

Abstract
Poly-lactic-co-glycolic acid (PLGA) has been the most successful polymeric biomaterial for use in controlled drug delivery systems. There are several different chemical and physical properties of PLGA that impact the release behavior of drugs from PLGA delivery devices. These properties must be considered and optimized in drug release device formulation. Mathematical modeling is a useful tool for identifying, characterizing, and predicting the mechanisms of controlled release. The advantages and limitations of poly (lactic-co-glycolic acid) for controlled release are reviewed, followed by a review of current approaches in controlled release technology that utilize PLGA. Mathematical modeling applied towards controlled release rates from PLGA-based devices will also be discussed to provide a complete picture of state of the art understanding of the control achievable with this polymeric system, as well as the limitations.
Keywords: PLGA, PLA, PGA, Controlled release, Mathematical modeling
I. CONTROLLED RELEASE TECHNOLOGY
Controlled release technology is used to prolong the release of a chemical from a polymeric carrier device. These devices are usually formed by incorporating the drug within a polymer matrix that acts as a carrier for the drug. This technology has found commercial success since the 1950’s when it was first developed for oral sustained release products (1). The first clinical use of polymeric devices for controlled release was introduced in the 1960’s, while the development of these devices has been widely researched since the early 1950’s (2,3). Modern applications of this technology are mainly focused on the delivery of pharmaceuticals, but the fields of agriculture, pesticides, food, cosmetics and household products have also found useful applications for controlled release (1,4–8). Controlled release of pharmaceuticals have resulted in several benefits to drug delivery:
eliminating the need for repetitive dosing to maintain therapeutic effects
improving patient comfort and compliance
providing better regulation and control over the release rate of a drug
reducing variability in drug – blood plasma concentrations that may lead to toxicity and efficacy concerns (Figure 1)
protecting active drug molecules from degradation and loss of therapeutic activity prior to delivery
gaining the use of active therapeutic agents that are ineffective with conventional delivery methods
Figure 1.

Comparing blood-plasma concentration variability between repetitive first order oral dosage and zero order controlled release (a) dosage schedule (b)blood-plasma concentration response
Several different routes of administration (oral, injectable, transdermal, implantable) and device morphologies (micro-/nano- spheres, hydrogels, films, pellets) have been investigated for controlled release devices. Injectable micro-/nano-spheres are the most popular of these forms. These types of advanced delivery systems continue to be developed for wide range of drugs, different biodegradable polymers, device morphologies, routes of administration, and complex device designs.
II. POLYMERS USED IN CONTROLLED RELEASE
Several different synthetic and natural polymers have been investigated for controlled release systems and several of these have been approved for commercial use by the FDA (2). Natural polymers include high molecular weight polysaccharides such as agarose, alginate, carrageenan, hyaluronic acid, chitosan and dextran, or proteins such as collagen, albumin, gelatin, elastin, and silk fibroin (2,9,10). However natural polymers are generally more limited in terms of their synthesis and processing than synthetic polymers. This disadvantage has focused more attention towards the use of synthetic polymers for controlled release systems. Synthetic polymers include cellulose derivatives, acrylic polymers, polaxamers, polaxamines, polyesters, polyanhydrides, polyamides, phosphorus based polymers and polyurethanes (2,4). Two important properties to consider among the many different synthetic polymers are biodegradability and biocompatibility. Biodegradable polymers are of interest because the need for surgery to remove implanted release devices is eliminated. The biocompatibility of the polymer is important so that complications with toxicity and inflammation are avoided. A number of different release mechanisms and release kinetics have been observed in studies with these polymers.
III. MECHANISMS OF RELEASE
The mechanism of release is the rate limiting step or series of rate limiting steps that control the rate of drug release from a device until release is exhausted. The major release mechanisms include: diffusion, solvent penetration/device swelling, degradation and erosion of the polymer matrix, or a combination of these mechanisms occurring on different time scales that leads to a more complex release process. Schematics of these individual release mechanisms are displayed in Figure 2.
Figure 2.

Schematic of common release mechanisms (a) Diffusion (b) Polymer degradation and erosion (c)solvent penetration/device swelling
The most desirable case is zero order release kinetics. In such a case the rate of drug release is independent of its dissolved concentration in the release medium and is delivered at a constant rate over time. This type of release is unachievable by current polymeric release systems. Diffusion (2a) is the most common release mechanism and is dependent on the concentration of the dissolved drug as described by Fick’s second Law. The rate of release for diffusion has a half ordered time dependency. Erodible delivery systems (2b) are also non-zero ordered and the rate of release is dependent on the degradation kinetics of the polymer used. Solvent penetration systems (2c) are also non-zero ordered and their rate is dependent upon the permeability of the polymer used. Mathematical equations that are used to model various release mechanisms are reviewed in section X of this article. Typically the polymer and the processing modes selected for the device formulation influence the mechanism of release. Internal diffusion to the surface of the delivery device is the most common release mechanism.
IV. PROPERTIES THAT EFFECT THE RELEASE MECHANISM
In controlled release systems there are several factors that influence the release mechanism and the kinetics of the release. Material properties may be related to the polymer, the releasing chemical, interactions between the polymer and releasing chemical, or properties of the processing methods chosen to fabricate the device. Examples of these properties are displayed in Figure 3. A list of these properties includes: molecular weight, crystallinity, monomer composition, solubility, hydrophobicity, pKa, permeability, porosity, mass load percent, and device morphology (9). Certain material properties may be manipulated and present themselves as design variables that may be optimized to fit the need of a specific release system. Understanding the effects of such material properties plays a major role in the development of a successful controlled release system. Factors that may be affected include: the polymers ability to be fabricated into a desired morphology or modality (i.e. microsphere, hydrogel); the kinetics of biodegradation; biocompatibility or toxicity of the polymer; and the ability of degradation products to be resorbed by the body. If these effects have been characterized it is easier to select the appropriate polymer(s) for the design of a controlled release device.
Figure 3.

Factors /material properties that affect the release mechanism
V. HISTORY OF PLGA IN CONTROLLED RELEASE
To date, poly-lactic-co-glycolic-acid (PLGA) is the most well known and widely applied polymer in controlled release systems. This synthetic polymer has found great success due to its biocompatibility, biodegradability, and favorable release kinetics, but also faces stability concerns for protein delivery (11). Poly-(glycolic acid) (PGA) and poly-(lactic acid) (PLA) along with the copolymer poly-(lactic-co-glycolic acid) (PLGA) are biodegradable synthetic polymers that were discovered as surgical sutures in the1960’s (12–15). The successful development of these polymers as surgical sutures led to the expansion of their use as polymeric biomaterials. Since then the copolymer has been established as the most successful and widely researched polymer for application in controlled release systems (16) and is considered the “gold standard” of biodegradable polymers for controlled delivery systems (9). PLGA has been used to release a wide range of small molecule drugs, peptides, and proteins, including fertility regulating hormones, growth hormones, steroid hormones, anti-inflammatory drugs, cytokines, chemotherapeutics, antibiotics, narcotic antagonists, insulin, and vaccines (7,17–22). In comparison to other polymers that have been investigated for controlled release, PLGA is relatively easy to process into different device morphologies such as injectable micro-/nanospheres (23).
VI. PROPERTIES OF PLGA
PLGA is an aliphatic polymer with a polyester backbone that is formed through the copolymerization of lactic and glycolic acid monomers (Figure 4). Because PLGA is a copolymer, it can be polymerized into many different formulations based on the composition of lactic and glycolic acid monomers. This copolymer composition may be used to manipulate the crystallinity, hydrophilicity, and glass transition temperature of the copolymer (14). The glass transition temperature typically will vary between 40–60°C depending on the copolymer composition and molecular weight, however all compositions of PLGA will result in a thermoplastic amorphous polymer matrix below the glass transition temperature (24). If lactic acid is used in a higher ratio than glycolic acid, a more hydrophobic copolymer is formed due to the higher hydrophobicity of lactic acid. Such a change can be used to reduce the rate of water penetration into a device. Also, by manipulating the copolymer composition of lactic acid the hydrophobicity of the drug and copolymer can be matched better to promote a more uniform dispersion of drug throughout the polymer matrix or help to stabilize encapsulated proteins (23). An increase in the ratio of glycolic acid in the copolymer composition can be used to produce a more crystalline polymer matrix (25). Glycolic acid does not form a racemic mixture like lactic acid. However, the lactic acid monomers that are used for the copolymerization of PLGA are often preferred in a racemic mixture of the D-PLA and L-PLA enantiomers over the stereo-regular form of either D-PLA or L-PLA. This is because the use of the racemic mixture yields a polymer product that exhibits more amorphous features in its structure to improve the homogeneous dispersion of drugs throughout the PLGA polymer matrix when fabricating delivery devices (23).
Figure 4.

Molecular structures of a) L-PLA b) D-PLA c) PGA and d) PLGA(x=number of lactic acid units, y=number of glycolic acid units)
VII. PLGA DEGRADATION
PLGA is degraded through the hydrolytic cleavage of its polyester backbone (26). The degradation kinetics of PLGA have been characterized by collecting data via size exclusion chromatography followed by modeling (27). The degradation studies characterized the polymer as bulk erosion and follow pseudo-first-order degradation kinetics that can be modeled using Eq. 1. X is the number of bond cleavages per initial number-average molecule, kd is the copolymer degradation rate constant, t is time. X is calculated by the measurements of the initial number average molecular weight and the number average molecular weight at degradation time t via size exclusion chromatography.
| (Eq. 1) |
The hydrolysis of the polyester can be catalyzed by an acid. Because carboxylic acid chain ends are products of the hydrolytic cleavage of the PLGA polymers, the degradation of the polymer may lead to autocatalysis (28). This autocatalytic behavior has lead to heterogeneous degradation of PLGA polymer matrix as a result of pH gradients within delivery devices (29). A pH gradient from the center to the surface of the device may form due to the internal diffusion of the acidic degradation products to the surface and away from the device.
Since PLGA is a bulk eroding polymer and not a surface eroding polymer PLGA degradation products, such as monomers and small PLGA fragments, will diffuse and erode away from the entire volume of the device and not just the surface of the device. The difference between these two erosion processes is shown in Figure 6. Because PLGA degradation is driven by hydrolysis, the process can be exploited in delivery systems (30). The degradation products, glycolic and lactic acid, are both non-toxic chemicals that can be metabolized by the body (31).
Figure 6.

Surface & Bulk Erosion Schematics
VIII. PLGA RELEASE MECHANISM
The degradation rate of the PLGA polymer is the most commonly discussed release mechanism for PLGA based release devices (2,32–38). However, depending on the properties of the device other processes may also contribute to the release mechanism, including: water penetration and solubilization when the device is first submerged in an aqueous environment; erosion and diffusion of PLGA polymer fragments; and the rate of diffusion of the releasing drug. Because these processes can occur on similar timescales the release mechanism may be complex, and understanding differences that are observed among release data may therefore be difficult to interpret. To describe a generalized release mechanism from a PLGA matrix we first consider the rate of water penetration and hydration of the PLGA matrix. Initially water penetrates from the surface to the center of the release device which in turn activates the processes of hydrolytic PLGA polymer degradation and diffusion of the drug encapsulated within the polymer matrix. As polymer degradation proceeds, small fragments and monomers from PLGA begin to erode away from the device, which will in turn accelerate the diffusion rate of the releasing drug. If a pH gradient is formed the autocatalytic nature of PLGA hydrolysis will accelerate the diffusion and erosion processes of the drug and polymer even more at the center of the device. Once enough mass has eroded away from the center of the device the pH gradient will begin to dissipate counterbalancing the autocatalytic hydrolysis until the exhaustion of the release drug from the device is complete. For small molecule drugs that will diffuse through the PLGA matrix much faster it may be expected that the exhaustion of the release device will occur before the total degradation and erosion of the PLGA polymer matrix. A schematic of the PLGA release mechanism is depicted in Figure 7.
Figure 7.

Schematic of processes that contribute to PLGA device release mechanism: a) Initial drug loaded device b) Water penetration and drug diffusion c) Bulk degradation and erosion d) Autocatalytic PLGA degradation and accelerated drug diffusion e) Total degradation and exhaustion of drug release
IX. CURRENT PLGA CONTROLLED RELEASE RESEARCH
Several research areas have emerged that are focused towards enhancing the performance of PLGA based release devices. These areas can be classified as: the blending of PLGA with other polymers, modifying the surface of PLGA release devices, increasing drug stability with the use of excipients, using novel processing techniques, or applying PLGA release in new fields such as tissue engineering.
A. PLGA Blended with Other Polymers
Several methods of incorporating other polymers with PLGA have been investigated to combine the benefits of the different polymers. The majority of this work has been focused towards copolymerizing PLGA with other polymers or forming complex device designs such as the encapsulation of a PLGA device within another polymer. Embedding drug loaded PLGA micro-particles within hydrogels is an approach towards developing novel PLGA delivery systems that has gained popularity. Hydrogels are attractive biomaterials due to their ability to provide a soft, permeable, and hydrophilic interface with body tissue. However their highly hydrophilic nature limits their ability to release hydrophobic drugs or to achieve zero order release over extended periods. These obstacles are overcome by embedding PLGA particles in hydrogel systems because they utilize the capabilities of controlled release from PLGA while providing the benefits of hydrogels. Some hydrogels that have been investigated include poly-vinyl-alcohol, alginate, chitosan, and gelatin (39–41). PLGA has also been copolymerized with other synthetic polymers. Thermosensitive PEG-PLGA-PEG tri-block copolymers have been developed that can exist as free flowing liquids at room temperature but form gels at body temperature (42). PEGylated PLGA nano-particles were shown to be more effective carriers for protein and peptide drugs due to their ability to extend the half life of the loaded drug (43).
B. Surface Modification and Coatings
The coating or surface modification of PLGA particles with other biodegradable polymers, and/or chemical components is another area of research. The goal of these studies is to expand the range of functionality of PLGA micro-/nano-particle release systems. The introduction of a surface coating has overcome some obstacles of PLGA delivery for enhanced drug release. The blood brain barrier poses an obstacle when attempting the delivery of drugs to the brain. Trimethylated chitosan surface coatings facilitates the active transport of PLGA nano-particles across the blood brain barrier due to its properties as a cationic ligand (44). PLGA micro-/nano-particles that combine PLGA particles with the beneficial properties of liposomal delivery devices have been developed by layering PLGA nano-particles with a soybean lethicin monolayer and a polyethylene glycol shell (45). Targeted controlled release of antibiotics to bone tissues using hydroxyapatite, a major component in bone, may be achieved by hydroxyapatite-coated PLGA microspheres (46). Coated PLGA particle delivery systems continue to be investigated and their success provides opportunities for novel targeted delivery systems that may also maintain controlled or sustained release. Also for embedded PLGA particles within hydrogels, low levels of surface active polyacids have been introduced to achieve the erosion of the embedded PLGA particles (40).
C. Increased Stability Formulations
The incomplete release of native protein as a result of protein instability is one of the largest obstacles facing protein delivery from PLGA controlled delivery devices. The denaturation and aggregation of native protein due to instability not only causes loss of therapeutic activity but also may cause complications regarding immunogenicity and toxicity (47). Several approaches have found limited success in overcoming this problem. Processing may be optimized for the most favorable conditions that minimize the degradation or denaturation of active protein. Introducing stabilizing excipients into PLGA formulations may prevent denaturing interactions between the polymer and protein. Reversible protein aggregation prior to PLGA encapsulation may be used to minimize the destabilizing polymer-protein interactions (48). Prior approaches towards protein stabilization using chemical modifications such as PEGylation may be used to create a more stable protein (49). Protein degradation that is due to the presence of acidic PLGA degradation products may be limited by tailoring the properties of the PLGA polymer to delay degradation or by incorporating basic salts in formulations to neutralize the acidic environment (48).
D. Material Formats & Processing
Some materials science research has been centered on improving and optimizing the processing steps of PLGA systems, mainly for the emulsion process of fabricating PLGA microand nano-particles. Organic solvent selection, polymer molecular weight, and copolymer composition are processing variables of the emulsion fabrication method of PLGA microspheres that may be optimized for particle formation and release behavior (50). Also a more uniform size distribution of PLGA particles may be achieved through advanced emulsification techniques such as membrane emulsification instead of the common freeze-thaw emulsion method (51).
E. PLGA Tissue Engineering Scaffolds
The field of tissue engineering has also been utilizing the well characterized release behavior of PLGA in the formulation of advanced tissue engineering scaffolds. Blends of PLGA with other polymers have been investigated towards biomaterials (52,53). Analogous to the delivery devices previously mentioned, some tissue engineering scaffold systems have taken advantage of embedding loaded PLGA micro-particles to enhance release rates of important active agents such as growth factors from the scaffolds to the cells (54,55). For these scaffolds PLGA particles provide the controlled delivery of important chemical factors to the cells of the scaffold while a better base polymer is used to provide greater mechanical support and cell adhesion over PLGA. Therefore the benefits of two systems can be combined.
X. MATHEMATICAL MODELING OF PLGA RELEASE SYSTEMS
Mathematical modeling is a useful tool that can be used to identify release mechanisms, characterize the significant transport processes involved, estimate unknown parameters (i.e. diffusion coefficient), reduce experimentation, and provide predictive capabilities (56,57). From prior work, several mechanistic and empirical models have been developed for controlled release systems.
Empirical models have been more popular in the fitting of controlled release data and are simpler to develop, however in terms of predictive power and comprehension of the physics of the release system they are much weaker. Because mechanistic models are developed as systems of differential equations that describe the mass transport phenomenon that control drug release and because of their stronger predictive capabilities over empirical models mechanistic models should be used over empirical models whenever possible. Of the several models that have been developed, the majority can be grouped into a few categories:
I: One-dimensional Fickian diffusion models analytically solved for several simple geometries
II: Dissolution based models for drugs of low water solubility
III: Generalized empirical models describing nth ordered release kinetics 0 < n < 1
IV: Empirical models describing polymer degradation and erosion
V: Diffusion-reaction based mechanistic models developed by Monte-Carlo simulation (58–60).
Both mechanistic and empirical models have been applied towards PLGA release systems. The need for robust mechanistic models of controlled release systems gained attention in the 1980’s after a large amount of empirical in vitro and in vivo release data had been collected for PLGA (61). Early mechanistic modeling of PLGA has focused attention towards characterizing the processes of polymer degradation coupled with drug diffusion. With the further characterization of PLGA degradation kinetics came updated classic models that accounted for degradation.
The classic Higuchi equation was updated to incorporate a time dependent permeability that would account for the effects of a degrading PLGA polymer matrix (Eq. 2).
| (Eq. 2) |
Where Qt is the cumulative mass of drug release per unit release area at time t, C* w is the solubility of the drug in the release medium, P is the time dependent permeability, A is the drug loading, L is the half thickness of the matrix, and Qinf is the drug loading per unit release area (62). This model had the advantage of being able to account for the changing porosity of a PLGA release device that was observed through scanning electron microscopy of degrading PLGA particles. The typical Higuchi that assumes a constant permeability coefficient is not enough to approximate release from PLGA devices. A release model based on Fick’s second law for a spherical geometry depends on a changing polymer molecular weight (Eq. 3).
| (Eq. 3) |
Where Mt is the cumulative mass released at time t, r is the radius of the micro particle, C0 is the initial drug concentration within the polymer matrix, Cs is the solubility of the drug in the medium, D is the diffusion coefficient (63). In this model the changing molecular weight is accounted for by the diffusion coefficient. However this model is limited to the spherical particle format and must be resolved for non-spherical device morphologies.
Although the majority of modeling efforts for PLGA release has focused on a complex release mechanism there are studies that have focused exclusively on a diffusion based release mechanism. The Roseman-Higuchi model (Eq. 4) was applied to small molecule release data and suggested a strictly diffusion based release mechanism.
| (Eq. 4) |
F is the fraction of drug release at time t, ao is the radius of the cylinder, C is the solubility of the drug in the polymer, D is the diffusivity, and A is the total initial content of drug in polymer (64). The use of this equation is limited with the knowledge that PLGA is a degrading polymer. Formulations of PLGA that degrade much more slowly than the typical 50:50 PLGA formulation that is used may find this model useful because degradation of the polymer will have less of an impact on release in such as case. The model proposed by Heller et al. (1980) describes a diffusion mechanism that accounts for the changing porosity of the polymer matrix due to homogeneous degradation (Eq. 5). However, after the fitting of this model to release data, it was found to have limited success compared to the Higuchi models (42).
| (Eq. 5) |
In this model C is a parameter that accounts for the surface area, permeability coefficient, and the initial concentration (65).
A modified version of the Heller equation that incorporated a diffusion term (Eq. 6) successfully described release of small hydrophobic molecules from PEG-PLGA-PEG tri-block copolymers microspheres.
| (Eq. 6) |
B and k are functions of the initial drug concentration, the diffusion coefficient, and the device thickness (42). However application of this model to typical PLGA systems has not been reported.
A release model for a complex device that consisted of spherical drug particles dispersed within a PLGA disc was created to describe a burst phase release from the surface of the device as well as a polymer degradation controlled release (Eq. 7). The burst phase was described as a first ordered surface dissolution. This equation is useful when burst release is a major contributing factor to the release. This is typically the case when a considerable amount of the releasing drug has settled on the surface of the device and instantaneously dissolves at the beginning of release.
| (Eq. 7) |
In this equation, Ftot is the total fraction of drug released at time t, Kb is the release rate constant, k is the rate constant, and Tmax is the time to maximum release (66).
The classic Fickian diffusion model was modified for PLGA drug release to incorporate a time dependent diffusion coefficient that is dependent on the changing polymer molecular weight as degradation occurs (Eq. 8–10).
| (Eq. 8) |
| (Eq. 9) |
| (Eq. 10) |
Mt is the cumulative mass released at time t, Minf is the cumulative mass released at infinite time, r is the radius of the microsphere, D is the diffusion coefficient, MW is the polymer molecular weight, D0 is the initial drug diffusivity, k is a constant, and kdegr is the first order polymer degradation rate constant (32). This system of equations will not extrapolate well across various PLGA device formulations. The coefficients that describe the effect of degradation on the diffusivity must be recalibrated in each case.
Recent developments in mathematical modeling of controlled release systems have presented deterministic models solved with numerical integration for the prediction of bulk eroding biodegradable polymer matrices such as PLGA (67). In recent PLGA release modeling work, numerical approaches using finite element solutions have been used to develop a internal Fickian diffusion model with a time dependent porosity term that accounts for a pseudo-first order degradation rate of the PLGA polymer matrix (Eq. 11–14).
| (Eq. 11) |
| (Eq. 12) |
| (Eq. 13) |
| (Eq. 14) |
In this model Ca is the agent concentration within the matrix, Deff is the effective diffusion coefficient, D is the diffusion coefficient, e is the porosity at time t, tau is the mean time for pore formation,σ2 is the variance in time required to form pores, kCw is the average pseudo first order degradation rate, Mwo is the initial molecular weight of the polymer, and Mwr is the average polymer molecular weight in a differential volume of matrix that permits diffusion of the encapsulated agent. Simulation of this model has successfully predicted a wide range of therapeutically relevant release behaviors.
Even though more complex models and modeling techniques have been developed, classic empirical relationships such as the exponential model and Fickian diffusion model remain the most popular in modeling PLGA release data (68). Whether for the development of novel mathematical models and modeling approaches or for the characterization of release systems through the application of existing release models, mathematical modeling of controlled release data should be preformed whenever possible. Design and research of polymeric release systems greatly benefit from the physical characterization that modeling provides and helps improve reliability and reproducibility of a designed system.
XI. DISCUSSION
The field of controlled drug release is well established and continually presents many promising technologies for future drug delivery. A variety of concepts that pertain to the development of polymeric controlled drug delivery systems have been discussed, such as the most relevant formulation variables, polymers that may be used to fabricate such systems, and morphologies or geometries that may be used. PLGA is the most successful and most characterized polymer for controlled release drug delivery systems. It is favored because of its biocompatibility, biodegradability and mechanical strength and continues to be used to develop new controlled release systems. However several obstacles remain for PLGA in its use in controlled release systems. Due to the limited success of protein release from PLGA matrices due to protein instability, even for new formulation approaches and the design of more complex devices it is likely that more hydrophilic polymers may be a better option.
Also the use of chemically modified PLGA in delivery systems such as functionalizing PLGA to create surface modified particles has generated new ideas for targeted delivery. However the chemical modifications of PLGA are extensive. These modifications may compromise the mechanical strength of the polymer as well as the loss of receptor binding ability resulting in a loss of targeting (69).
With regard to modeling of PLGA, insufficient emphasis has been placed on the benefits of this approach. There exists many experimental release studies leading to an ocean of data however not enough of the release studies take advantage of modeling. Emphasis should also be placed on the use of mechanistic models over empirical models so that the field can gain a higher level of extrapolation for different systems. Also, for the development of more complex PLGA delivery systems there will be an increased need for the development of novel models and modeling methodologies to describe these systems. For these new systems the application of available models will not suffice. Thus, there is room for new approaches to modeling to provide a better tool kit in the directed or predictable outcomes desired to avoid trial and error experimental approaches.
The use of PLGA for responsive polymeric delivery systems is intriguing. These types of delivery systems are designed to adjust drug release based changes in the physiologic environment or through external stimuli. For example insulin release devices may be able to respond to changes in glucose concentration or external stimuli such as magnets, or ultrasound may be supplemented to control the rate of drug release (70). The strengths and limitations of PLGA towards developing such systems will be a factor that will contribute to its future uses in controlled release.
Figure 5.
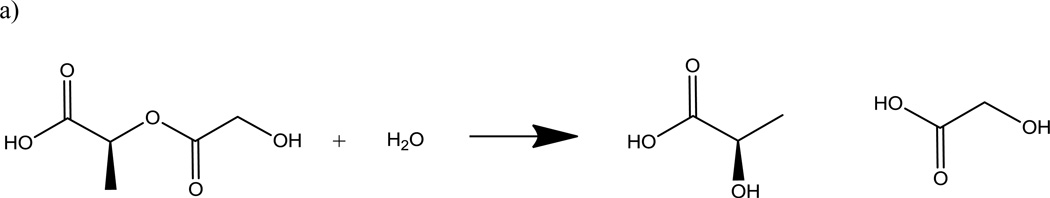

Schematic of PLGA degradation kinetics: a) passive hydrolysis b) autocatalysis
Table 1.
Common Controlled Release Models
| Model Name & Variables | Equation | Cat. | |
|---|---|---|---|
| Fickian Film Diffusion Model(58) (mass released at time t, Mt; mass released at infinite time, M∞; diffusivity, D; thickness, L) |
I | ||
| Higuchi Model (71) (mass released at time t per unit area, Q; diffusivity, D; initial drug concentration, C0; drug solubility, Cs) |
II | ||
| Hopfenberg model(72) (mass released at time t, Mt; mass released at infinite time, M∞; erosion rate constant, k0; initial drug concentration, C0; characteristic length, a0; shape factor, n) |
IV | ||
| Zero-order release rate(73) (mass released at time t, Qt; initial mass, Q0; rate constant, k0) |
Qt = Q0 + k0t | III | |
| First order release rate(74) (mass released at time t, Qt; initial mass, Q0; rate constant, k1) |
Qt = Q0 · e−k1t | III | |
| Hixon-Crowell Model(75) (mass fraction dissolved into solution at time t, ft; release constant, Kβ) |
(1 − ft)1/3 = 1 − Kβt | II | |
| Weibull Model(76) (mass released at time t, Mt; mass released at infinite time, M∞; location parameter (lag time) Ti; shape parameter, b; time scale parameter, a) |
III | ||
| Exponential Model(58) (mass released at time t, Mt; mass released at infinite time, M∞; rate constant, k; exponential factor, n) |
III | ||
| Exponential model with lag time(77) (mass released at time t after lag phase, Mt-tlag; mass released at infinite time, M∞; rate constant, k; exponential factor, n; lag time, tlag) |
III | ||
| Exponential model with burst effect (mass released at time t after lag phase, Mt-tlag; mass released at infinite time, M∞; rate constant, k; exponential factor, n; mass fraction released as burst, b) |
III |
ACKNOWLEGMENTS
We thank the NIH (EB002520) for support of these studies.
Contributor Information
Daniel J. Hines, Email: daniel.hines@tufts.edu.
David L. Kaplan, Email: david.kaplan@tufts.edu.
REFERENCES
- 1.Hui HW, Robinson JR, Lee VHL. Design and fabrication of oral controlled release drug delivery systems. In: Robinson JR, Lee VHL, editors. Controlled drug delivery: Fundamentals and applications. New York: Marcel Dekker; 1987. [Google Scholar]
- 2.Uhrich KE, Cannizzaro SM, Langer RS, Shakesheff KM. Polymeric Systems for Controlled Drug Release. Chem Rev. 1999;99(11):3181–3198. doi: 10.1021/cr940351u. 11/01 2012/02; [DOI] [PubMed] [Google Scholar]
- 3.Drake C, Arch A. Standard Telephones and Cables Public Limited Company, assignee. Controlled release system. 1989 Sep; inventors. 486609712. [Google Scholar]
- 4.Langer R, Peppas N. Chemical and Physical Structure of Polymers as Carriers for Controlled Release of Bioactive Agents: A Review. Journal of Macromolecular Science, Part C: Polymer Reviews. 1983;23(1):61–126. 01/01 2012/02; [Google Scholar]
- 5.Shaviv A, Mikkelsen R. Controlled-release fertilizers to increase efficiency of nutrient use and minimize environmental degradation - A review. Nutrient Cycling in Agroecosystems. 1993;35(1):1–12. [Google Scholar]
- 6.Madene A, Jacquot M, Scher J, Desobry S. Flavour encapsulation and controlled release ? a review. Int J Food Sci Tech. 2006;41(1):1–21. [Google Scholar]
- 7.Gerstl Z, Nasser A, Mingelgrin U. Controlled Release of Pesticides into Water from Clay-Polymer Formulations. J Agric Food Chem. 1998;46(9):3803–3809. 09/01 2012/07; [Google Scholar]
- 8.Lisa Brannon-Peppas. Controlled Release in the Food and Cosmetics Industries. Polymeric Delivery Systems: American Chemical Society. 1993:42–52. [Google Scholar]
- 9.Pillai O, Panchagnula R. Polymers in drug delivery. Curr Opin Chem Biol. 2001;5(4):447–451. doi: 10.1016/s1367-5931(00)00227-1. 8/1; [DOI] [PubMed] [Google Scholar]
- 10.Herrero-Vanrell R, Rincón AC, Alonso M, Reboto V, Molina-Martinez IT, Rodríguez-Cabello JC. Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. J Controlled Release. 2005;102(1):113–122. doi: 10.1016/j.jconrel.2004.10.001. 1/20; [DOI] [PubMed] [Google Scholar]
- 11.Crotts G, Park TG. Protein delivery from poly(lactic-co-glycolic acid) biodegradable microspheres: Release kinetics and stability issues. J Microencapsul. 1998;15(6):699–713. doi: 10.3109/02652049809008253. 01/01 2012/02; [DOI] [PubMed] [Google Scholar]
- 12.Cutright DE, Beasley JD, III, Perez B. Histologic comparison of polylactic and polyglycolic acid sutures. Oral Surgery, Oral Medicine, Oral Pathology. 1971;32(1):165–173. doi: 10.1016/0030-4220(71)90265-9. 7; [DOI] [PubMed] [Google Scholar]
- 13.Frazza EJ, Schmitt EE. A new absorbable suture. J Biomed Mater Res. 1971;5(2):43–58. doi: 10.1002/jbm.820050207. [DOI] [PubMed] [Google Scholar]
- 14.Middleton JC, Tipton AJ. Synthetic biodegradable polymers as orthopedic devices. Biomaterials. 2000;21(23):2335–2346. doi: 10.1016/s0142-9612(00)00101-0. 12/1; [DOI] [PubMed] [Google Scholar]
- 15.Racey GL, Wallace WR, Cavalaris CJ, Marguard JV. Comparison of a polyglycolicpolylactic acid suture to black silk and plain catgut in human oral tissues. J Oral Surg. 1978 Oct;36(10):766–770. [PubMed] [Google Scholar]
- 16.Freiberg S, Zhu XX. Polymer microspheres for controlled drug release. Int J Pharm. 2004;282(1–2):1–18. doi: 10.1016/j.ijpharm.2004.04.013. 9/10; [DOI] [PubMed] [Google Scholar]
- 17.Kitchell JP, Wise DL. Methods in Enzymology. Academic Press; [32] Poly(lactic/glycolic acid) biodegradable drug—polymer matrix systems; pp. 436–448. [DOI] [PubMed] [Google Scholar]
- 18.Chen L, Apte RN, Cohen S. Characterization of PLGA microspheres for the controlled delivery of IL-1α for tumor immunotherapy. J Controlled Release. 1997;43(2–3):261–272. 1/18; [Google Scholar]
- 19.Han M. Biodegradable membranes for the controlled release of progesterone. 1. Characterization of membrane morphologies coagulated from PLGA/progesterone/DMF solutions. J Appl Polym Sci. 2000;75(1):60–67. [Google Scholar]
- 20.Wang YM, Sato H, Horikoshi I. In vitro and in vivo evaluation of taxol release from poly(lactic-co-glycolic acid) microspheres containing isopropyl myristate and degradation of the microspheres. J Controlled Release. 1997;49(2–3):157–166. 12/15; [Google Scholar]
- 21.Kawashima Y, Yamamoto H, Takeuchi H, Fujioka S, Hino T. Pulmonary delivery of insulin with nebulized dl-lactide/glycolide copolymer (PLGA) nanospheres to prolong hypoglycemic effect. J Controlled Release. 1999;62(1–2):279–287. doi: 10.1016/s0168-3659(99)00048-6. 11/1; [DOI] [PubMed] [Google Scholar]
- 22.Price JS, Tencer AF, Arm DM, Bohach GA. Controlled release of antibiotics from coated orthopedic implants. J Biomed Mater Res. 1996;30(3):281–286. doi: 10.1002/(SICI)1097-4636(199603)30:3<281::AID-JBM2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 23.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactideco-glycolide) (PLGA) devices. Biomaterials. 2000;21(23):2475–2490. doi: 10.1016/s0142-9612(00)00115-0. 12/1; [DOI] [PubMed] [Google Scholar]
- 24.Champion JA, Katare YK, Mitragotri S. Particle shape: A new design parameter for micro- and nanoscale drug delivery carriers. J Controlled Release. 2007;121(1–2):3–9. doi: 10.1016/j.jconrel.2007.03.022. 8/16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson JM, Shive MS. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28(1):5–24. doi: 10.1016/s0169-409x(97)00048-3. 10/13; [DOI] [PubMed] [Google Scholar]
- 26.Houchin ML, Topp EM. Chemical degradation of peptides and proteins in PLGA: A review of reactions and mechanisms. J Pharm Sci. 2008;97(7):2395–2404. doi: 10.1002/jps.21176. [DOI] [PubMed] [Google Scholar]
- 27.Kenley RA, Lee MO, Mahoney TR, Sanders LM. Poly(lactide-co-glycolide) decomposition kinetics in vivo and in vitro. Macromolecules. 1987;20(10):2398–2403. 10/01 2012/07; [Google Scholar]
- 28.Siepmann J, Elkharraz K, Siepmann F, Klose D. How Autocatalysis Accelerates Drug Release from PLGA-Based Microparticles: A Quantitative Treatment. Biomacromolecules. 2005;6(4):2312–2319. doi: 10.1021/bm050228k. 07/01 2012/07; [DOI] [PubMed] [Google Scholar]
- 29.Vert M, Mauduit J, Li S. Biodegradation of PLA/GA polymers: increasing complexity. Biomaterials. 1994;15(15):1209–1213. doi: 10.1016/0142-9612(94)90271-2. 12; [DOI] [PubMed] [Google Scholar]
- 30.Lin Y, Feng C, Ye S, Lin Y, Chen C, Liao C, et al. An In Vivo Study on the Biocompatibility of a Bioresorbable Poly(L-lactide-co-glycolide) Pin for Bone Fixation. Journal of Medical and Biological Engineering. 2001;21(4):233. [Google Scholar]
- 31.Siegel SJ, Kahn JB, Metzger K, Winey KI, Werner K, Dan N. Effect of drug type on the degradation rate of PLGA matrices. European Journal of Pharmaceutics and Biopharmaceutics. 2006;64(3):287–293. doi: 10.1016/j.ejpb.2006.06.009. 11; [DOI] [PubMed] [Google Scholar]
- 32.Faisant N, Siepmann J, Benoit JP. PLGA-based microparticles: elucidation of mechanisms and a new simple mathematical model quantifying drug release. European Journal of Pharmaceutical Sciences. 2002;15(4):355–366. doi: 10.1016/s0928-0987(02)00023-4. 5; [DOI] [PubMed] [Google Scholar]
- 33.Varde NK. Elucidation of drug release mechanisms in PLGA microspheres. ProQuest. 2007 [Google Scholar]
- 34.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers. 2011;3(3):1377–1397. doi: 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Friess W, Schlapp M. Release mechanisms from gentamicin loaded poly (lactic-co-glycolic acid)(PLGA) microparticles. J Pharm Sci. 2002;91(3):845–855. doi: 10.1002/jps.10012. [DOI] [PubMed] [Google Scholar]
- 36.Herrera LC, Tesoriero MVD, Hermida LG, Murtaza G, Ahmad M, Khan SA, et al. In Vitro Release Testing of PLGA Microspheres with Franz Diffusion Cells. In Vitro. 2012;19(2) [Google Scholar]
- 37.Klose D, Siepmann F, Elkharraz K, Krenzlin S, Siepmann J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int J Pharm. 2006;314(2):198–206. doi: 10.1016/j.ijpharm.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 38.Park TG, Yong Lee H, Sung Nam Y. A new preparation method for protein loaded poly (D, L-lactic-co-glycolic acid) microspheres and protein release mechanism study. J Controlled Release. 1998;55(2):181–191. doi: 10.1016/s0168-3659(98)00050-9. [DOI] [PubMed] [Google Scholar]
- 39.Joung YK. PLGA microparticle-embedded thermosensitive hydrogels for sustained release of hydrophobic drugs. Biomedical Materials. 2007;2(4):269. doi: 10.1088/1748-6041/2/4/010. [DOI] [PubMed] [Google Scholar]
- 40.Galeska I, Kim T, Patil S, Bhardwaj U, Chatttopadhyay D, Papadimitrakopoulos F, et al. Controlled release of dexamethasone from PLGA microspheres embedded within polyacid-containing PVA hydrogels. The AAPS Journal. 2005;7(1):E231–E240. doi: 10.1208/aapsj070122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Zhang S, Chen P, Cheng L, Zhou W, Tang W, et al. Controlled release of insulin from PLGA nanoparticles embedded within PVA hydrogels. Journal of Materials Science: Materials in Medicine. 2007;18(11):2205–2210. doi: 10.1007/s10856-007-3010-0. [DOI] [PubMed] [Google Scholar]
- 42.Jeong B, Bae YH, Kim SW. Drug release from biodegradable injectable thermosensitive hydrogel of PEG–PLGA–PEG triblock copolymers. J Controlled Release. 2000;63(1–2):155–163. doi: 10.1016/s0168-3659(99)00194-7. 1/3; [DOI] [PubMed] [Google Scholar]
- 43.Li Y, Pei Y, Zhang X, Gu Z, Zhou Z, Yuan W, et al. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J Controlled Release. 2001;71(2):203–211. doi: 10.1016/s0168-3659(01)00218-8. 4/2; [DOI] [PubMed] [Google Scholar]
- 44.Wang ZH, Wang ZY, Sun CS, Wang CY, Jiang TY, Wang SL. Trimethylated chitosan-conjugated PLGA nanoparticles for the delivery of drugs to the brain. Biomaterials. 2010;31(5):908–915. doi: 10.1016/j.biomaterials.2009.09.104. 2; [DOI] [PubMed] [Google Scholar]
- 45.Chan JM, Zhang L, Yuet KP, Liao G, Rhee J, Langer R, et al. PLGA–lecithin–PEG core–shell nanoparticles for controlled drug delivery. Biomaterials. 2009;30(8):1627–1634. doi: 10.1016/j.biomaterials.2008.12.013. 3; [DOI] [PubMed] [Google Scholar]
- 46.Xu Q, Czernuszka JT. Controlled release of amoxicillin from hydroxyapatite-coated poly(lactic-co-glycolic acid) microspheres. J Controlled Release. 2008;127(2):146–153. doi: 10.1016/j.jconrel.2008.01.017. 4/21; [DOI] [PubMed] [Google Scholar]
- 47.van de Weert M, Hennink WE, Jiskoot W. Protein Instability in Poly(Lactic-co-Glycolic Acid) Microparticles. Pharmaceutical Research. 2000;17(10):1159–1167. doi: 10.1023/a:1026498209874. [DOI] [PubMed] [Google Scholar]
- 48.Giteau A, Venier-Julienne MC, Aubert-Pouëssel A, Benoit JP. How to achieve sustained and complete protein release from PLGA-based microparticles? Int J Pharm. 2008;350(1–2):14–26. doi: 10.1016/j.ijpharm.2007.11.012. 2/28; [DOI] [PubMed] [Google Scholar]
- 49.Taluja A, Bae YH. Role of a novel multifunctional excipient poly(ethylene glycol)-block-oligo(vinyl sulfadimethoxine) in controlled release of lysozyme from PLGA microspheres. Int J Pharm. 2008 Jun 24;358(1–2):50–59. doi: 10.1016/j.ijpharm.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 50.Mittal G, Sahana DK, Bhardwaj V, Ravi Kumar MNV. Estradiol loaded PLGA nanoparticles for oral administration: Effect of polymer molecular weight and copolymer composition on release behavior in vitro and in vivo. J Controlled Release. 2007;119(1):77–85. doi: 10.1016/j.jconrel.2007.01.016. 5/14; [DOI] [PubMed] [Google Scholar]
- 51.Gasparini G, Kosvintsev SR, Stillwell MT, Holdich RG. Preparation and characterization of PLGA particles for subcutaneous controlled drug release by membrane emulsification. Colloids and Surfaces B: Biointerfaces. 2008;61(2):199–207. doi: 10.1016/j.colsurfb.2007.08.011. 2/15; [DOI] [PubMed] [Google Scholar]
- 52.Jose MV, Thomas V, Johnson KT, Dean DR, Nyairo E. Aligned PLGA/HA nanofibrous nanocomposite scaffolds for bone tissue engineering. Acta Biomaterialia. 2009;5(1):305–315. doi: 10.1016/j.actbio.2008.07.019. 1; [DOI] [PubMed] [Google Scholar]
- 53.Jose MV, Thomas V, Dean DR, Nyairo E. Fabrication and characterization of aligned nanofibrous PLGA/Collagen blends as bone tissue scaffolds. Polymer. 2009;50(15):3778–3785. 7/17; [Google Scholar]
- 54.Wenk E, Meinel AJ, Wildy S, Merkle HP, Meinel L. Microporous silk fibroin scaffolds embedding PLGA microparticles for controlled growth factor delivery in tissue engineering. Biomaterials. 2009;30(13):2571–2581. doi: 10.1016/j.biomaterials.2008.12.073. 5; [DOI] [PubMed] [Google Scholar]
- 55.Mondalek FG, Lawrence BJ, Kropp BP, Grady BP, Fung K, Madihally SV, et al. The incorporation of poly(lactic-co-glycolic) acid nanoparticles into porcine small intestinal submucosa biomaterials. Biomaterials. 2008;29(9):1159–1166. doi: 10.1016/j.biomaterials.2007.11.020. 3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klose D, Siepmann F, Elkharraz K, Siepmann J. PLGA-based drug delivery systems: Importance of the type of drug and device geometry. Int J Pharm. 2008;354(1–2):95–103. doi: 10.1016/j.ijpharm.2007.10.030. 4/16; [DOI] [PubMed] [Google Scholar]
- 57.Göpferich A. Mechanisms of polymer degradation and erosion. Biomaterials. 1996;17(2):103–114. doi: 10.1016/0142-9612(96)85755-3. 1; [DOI] [PubMed] [Google Scholar]
- 58.Ritger PL, Peppas NA. A simple equation for description of solute release. I. Fickian and non-fickian release from non-swellable devices in the form of slabs, spheres, cylinders or discs. J Controlled Release. 1987;5(1):23–36. 6; [PubMed] [Google Scholar]
- 59.Ritger PL, Peppas NA. A simple equation for description of solute release. II. Fickian and anomalous release from swellable devices. J Controlled Release. 1987;5(1):37–42. 6; [PubMed] [Google Scholar]
- 60.Siepmann J, Peppas NA. Modeling of drug release from delivery systems based on hydroxypropyl methylcellulose (HPMC) Adv Drug Deliv Rev. 2001;48(2–3):139–157. doi: 10.1016/s0169-409x(01)00112-0. 6/11; [DOI] [PubMed] [Google Scholar]
- 61.Wise DL, Trantolo DJ, Marino RT, Kitchell JP. Opportunities and challenges in the design of implantable biodegradable polymeric systems for the delivery of antimicrobial agents and vaccines. Adv Drug Deliv Rev. 1987;1(1):19–40. 5; [Google Scholar]
- 62.Chiu LK, Chiu WJ, Cheng Y- Effects of polymer degradation on drug released — mechanistic study of morphology and transport properties in 50:50 poly(dl-lactide-co-glycolide) Int J Pharm. 1995;126(1–2):169–178. 12/29; [Google Scholar]
- 63.Faisant N, Siepmann J, Richard J, Benoit JP. Mathematical modeling of drug release from bioerodible microparticles: effect of gamma-irradiation. European Journal of Pharmaceutics and Biopharmaceutics. 2003;56(2):271–279. doi: 10.1016/s0939-6411(03)00104-8. 9; [DOI] [PubMed] [Google Scholar]
- 64.Hsu Y, Gresser JD, Trantolo DJ, Lyons CM, Gangadharam PRJ, Wise DL. In vitro controlled release of isoniazid from poly (lactide-co-glycolide) matrices. J Controlled Release. 1994;31(3):223–228. 10; [Google Scholar]
- 65.Baker RW. Controlled Release Society; Controlled release of bioactive materials: based on the symposium held at the 6th international meeting of the Controlled Release Society; August 1979; New Orleans, Louisiana. Academic Press; 1980. [Google Scholar]
- 66.Gallagher KM, Corrigan OI. Mechanistic aspects of the release of levamisole hydrochloride from biodegradable polymers. J Controlled Release. 2000;69(2):261–272. doi: 10.1016/s0168-3659(00)00305-9. 11/3; [DOI] [PubMed] [Google Scholar]
- 67.Rothstein SN, Federspiel WJ, Little SR. A simple model framework for the prediction of controlled release from bulk eroding polymer matrices. J Mater Chem. 2008;18(16):1873–1880. [Google Scholar]
- 68.Budhian A, Siegel SJ, Winey KI. Controlling the in vitro release profiles for a system of haloperidol-loaded PLGA nanoparticles. Int J Pharm. 2008;346(1–2):151–159. doi: 10.1016/j.ijpharm.2007.06.011. 1/4; [DOI] [PubMed] [Google Scholar]
- 69.Lim T, Poh C, Wang W. Poly (lactic-co-glycolic acid) as a controlled release delivery device. Journal of Materials Science: Materials in Medicine. 2009;20(8):1669–1675. doi: 10.1007/s10856-009-3727-z. [DOI] [PubMed] [Google Scholar]
- 70.Kost J, Langer R. Responsive polymeric delivery systems. Adv Drug Deliv Rev. 2001;46(1–3):125–148. doi: 10.1016/s0169-409x(00)00136-8. 3/1; [DOI] [PubMed] [Google Scholar]
- 71.Higuchi T. Rate of release of medicaments from ointment bases containing drugs in suspension. J Pharm Sci. 1961;50(10):874–875. doi: 10.1002/jps.2600501018. [DOI] [PubMed] [Google Scholar]
- 72.Controlled release polymeric formulations : a symposium. Washington: The Society; 1976. [Google Scholar]
- 73.Varelas CG, Dixon DG, Steiner CA. Zero-order release from biphasic polymer hydrogels. J Controlled Release. 1995;34(3):185–192. doi: 10.1016/s0168-3659(97)00182-x. 6; [DOI] [PubMed] [Google Scholar]
- 74.Gibaldi M, Feldman S. Establishment of sink conditions in dissolution rate determinations. Theoretical considerations and application to nondisintegrating dosage forms. J Pharm Sci. 1967;56(10):1238–1242. doi: 10.1002/jps.2600561005. [DOI] [PubMed] [Google Scholar]
- 75.Hixson AW, Crowell JH. Dependence of Reaction Velocity upon surface and Agitation. Ind Eng Chem. 1931;23(8):923–931. 08/01 2012/02; [Google Scholar]
- 76.Langenbucher F. Letters to the Editor: Linearization of dissolution rate curves by the Weibull distribution. J Pharm Pharmacol. 1972;24(12):979–981. doi: 10.1111/j.2042-7158.1972.tb08930.x. [DOI] [PubMed] [Google Scholar]
- 77.Harland RS, Gazzaniga A, Sangalli ME, Colombo P, Peppas NA. Drug/Polymer Matrix Swelling and Dissolution. Pharmaceutical Research. 1988;5(8):488–494. doi: 10.1023/a:1015913207052. [DOI] [PubMed] [Google Scholar]