespan in the setting of ongoing DNA damage120. However, it is not clear whether the activation of these longevity pathways in the context of DNA damage leads to the same transcriptional and cellular changes as seen in long-lived carriers of mutations in these pathways. For example, the prolonged activation of AMPK seen under energy stress leads to p53-dependent cellular senescence and apoptosis, which indicates that AMPK activation can accelerate cellular ageing under specific conditions52. Similarly, IGF1 concentration decreases with age, and this has been linked to functional decline in different stem cells121. These findings further support the notion that the effects of classical ageing pathways might be context dependent.
The model presented here acknowledges the existence of other, yet to be identified pathways that are involved in mediating mitochondrial and metabolic compromise, as p53 deficiency in mice with dysfunctional telomeres only partially restores PGC levels and ameliorates mitochondrial defects. Other p53 family members are prime candidates, in particular p63, which has been shown to be involved in organismal ageing and cellular senescence122,123. Sirtuins may also have a role, as they associate with telomeres and regulate p53 and PGC1α124. Indeed, SIRT1 activity has been found to decrease in aged tissues, and this might contribute to the increased p53 activity and suppressed PGC1α activity seen in aged mouse and human tissues7,106. However, recent reports have questioned the relevance of SIRT1 in lifespan regulation125, which emphasizes the need for studies that focus on the role of sirtuins and their linkage to this axis in ageing and age-related pathologies. Another potential candidate is the BMI–p16 pathway, as it is highly implicated in ageing, and loss of BMI impairs mitochondrial function107. Finally, p21-dependent signalling has been suggested to induce mitochondrial dysfunction in cultured human fibroblasts with critically short telomeres, although it should be noted that fibroblasts depend less on mitochondria and OXPHOS for ATP production and mainly use glycolysis to generate ATP86. In this context, it is important to note here that telomere-dysfunctional yeast display increased expression of OXPHOS genes and proliferation of mitochondria (although the functionality of the mitochondria was not tested), and senescent fibroblasts with dysfunctional telomeres have been reported to have increased mitochondrial biogenesis126,127. These studies highlight not only the cell-specific effects of telomere dysfunction on mitochondrial biology but also the differences between mice and yeast, which might be related to growth conditions and yeast-specific regulation after telomere dysfunction.
In this model, the cellular phenotypes induced by mitochondrial dysfunction would range from functional impairment (for example, decreased ATP generation) to classical cellular phenotypes of growth arrest, senescence and apoptosis. The link between impaired mitochondrial function and cellular senescence has been demonstrated in vitro: decreasing the expression of Rieske Fe–S protein (RISP) of complex III or pharmacological inhibition of the electron transport chain and OXPHOS are sufficient to trigger senescence128. The combined effects of these non-exclusive phenotypes would include cellular and tissue compromise and functional failure.
“Deciphering these ageing networks could advance the development of therapeutic strategies”
Conclusion
The integrated model of ageing presented in this Opinion article focuses on the intersection of DNA damage and metabolic pathways and how they might converge on a common effector, mitochondria, to drive ageing. Although this model centres on mitochondria, other important effectors of ageing, such as dysregulated autophagy, translation and protein folding, no doubt conspire to bring about and/or reinforce the ageing process129-131. It remains to be established whether and how these different effectors are commonly used by DNA damage and metabolic pathways to drive ageing. Similarly, how these effectors are interconnected merits further studies in different ageing model systems.
It is largely unclear how mitochondria, p53 and other players of the ageing process can both extend and shorten lifespan. It will be crucial to establish the molecular mechanisms that determine different outcomes, which may involve cell-type-specific actions of p53 and/or various isoforms of the p53 family members. In this regard, it will be essential to characterize what other mitochondrial biochemical pathways (beyond ROS production and OXPHOS) are impaired and might contribute to cellular and organismal ageing.
Deciphering these ageing networks could yield biomarkers of ageing and advance the development of therapeutic strategies designed to rejuvenate both proliferating and quiescent tissues in the aged. These therapeutic strategies might include: stabilizing telomeres through transient telomerase reactivation; attenuation of p53 activation or neutralization of specific p53 targets that are specifically involved in ageing; and enhancing PGC activity to promote mitochondrial biogenesis and function. Along these lines, a small molecule activator of telomerase has been reported to prolong healthspan in female mice without increasing the risk of cancer132. Moreover, PGC1α overexpression in skeletal muscle ameliorates age-associated decline of muscular function in wild-type mice133. Similarly, other interventions known to improve mitochondrial biogenesis and function, such as physical activity or administration of resveratrol (which is a putative sirtuin activator), have been shown to improve age-associated decline134,135. It is intriguing that sirtuins can deacetylate p53 (which decreases its activity) and PGCs (which increases their activity), thereby differentially modulating two key components in the pathway that connects DNA damage signalling and mitochondrial decline. Beyond ageing, recent studies have also uncovered the importance of the telomere–p53–mitochondrion axis for cancer, which suggests that this pathway could be targeted for cancer therapy136.
The discovery of the molecular circuitry of ageing has positioned the field to develop rational strategies for ageing, a ‘disease’ with 100% penetrance and 100% mortality. Although it remains to be determined whether natural ageing can be blocked or even be reversed, as recently demonstrated in the premature ageing setting75, therapeutic manipulation of this pathway may hold promise for the reduction of age-related diseases, which have become more prevalent with marked increases in life expectancy worldwide.
Acknowledgments
The authors apologize to all their colleagues whose work could not be cited. Part of the work described in this article was supported by a fellowship from the Deutsche Forschungsgemeinschaft (to E.S.) and by R01 and U01 grants from the US National Institutes of Health (NIH) National Cancer Institute and the Robert A. and Renee E. Belfer Foundation. R.A.D. was supported by an Ellison Foundation for Medical Research Senior Scholar and an American Cancer Society Research Professor award.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Ergün Sahin, Email: esahin@bcm.edu, Huffington Center On Aging and the Department of Molecular Physiology and Biophysics, Baylor College of Medicine, One Baylor Plaza, Houston, Texas 77030, USA.
Ronald A. DePinho, Email: rdepinho@mdanderson.org, Department of Cancer Biology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.
References
- 1.Christensen K, Doblhammer G, Rau R, Vaupel JW. Ageing populations: the challenges ahead. Lancet. 2009;374:1196–1208. doi: 10.1016/S0140-6736(09)61460-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang RA. C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 3.Kenyon CJ. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- 4.Deelen J, et al. Gene set analysis of GWAS data for human longevity highlights the relevance of the insulin/IGF-1 signaling and telomere maintenance pathways. Age (Dordr) 2011 Nov 24; doi: 10.1007/s11357-011-9340-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ziv E, Hu D. Genetic variation in insulin/IGF-1 signaling pathways and longevity. Ageing Res Rev. 2011;10:201–204. doi: 10.1016/j.arr.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Guarente L. Mitochondria — a nexus for aging, calorie restriction, and sirtuins? Cell. 2008;132:171–176. doi: 10.1016/j.cell.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sahin E, Depinho RA. Linking functional decline of telomeres, mitochondria and stem cells during ageing. Nature. 2010;464:520–528. doi: 10.1038/nature08982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 9.Harley CB, Futcher AB, Greider CW. Telomeres shorten during ageing of human fibroblasts. Nature. 1990;345:458–460. doi: 10.1038/345458a0. [DOI] [PubMed] [Google Scholar]
- 10.Allsopp RC, et al. Telomere length predicts replicative capacity of human fibroblasts. Proc Natl Acad Sci USA. 1992;89:10114–10118. doi: 10.1073/pnas.89.21.10114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hastie ND, et al. Telomere reduction in human colorectal carcinoma and with ageing. Nature. 1990;346:866–868. doi: 10.1038/346866a0. [DOI] [PubMed] [Google Scholar]
- 12.Harley CB. Telomere loss: mitotic clock or genetic time bomb? Mutat Res. 1991;256:271–282. doi: 10.1016/0921-8734(91)90018-7. [DOI] [PubMed] [Google Scholar]
- 13.Vaziri H, Benchimol S. From telomere loss to p53 induction and activation of a DNA-damage pathway at senescence: the telomere loss/DNA damage model of cell aging. Exp Gerontol. 1996;31:295–301. doi: 10.1016/0531-5565(95)02025-x. [DOI] [PubMed] [Google Scholar]
- 14.Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- 15.Chin L, et al. p53 deficiency rescues the adverse effects of telomere loss and cooperates with telomere dysfunction to accelerate carcinogenesis. Cell. 1999;97:527–538. doi: 10.1016/s0092-8674(00)80762-x. [DOI] [PubMed] [Google Scholar]
- 16.Wong KK, et al. Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature. 2003;421:643–648. doi: 10.1038/nature01385. [DOI] [PubMed] [Google Scholar]
- 17.Ferron S, et al. Telomere shortening and chromosomal instability abrogates proliferation of adult but not embryonic neural stem cells. Development. 2004;131:4059–4070. doi: 10.1242/dev.01215. [DOI] [PubMed] [Google Scholar]
- 18.Flores I, Blasco MA. A p53-dependent response limits epidermal stem cell functionality and organismal size in mice with short telomeres. PLoS ONE. 2009;4:e4934. doi: 10.1371/journal.pone.0004934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrison DE, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiacchiera F, Simone C. The AMPK—FoxO3A axis as a target for cancer treatment. Cell Cycle. 2010;9:1091–1096. doi: 10.4161/cc.9.6.11035. [DOI] [PubMed] [Google Scholar]
- 22.Canto C, Auwerx J. PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daitoku H, Yamagata K, Matsuzaki H, Hatta M, Fukamizu A. Regulation of PGC-1 promoter activity by protein kinase B and the forkhead transcription factor FKHR. Diabetes. 2003;52:642–649. doi: 10.2337/diabetes.52.3.642. [DOI] [PubMed] [Google Scholar]
- 24.Olmos Y, et al. Mutual dependence of Foxo3a and PGC-1α in the induction of oxidative stress genes. J Biol Chem. 2009;284:14476–14484. doi: 10.1074/jbc.M807397200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puigserver P, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1α interaction. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 26.Luo J, et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 27.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 28.Feng Z, Levine AJ. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20:427–434. doi: 10.1016/j.tcb.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010;40:333–344. doi: 10.1016/j.molcel.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sadagurski M, et al. IRS2 increases mitochondrial dysfunction and oxidative stress in a mouse model of Huntington disease. J Clin Invest. 2011;121:4070–4081. doi: 10.1172/JCI46305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown-Borg HM, Johnson WT, Rakoczy SG. Expression of oxidative phosphorylation components in mitochondria of long-living Ames dwarf mice. Age (Dordr) 2011;34:43–57. doi: 10.1007/s11357-011-9212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128:325–339. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 33.Renault VM, et al. FoxO3 regulates neural stem cell homeostasis. Cell Stem Cell. 2009;5:527–539. doi: 10.1016/j.stem.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paik JH, et al. FoxOs cooperatively regulate diverse pathways governing neural stem cell homeostasis. Cell Stem Cell. 2009;5:540–553. doi: 10.1016/j.stem.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zid BM, et al. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in. Drosophila Cell. 2009;139:149–160. doi: 10.1016/j.cell.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Selman C, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canto C, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056–1060. doi: 10.1038/nature07813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 39.Kujoth GC, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 40.Dai DF, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dai DF, et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell. 2010;9:536–544. doi: 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez VI, et al. Is the oxidative stress theory of aging dead? Biochim Biophys Acta. 2009;1790:1005–1014. doi: 10.1016/j.bbagen.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hekimi S, Lapointe J, Wen Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011;21:569–576. doi: 10.1016/j.tcb.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Safdar A, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dillin A, et al. Rates of behavior and aging specified by mitochondrial function during development. Science. 2002;298:2398–2401. doi: 10.1126/science.1077780. [DOI] [PubMed] [Google Scholar]
- 46.Lee SS, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nature Genet. 2003;33:40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 47.Kirchman PA, Kim S, Lai CY, Jazwinski SM. Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae. Genetics. 1999;152:179–190. doi: 10.1093/genetics/152.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dell’agnello C, et al. Increased longevity and refractoriness to Ca2+-dependent neurodegeneration in Surf1 knockout mice. Hum Mol Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, et al. Evolutionary conservation of the clk-1-dependent mechanism of longevity: loss of mclk1 increases cellular fitness and lifespan in mice. Genes Dev. 2005;19:2424–2434. doi: 10.1101/gad.1352905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vigneron A, Vousden KH. p53, ROS and senescence in the control of aging. Aging (Albany NY) 2010;2:471–474. doi: 10.18632/aging.100189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sung JY, Woo CH, Kang YJ, Lee KY, Choi HC. AMPK induces vascular smooth muscle cell senescence via LKB1 dependent pathway. Biochem Biophys Res Commun. 2011;413:143–148. doi: 10.1016/j.bbrc.2011.08.071. [DOI] [PubMed] [Google Scholar]
- 52.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 53.Blackburn EH. Switching and signaling at the telomere. Cell. 2001;106:661–673. doi: 10.1016/s0092-8674(01)00492-5. [DOI] [PubMed] [Google Scholar]
- 54.Shay JW, Wright WE. Hallmarks of telomeres in ageing research. J Pathol. 2007;211:114–123. doi: 10.1002/path.2090. [DOI] [PubMed] [Google Scholar]
- 55.Chan SR, Blackburn EH. Telomeres and telomerase. Phil Trans R Soc Lond B. 2004;359:109–121. doi: 10.1098/rstb.2003.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim NW, et al. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994;266:2011–2015. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 57.Bodnar AG, et al. Extension of life-span by introduction of telomerase into normal human cells. Science. 1998;279:349–352. doi: 10.1126/science.279.5349.349. [DOI] [PubMed] [Google Scholar]
- 58.Aikata H, et al. Telomere reduction in human liver tissues with age and chronic inflammation. Exp Cell Res. 2000;256:578–582. doi: 10.1006/excr.2000.4862. [DOI] [PubMed] [Google Scholar]
- 59.Aubert G, Lansdorp PM. Telomeres and aging. Physiol Rev. 2008;88:557–579. doi: 10.1152/physrev.00026.2007. [DOI] [PubMed] [Google Scholar]
- 60.Chimenti C, et al. Senescence and death of primitive cells and myocytes lead to premature cardiac aging and heart failure. Circ Res. 2003;93:604–613. doi: 10.1161/01.RES.0000093985.76901.AF. [DOI] [PubMed] [Google Scholar]
- 61.Chen J. Hematopoietic stem cell development, aging and functional failure. Int J Hematol. 2011;94:3–10. doi: 10.1007/s12185-011-0856-1. [DOI] [PubMed] [Google Scholar]
- 62.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nature Rev Genet. 2005;6:611–622. doi: 10.1038/nrg1656. [DOI] [PubMed] [Google Scholar]
- 63.Calado RT, Young NS. Telomere diseases. N Engl J Med. 2009;361:2353–2365. doi: 10.1056/NEJMra0903373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martin GM. Genetic modulation of senescent phenotypes in Homo sapiens. Cell. 2005;120:523–532. doi: 10.1016/j.cell.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 65.Armanios M. Syndromes of telomere shortening. Annu Rev Genom Hum Genet. 2009;10:45–61. doi: 10.1146/annurev-genom-082908-150046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Flores I, et al. The longest telomeres: a general signature of adult stem cell compartments. Genes Dev. 2008;22:654–667. doi: 10.1101/gad.451008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hewitt G, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nature Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hemann MT, Strong MA, Hao LY, Greider CW. The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell. 2001;107:67–77. doi: 10.1016/s0092-8674(01)00504-9. [DOI] [PubMed] [Google Scholar]
- 69.Hao LY, et al. Short telomeres, even in the presence of telomerase, limit tissue renewal capacity. Cell. 2005;123:1121–1131. doi: 10.1016/j.cell.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 70.Tomas-Loba A, et al. Telomerase reverse transcriptase delays aging in cancer-resistant mice. Cell. 2008;135:609–622. doi: 10.1016/j.cell.2008.09.034. [DOI] [PubMed] [Google Scholar]
- 71.Lee HW, et al. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 72.Rudolph KL, et al. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 73.Du X, et al. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang S, et al. Essential role of limiting telomeres in the pathogenesis of Werner syndrome. Nature Genet. 2004;36:877–882. doi: 10.1038/ng1389. [DOI] [PubMed] [Google Scholar]
- 75.Jaskelioff M, et al. Telomerase reactivation reverses tissue degeneration in aged telomerase-deficient mice. Nature. 2011;469:102–106. doi: 10.1038/nature09603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vulliamy TJ, Dokal I. Dyskeratosis congenita: the diverse clinical presentation of mutations in the telomerase complex. Biochimie. 2008;90:122–130. doi: 10.1016/j.biochi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 77.Muftuoglu M, et al. The clinical characteristics of Werner syndrome: molecular and biochemical diagnosis. Hum Genet. 2008;124:369–377. doi: 10.1007/s00439-008-0562-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perlman S, Becker-Catania S, Gatti RA. Ataxia-telangiectasia: diagnosis and treatment. Semin Pediatr Neurol. 2003;10:173–182. doi: 10.1016/s1071-9091(03)00026-3. [DOI] [PubMed] [Google Scholar]
- 79.Sharpless NE, DePinho RA. Telomeres, stem cells, senescence, and cancer. J Clin Invest. 2004;113:160–168. doi: 10.1172/JCI20761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choudhury AR, et al. Cdkn1a deletion improves stem cell function and lifespan of mice with dysfunctional telomeres without accelerating cancer formation. Nature Genet. 2007;39:99–105. doi: 10.1038/ng1937. [DOI] [PubMed] [Google Scholar]
- 81.Leri A, et al. Ablation of telomerase and telomere loss leads to cardiac dilatation and heart failure associated with p53 upregulation. EMBO J. 2003;22:131–139. doi: 10.1093/emboj/cdg013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee JW, Harrigan J, Opresko PL, Bohr VA. Pathways and functions of the Werner syndrome protein. Mech Ageing Dev. 2005;126:79–86. doi: 10.1016/j.mad.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 83.Yang DQ, Halaby MJ, Li Y, Hibma JC, Burn P. Cytoplasmic ATM protein kinase: an emerging therapeutic target for diabetes, cancer and neuronal degeneration. Drug Discov Today. 2011;16:332–338. doi: 10.1016/j.drudis.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 84.Basel-Vanagaite L, et al. Expanding the clinical phenotype of autosomal dominant dyskeratosis congenita caused by TERT mutations. Haematologica. 2008;93:943–944. doi: 10.3324/haematol.12317. [DOI] [PubMed] [Google Scholar]
- 85.Mercer JR, et al. DNA damage links mitochondrial dysfunction to atherosclerosis and the metabolic syndrome. Circ Res. 2010;107:1021–1031. doi: 10.1161/CIRCRESAHA.110.218966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Passos JF, et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010;6:347. doi: 10.1038/msb.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kovalenko OA, et al. A mutant telomerase defective in nuclear-cytoplasmic shuttling fails to immortalize cells and is associated with mitochondrial dysfunction. Aging Cell. 2010;9:203–219. doi: 10.1111/j.1474-9726.2010.00551.x. [DOI] [PubMed] [Google Scholar]
- 88.Haendeler J, et al. Mitochondrial telomerase reverse transcriptase binds to and protects mitochondrial DNA and function from damage. Arterioscler Thromb Vasc Biol. 2009;29:929–935. doi: 10.1161/ATVBAHA.109.185546. [DOI] [PubMed] [Google Scholar]
- 89.Guo N, et al. Short telomeres compromise β-cell signaling and survival. PLoS ONE. 2011;6:e17858. doi: 10.1371/journal.pone.0017858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sarin KY, et al. Conditional telomerase induction causes proliferation of hair follicle stem cells. Nature. 2005;436:1048–1052. doi: 10.1038/nature03836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee J, et al. TERT promotes cellular and organismal survival independently of telomerase activity. Oncogene. 2008;27:3754–3760. doi: 10.1038/sj.onc.1211037. [DOI] [PubMed] [Google Scholar]
- 92.Choi J, et al. TERT promotes epithelial proliferation through transcriptional control of a Myc- and Wntrelated developmental program. PLoS Genet. 2008;4:e10. doi: 10.1371/journal.pgen.0040010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cong Y, Shay JW. Actions of human telomerase beyond telomeres. Cell Res. 2008;18:725–732. doi: 10.1038/cr.2008.74. [DOI] [PubMed] [Google Scholar]
- 94.Flores I, Cayuela ML, Blasco MA. Effects of telomerase and telomere length on epidermal stem cell behavior. Science. 2005;309:1253–1256. doi: 10.1126/science.1115025. [DOI] [PubMed] [Google Scholar]
- 95.Vidal-Cardenas SL, Greider CW. Comparing effects of mTR and mTERT deletion on gene expression and DNA damage response: a critical examination of telomere length maintenance-independent roles of telomerase. Nucleic Acids Res. 2010;38:60–71. doi: 10.1093/nar/gkp855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Santos JH, Meyer JN, Skorvaga M, Annab LA, Van Houten B. Mitochondrial hTERT exacerbates free-radical-mediated mtDNA damage. Aging Cell. 2004;3:399–411. doi: 10.1111/j.1474-9728.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 97.Sharma NK, et al. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2011;40:712–725. doi: 10.1093/nar/gkr758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ahmed S, et al. Telomerase does not counteract telomere shortening but protects mitochondrial function under oxidative stress. J Cell Sci. 2008;121:1046–1053. doi: 10.1242/jcs.019372. [DOI] [PubMed] [Google Scholar]
- 99.Park JI, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature. 2009;460:66–72. doi: 10.1038/nature08137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Strong MA, et al. Phenotypes in mTERT+/− and mTERT−/− mice are due to short telomeres, not telomere-independent functions of telomerase reverse transcriptase. Mol Cell Biol. 2011;31:2369–2379. doi: 10.1128/MCB.05312-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Patel AY, McDonald TM, Spears LD, Ching JK, Fisher JS. Ataxia telangiectasia mutated influences cytochrome c oxidase activity. Biochem Biophys Res Commun. 2011;405:599–603. doi: 10.1016/j.bbrc.2011.01.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ambrose M, Goldstine JV, Gatti RA. Intrinsic mitochondrial dysfunction in ATM-deficient lymphoblastoid cells. Hum Mol Genet. 2007;16:2154–2164. doi: 10.1093/hmg/ddm166. [DOI] [PubMed] [Google Scholar]
- 103.Pallardo FV, et al. Mitochondrial dysfunction in some oxidative stress-related genetic diseases: ataxia-telangiectasia, down syndrome, Fanconi anaemia and Werner syndrome. Biogerontology. 2010;11:401–419. doi: 10.1007/s10522-010-9269-4. [DOI] [PubMed] [Google Scholar]
- 104.Varela I, et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 105.Lombard DB, et al. DNA repair, genome stability, and aging. Cell. 2005;120:497–512. doi: 10.1016/j.cell.2005.01.028. [DOI] [PubMed] [Google Scholar]
- 106.Wenz T. Mitochondria and PGC-1α in aging and age-associated diseases. J Aging Res. 2011;2011:810619. doi: 10.4061/2011/810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu J, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459:387–392. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Salih DA, Brunet A. FoxO transcription factors in the maintenance of cellular homeostasis during aging. Curr Opin Cell Biol. 2008;20:126–136. doi: 10.1016/j.ceb.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 110.von Zglinicki T. Oxidative stress shortens telomeres. Trends Biochem Sci. 2002;27:339–344. doi: 10.1016/s0968-0004(02)02110-2. [DOI] [PubMed] [Google Scholar]
- 111.Passos JF, Saretzki G, von Zglinicki T. DNA damage in telomeres and mitochondria during cellular senescence: is there a connection? Nucleic Acids Res. 2007;35:7505–7513. doi: 10.1093/nar/gkm893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sablina AA, et al. The antioxidant function of the p53 tumor suppressor. Nature Med. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matoba S, et al. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 114.Bae BI, et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 115.Saleem A, Adhihetty PJ, Hood DA. Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genom. 2009;37:58–66. doi: 10.1152/physiolgenomics.90346.2008. [DOI] [PubMed] [Google Scholar]
- 116.Mendrysa SM, et al. Tumor suppression and normal aging in mice with constitutively high p53 activity. Genes Dev. 2006;20:16–21. doi: 10.1101/gad.1378506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Matheu A, et al. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007;448:375–379. doi: 10.1038/nature05949. [DOI] [PubMed] [Google Scholar]
- 118.Ventura N, et al. p53/CEP-1 increases or decreases lifespan, depending on level of mitochondrial bioenergetic stress. Aging Cell. 2009;8:380–393. doi: 10.1111/j.1474-9726.2009.00482.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Niedernhofer LJ, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 120.Hinkal G, Donehower LA. How does suppression of IGF-1 signaling by DNA damage affect aging and longevity? Mech Ageing Dev. 2008;129:243–253. doi: 10.1016/j.mad.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Devin JK, Young PP. The effects of growth hormone and insulin-like growth factor-1 on the aging cardiovascular system and its progenitor cells. Curr Opin Investig Drugs. 2008;9:983–992. [PubMed] [Google Scholar]
- 122.Keyes WM, Mills AA. p63: a new link between senescence and aging. Cell Cycle. 2006;5:260–265. doi: 10.4161/cc.5.3.2415. [DOI] [PubMed] [Google Scholar]
- 123.Su X, Flores ER. TAp63: the fountain of youth. Aging (Albany NY) 2009;1:866–869. doi: 10.18632/aging.100095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Burnett C, et al. Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature. 2011;477:482–485. doi: 10.1038/nature10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Passos JF, et al. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007;5:e110. doi: 10.1371/journal.pbio.0050110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nautiyal S, DeRisi JL, Blackburn EH. The genome-wide expression response to telomerase deletion in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 2002;99:9316–9321. doi: 10.1073/pnas.142162499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Moiseeva O, Bourdeau V, Roux A, Deschenes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009;29:4495–4507. doi: 10.1128/MCB.01868-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146:682–695. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 130.McCormick MA, Tsai SY, Kennedy BK. TOR and ageing: a complex pathway for a complex process. Phil Trans R Soc B. 2011;366:17–27. doi: 10.1098/rstb.2010.0198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–332. doi: 10.1038/nature10317. [DOI] [PubMed] [Google Scholar]
- 132.de Jesus BB, et al. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell. 2011;10:604–621. doi: 10.1111/j.1474-9726.2011.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wenz T, Rossi SG, Rotundo RL, Spiegelman BM, Moraes CT. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc Natl Acad Sci USA. 2009;106:20405–20410. doi: 10.1073/pnas.0911570106. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 134.Little JP, Safdar A, Bishop D, Tarnopolsky MA, Gibala MJ. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1303–R1310. doi: 10.1152/ajpregu.00538.2010. [DOI] [PubMed] [Google Scholar]
- 135.Timmers S, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011;14:612–622. doi: 10.1016/j.cmet.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hu J, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148:651–663. doi: 10.1016/j.cell.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hardie DG. Signal transduction: How cells sense energy. Nature. 2011;472:176–177. doi: 10.1038/472176a. [DOI] [PubMed] [Google Scholar]