

Abstract
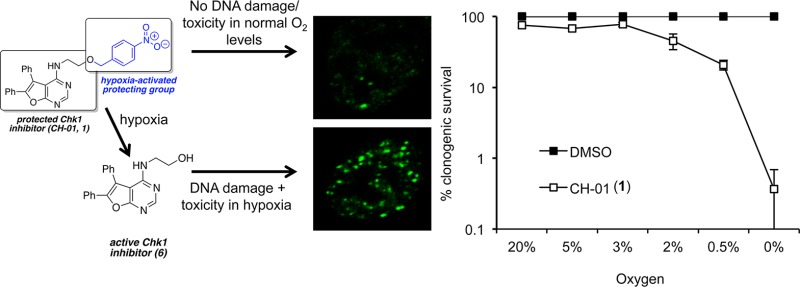
The increased resistance of hypoxic cells to all forms of cancer therapy presents a major barrier to the successful treatment of most solid tumors. Inhibition of the essential kinase Checkpoint kinase 1 (Chk1) has been described as a promising cancer therapy for tumors with high levels of hypoxia-induced replication stress. However, as inhibition of Chk1 affects normal replication and induces DNA damage, these agents also have the potential to induce genomic instability and contribute to tumorigenesis. To overcome this problem, we have developed a bioreductive prodrug, which functions as a Chk1/Aurora A inhibitor specifically in hypoxic conditions. To achieve this activity, a key functionality on the Chk1 inhibitor (CH-01) is masked by a bioreductive group, rendering the compound inactive as a Chk1/Aurora A inhibitor. Reduction of the bioreductive group nitro moiety, under hypoxic conditions, reveals an electron-donating substituent that leads to fragmentation of the molecule, affording the active inhibitor. Most importantly, we show a significant loss of viability in cancer cell lines exposed to hypoxia in the presence of CH-01. This novel approach targets the most aggressive and therapy-resistant tumor fraction while protecting normal tissue from therapy-induced genomic instability.
It is clear that the identification and exploitation of the differences between cancer and normal cells is essential for the design of effective therapeutics. The replicative stress response (RSR), which is characterized by Ataxia telangiectasia mutated rad3 related kinase-Checkpoint kinase 1 (ATR-Chk1) signaling, is elevated in numerous cancer cell types.1 Targeting ATR/Chk1 has been proposed to sensitize cancer cells to DNA damage and to be particularly effective in those that have lost p53-mediated control of the G1 checkpoint.2 Recently, targeting the RSR through inhibition of the ATR-Chk1 pathway was proposed as an effective means of treating tumors with high levels of oncogene-mediated replication stress.3,4 For example, tumors with elevated MYC levels have been shown to be sensitive to ATR/Chk1 inhibition.3,5−8 Both ATR and Chk1 inhibitors have been developed, although ATR inhibition has yet to be evaluated in the clinic.9−14 Clinical studies using Chk1 inhibitors in combination with standard DNA damaging chemotherapeutics have shown some success in combination with, for example, gemcitabine, irinotecan, and paclitaxel.15,16 Recently, studies demonstrating that cancer cells with high levels of oncogene-mediated replication stress or inherent DNA damage show increased sensitivity to Chk1 inhibition have raised the possibility of using Chk1 inhibitors as single agents.7 The enthusiasm for inhibiting kinases with roles in the cell cycle is somewhat tempered due to their roles in normal, unperturbed, replication.17 It cannot be forgotten, for example, that Chk1 is an essential gene, the loss of which leads to embryonic lethality and which perhaps more significantly has been found to be altered in human cancers.18 Indeed, a recent study demonstrated that while significant suppression of ATR activity led to loss of cell viability, ATR-haploinsufficiency promoted tumorigenesis.4
In addition to tolerating high levels of oncogene-mediated replication stress, tumors exist and thrive in conditions of low oxygen concentration (hypoxia). The degree of tumor hypoxia correlates well with resistance to therapy including radio/chemotherapy and surgery as well as an increased likelihood of metastasis.19,20 In conditions of severe hypoxia (<0.1% O2) a unique DNA damage response (DDR) occurs, which is characterized by both ATR and Ataxia telangiectasia mutated kinase (ATM) activity in the absence of detectable DNA damage.21,22 In response to these conditions the levels of nucleotides rapidly fall, and this correlates with a complete replication arrest. The RSR initiated in severe hypoxia includes Chk1, and loss/inhibition of Chk1 has been demonstrated to sensitize cells to hypoxia/reoxygenation.23
An elegant approach to exploiting the low levels of oxygenation in tumors is through the use of agents that are activated by these conditions, commonly known as bioreductive prodrugs or hypoxic cytotoxins (recently reviewed in ref (24)). These compounds contain functional groups that are susceptible to in vivo reduction under conditions of low oxygen concentration. Although there are five chemical moieties that have been demonstrated to undergo metabolism in hypoxia, the most common approach employs nitroaromatic derivatives, such as the 4-nitrobenzyl, 4-nitrofuryl, and 2-nitroimidazole groups.24 The nitro group undergoes nitroreductase-mediated one electron reduction to a radical anion in vivo, which is rapidly oxidized by molecular oxygen under normoxic conditions, forming superoxide and rendering this pathway unproductive. Under hypoxic conditions, the radical anion is not reoxidized but undergoes further reduction to form a nitroso group, hydroxylamine, or an amine. While the nitro group has no available lone pair and is mesomerically and inductively electron-withdrawing, the nitroso group, the hydroxylamine, and the amine groups have an available lone pair and hence are mesomerically electron-donating. This reversal in reactivity has been harnessed to activate compounds selectively under hypoxic conditions. The majority of these compounds are based on increasing the electrophilicity of alkylating agents that then confer general toxicity in the hypoxic region. A number of these agents have been described, including tirapazamine (TPZ), AQ4N, PR-104A, CEN-209, RH-1, and TH-302.25−28 TPZ has been tested in a number of clinical trials including a recent large randomized multicenter phase III trial combined with radiotherapy for head and neck cancers. This trial reported no benefit, although there were major deficiencies in the treatment of a subset of patients that could be responsible for this.29,30 Subsequently, analogues of TPZ have been described, one of which, CEN-209, is likely to be tested clinically in the near future.31 More recently, TH-302 has undergone extensive preclinical testing and has been tested in a phase II trial for advanced pancreatic cancer and a phase III soft-tissue sarcoma trial.32 Several factors are critical to the future success of bioreductive prodrugs, including the challenges of delivering such agents to hypoxic tumor cells and the need to identify biomarkers, which predict those tumors most likely to respond.
The majority of the bioreductive prodrugs described to date are designed to release a DNA damaging cytotoxin and therefore, once activated, act similarly to conventional chemotherapeutic agents. This approach raises the possibility of overlapping toxicities when these agents are combined with standard therapies. An alternative application of a hypoxia-activated group is to mask a drug, which acts as a protein ligand, to prevent binding to its target. This would render the compound inactive until the bioreductive group is removed under hypoxic conditions. Given that this strategy potentially allows targeting of promising cancer therapies to hypoxic tumors, it is surprising that it has not been more widely employed, although there are a few reports in the literature. Zhang et al. applied this strategy to the synthesis of three hypoxia-activated derivatives of 20(S)-camptothecin.33 They demonstrated that a 4-nitrobenzyl derivative conferred some selectivity for hypoxic cells over normal cells. Granchi et al. described nitrobenzyl and nitrofuryl bioreductive prodrugs that release an inhibitor of the lysyl oxidase (LOX) protein in hypoxia.34 In this instance the approach was beneficial as the released compound, BAPN, is a relatively nonselective LOX inhibitor with multiple biological interactions. Zhu et al. synthesized 4-nitrobenzyl derivatives of O6-benzylguanine, which is an inhibitor of the resistance protein O6-alkylguanine alkyltransferase (AGT).35 It was demonstrated that the gem-dimethyl-4-nitrobenzyl analogue was effective in sensitizing laromustine-resistant DU145 human prostate carcinoma cells to laromustine under hypoxic conditions. Here, we describe CH-01, which is a proof-of-concept compound that we propose is activated as a Chk1 and Aurora kinase A inhibitor after the hypoxia-promoted loss of the 4-nitrobenzyl group. Clonogenic survival assays demonstrate that CH-01 (1) had little or no effect on cells in normal oxygen conditions; conversely, hypoxic cells were extremely sensitive to CH-01. Of the tumor cell lines tested, the sensitivity to CH-01 correlated with the baseline levels of DNA damage/replication stress. Therefore this strategy allows us to concentrate Chk1/Aurora kinase A inhibition in hypoxic cells, targeting the most aggressive tumor fraction, while protecting normal tissue.
Results and Discussion
We have shown that depletion or inhibition of Chk1 sensitizes cells to hypoxia/reoxygenation.22,36 The biological reasons behind this observation include our finding that Chk1 has a role to play in reoxygenation-induced replication restart, as well as normal replication.23 In order to further validate Chk1 as a molecular target in hypoxic conditions, we have considered its autophosphorylation site serine 296 and the ATR-mediated phosphorylations on serine residues 317 and 345 as well as the total levels of Chk1. Chk1 was rapidly phosphorylated at all tested residues (Figure 1a). To verify that the hypoxia-mediated phosphorylation of Chk1 correlated with Chk1 activity, the levels of the Chk1 target Tousled-like kinase 1 (TLK1) are also shown.37 As described previously, the total levels of Chk1 decrease during increasing exposure to hypoxia.23 It is clear caution is warranted when considering the inhibition of essential genes such as Chk1. To demonstrate this point we exposed nontransformed human fetal lung fibroblasts (WI38) to a well-characterized Chk1 inhibitor, Gö6976, in the absence of additional stress. Increasing exposure to Gö6976 led to a significant accumulation of cells with more than six 53 binding protein 1 (53BP1) foci indicating that prolonged exposure to a Chk1 inhibitor leads to accumulation of DNA damage, which could in turn affect genome stability (Figure 1b). In order to target Chk1 kinase inhibition to the hypoxic regions of tumors, we synthesized a bioreductive Chk1 inhibitor, CH-01 (1). CH-01 is based on the Chk1 inhibitor 6 (Figure 2a) reported by Foloppe et al.38 During the course of this work, compound 6 was also shown to inhibit Aurora kinase A.39 Inhibitors of Aurora kinases have been tested clinically and show some promise; however, these agents have not been tested specifically in hypoxic conditions.40 In order to investigate the potential benefit of Aurora A inhibition in hypoxic cells, we incubated RKO cells with the known selective Aurora A inhibitor MLN8237 in both normoxic and hypoxic conditions. The colony survival assay shown demonstrates that cells in both normoxia and hypoxia are sensitive to inhibition of Aurora A (Figure 1c).
Figure 1.

Targeting Chk1/Aurora A inhibition to hypoxic cells. (a) Chk1 is phosphorylated and active in hypoxic conditions. RKO cells were exposed to hypoxia (≤0.02% O2) for the times indicated, and Western blotting was carried out. (b) Increasing exposure time to Gö6976 (100 nM) led to an accumulation of cells with >6 nuclear 53BP1 foci in WI38 cells. The inset shows an example of the 53BP1 foci observed. (c) RKO cells were treated with the Aurora A kinase inhibitor MLN8237 at the concentrations indicated in either normoxic or hypoxic (≤0.02% O2) conditions for 16 h. A colony survival assay is shown.
Figure 2.

The concept of the hypoxia-activated Chk1 inhibitor CH-01 and its synthesis. (a) Attachment of the 4-nitrobenzyl group to the terminal hydroxyl group renders the Chk1 inhibitor 6 inactive. Under hypoxic conditions, the nitro group is reduced forming an electron-donating substituent, which ejects the active Chk1 inhibitor 6. (b) Reagents and conditions: (a) malononitrile, Et2NH, dioxane, reflux, 16 h, 75%; (b) acetic formic anhydride, 85 °C, 6 h; (c) neat, 220 °C, 30 min, 51% over two steps; (d) POCl3, 55 °C, 2 h, 81%; (e) Et3N, DMF, 80 °C, 6 h, 93%.
Synthesis of a Bioreductive Chk1/Aurora A Inhibitor
Compound 6 was selected for its chemical simplicity and the well-defined Chk1 structure–activity relationships (SAR) reported around this scaffold. Compound 6 inhibits Chk1 kinase in an ATP competitive manner with a reported IC50 value of 20.9 μM, and the SAR showed that addition of a large substituent in place of the hydroxyl group resulted in a significant reduction in Chk1 affinity. Examination of an X-ray crystal structure of 6 bound to the ATP binding site of Chk1 reveals that the hydroxyl group binds oriented into a pocket, which is too small to accommodate a substituent such as the 4-nitrobenzyl group. This observation was corroborated by docking studies (see Supplementary Figure S1). Consequently we designed compound 1, which we predicted to be inactive as a Chk1 inhibitor until the 4-nitrobenzyl group is removed in hypoxia (Figure 2a). Structure–activity studies have shown that compound 6 inhibits Aurora kinase A with an IC50 value of 309 nM.39 In addition, these studies showed that larger groups, including 4-aminophenyl derivatives, were tolerated in place of the hydroxyl group. To predict whether addition of a 4-nitrobenzyl group would reduce the compound’s affinity for Aurora kinase A, we undertook docking studies. These studies suggested that although both compound 1 and compound 5 would bind to Aurora kinase A with a reduced affinity compared to that of compound 6, they could potentially still be accommodated in the ATP-binding site (see Supplementary Figure S2A and B). Compound 1 was synthesized using conditions similar to those reported by Foloppe (Figure 2b). Benzoin (7) was condensed with malononitrile to give 2-aminofuran 8. Reaction with acetic formic anhydride afforded the formamide 9, which cyclized to give 10 upon heating. Treatment of 10 with phosphorus oxychloride furnished the chloride 11, which underwent facile reaction with O-(4-nitrobenyl)ethanolamine (12) to give the final product (1). Analysis of compound 1 in a radioactive (33P-ATP) filter-binding assay revealed no activity against either Chk1 or Aurora kinase A at concentrations up to 100 μM (Supplementary Figure S3). Conversely, compound 6 showed IC50 values of 1.75 and 0.81 μM against Chk1 and Aurora kinase A, respectively.
Mechanism of CH-01 Action
To determine whether 1 fragmented under reducing conditions as predicted, we carried out two reductions in progressively more biologically relevant conditions. Shigenaga et al. have previously employed zinc in aqueous ammonium chloride to reduce a 4-nitrobenzyl group to a 4-aminobenzyl group and hence demonstrate hypoxic activation of a peptide.41 Using similar conditions of zinc and ammonium chloride in N,N-dimethylformamide (DMF), we demonstrated that 1 was reduced to give the nitroso compound 4 (confirmed by mass spectrometry) and the amine 5 after a period of 60 min (Supplementary Figure S4a). Upon exposure to aqueous conditions (potassium phosphate buffer, pH 7.4), HPLC analysis indicated that compounds 4 and 5 fragmented to give the active kinase inhibitor 6 (Figure 3a and Supplementary Figure S4b). Encouraged that we had proved reduction of 1 could induce fragmentation in buffer, we treated 1 with bactosomal human NADPH-CYP reductase in potassium phosphate buffer with the exclusion of oxygen. Under these conditions reduction of the 4-nitrobenzyl group of 1 to the 4-aminobenzyl derivative 5 was observed and fragmentation to the active inhibitor 6 occurred. When this experiment was repeated in the presence of oxygen, no reduction or production of compound 6 was observed (Figure 3b and data not shown). These results indicate that the nitrobenzyl group is reduced under purely chemical conditions and by reductase enzymes in hypoxic conditions. The reduced products fragment to give the active kinase inhibitor 6 in a manner consistent with the proposed in vitro mode of activation for compound 1.
Figure 3.
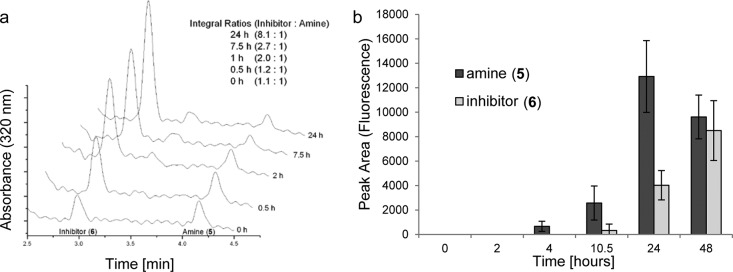
CH-01 is reduced and fragments in hypoxic conditions. CH-01 was subjected to different reduction conditions, and the resulting metabolites were analyzed by HPLC. A combination of photodiode array spectrophotometer, mass spectrometer, and fluorescence spectrophotometer (λex 320 nm, λem 380 nm) was used to detect and characterize the metabolites. (a) CH-01 was subjected to zinc reduction for 1 h, and this solution was injected into potassium phosphate buffer pH 7.4. Incubation at 37 °C and analysis of the supernatant reveal loss of the amine 5 and formation of the active inhibitor 6. (b) Bactosomal human NADPH-CYP reductase reduces CH-01 (1) to the amine 5 under hypoxic conditions (≤0.02% O2). Over 24 h amine 5 accumulates and fragments to release the active inhibitor 6.
In Hypoxic Conditions CH-01 Leads to Loss of Cell Viability
Loss or inhibition of Chk1 has been shown to induce DNA damage due to impaired replisome stability and DNA repair.42 Therefore, we investigated whether CH-01 (1) induced DNA damage in normoxia, hypoxia, and after reoxygenation. Initially, we carried out Western blotting for γH2AX and determined that the presence of 1 in hypoxic conditions led to an increase in the hypoxia-induced γH2AX signal (Figure 4a). As the robust induction of γH2AX by hypoxia alone makes this signal difficult to quantify we used the formation of 53BP1 foci as an alternative measure of DNA damage. As shown previously, hypoxia alone did not induce an accumulation of 53BP1 foci, although subsequent reoxygenation did cause DNA damage.21 In contrast, exposure to 1 in hypoxia alone led to the accumulation of 53BP1 foci in over 50% of the cells (Figure 4b). These data indicate that CH-01 leads to the accumulation of DNA damage only in hypoxic conditions.
Figure 4.

CH-01 induces DNA damage and is toxic in hypoxic conditions. (a) RKO cells were exposed to normoxia or hypoxia (≤0.02% O2) for 6 h in the presence or absence of 25 μM CH-01 (1). Western blots for γH2AX and actin are shown. (b) RKO cells were exposed to normoxia or hypoxia (≤0.02% O2) for 6 h and hypoxia followed by 18 h of reoxygenation ± 1 (25 μM). The graph shows the percentage of cells with >6 nuclear 53BP1 foci. Significance values: * p < 0.05; **p < 0.0001. (c) RKO cells were exposed to hypoxia for the time periods indicated with either 1 (25 μM), MLN8237 (500 nM) or, as a control, DMSO. The levels of phosphorylated histone 3 (pH3 Ser10) were determined by Western blotting. Histone 3 (H3) is shown as loading control. (d) Clonogenic assays were carried out on RKO cells exposed to the oxygen tensions indicated for 24 h in the presence of 25 μM 1 or DMSO.
To confirm that 6 inhibits Aurora A/B, we treated RKO cells with CH-01 (1) in hypoxic conditions and then carried out Western blotting for histone-3 phosphorylated at serine 10, which is a characterized target of Aurora A/B.43 Treatment of RKO cells in normoxia with MLN8237 demonstrated that phosphorylation of pH3 at ser10 can be inhibited through Aurora A inhibition. In contrast, 1 has no effect on this signal in normoxic conditions, indicating that this compound does not inhibit Aurora A/B. In response to hypoxia the pH3 ser10 signal decreased, and this decrease was exacerbated in the presence of 1. After subsequent reoxygenation a clear and significant reduction of pH3 Ser10 was observed when RKO cells were incubated with 1 during hypoxia prior to reoxygenation (Figure 4c). These data suggest that 1 is reduced and fragmented to give 6, which inhibits both Chk1 and Aurora A/B. This dual inhibition is beneficial as both Chk1 and Aurora A are involved in cell cycle progression and are therapeutic targets. Next, we exposed RKO cells to 1 and incubated the cells in oxygen concentrations ranging from 20% to ≤0.02% O2 (Figure 4d). A colony survival assay was carried out at each of the oxygen tensions indicated and demonstrates a significant oxygen-dependent loss of viability. Most importantly, at oxygen levels associated with normal tissues (3% O2 and above) there was little or no effect on cell viability.
It was important to determine that the biological effects observed were due to the release of 6 and not the bioreductive cage. Cells were exposed to the nonbioreductive inhibitor 6, compound 13, which releases EtOH under hypoxic conditions, and CH-01 (1, Figure 5a,b). As expected, RKO cells were sensitive to Chk1 inhibitor 6 ,and this sensitivity was increased in hypoxic conditions as previously reported. In contrast, 1 had no significant effect on the normoxic cells but significantly increased the sensitivity to hypoxia/reoxygenation. Importantly, compound 13 did not decrease cell viability, suggesting that the release of the reduced nitrobenzyl group side product alone is not cytotoxic.
Figure 5.

The biological activity of CH-01 is not associated with the bioreductive group. (a) Clonogenic assays were carried out using RKO cells exposed to DMSO, 25 μM inhibitor 6, 25 μM CH-EtOH 13 or 25 μM CH-01 1 for 24 h in either normoxia or hypoxia (≤0.02% O2). (b) The structure of compound 13, which ejects EtOH under hypoxic conditions.
Up to this point our studies had been restricted to the RKO cell line. However, we proposed that different cells lines would show varying degrees of sensitivity to 1. To investigate this hypothesis further we used three lung cancer cell lines (A549, H1299, and H1975) to determine the effect of 1 in normoxia and hypoxia. Recent, reports suggest that the baseline levels of DNA damage and/or replication stress contribute to the relative sensitivity to Chk1 inhibitors. We proposed that this might also be the case for inhibitors of alternative kinases involved in the cell cycle and so might predict sensitivity to inhibition of Chk1 and Aurora A by 1. Therefore, before testing the effect of 1 on cell viability in hypoxic conditions, we measured the basal levels of DNA damage and replication stress in these 3 cell lines. This measurement was achieved by staining for both 53BP1 and γH2AX, which are markers of DNA damage and/or replication stress (Figure 6a,b). In each cell line a significant proportion of the cells were found to be positive for these markers, although both the H1299 and H1975 cell line had higher levels of 53BP1/γH2AX positive cells than the A549s. Interestingly, large nuclear bodies were seen in the H1299 cell line, which were reminiscent of the recently described Oct-1, PTF, transcription (OPT) domains.44 These data suggest that the A549 cell line should show the least sensitivity to 1 in hypoxic conditions. To test this hypothesis, the cell lines were exposed to 1 for 24 h in either normoxia or hypoxia (≤0.02% O2). As predicted all three-cell lines were sensitive to 1 in hypoxic conditions, and of the three the A549 cells were the least sensitive. However, despite the relatively similar levels of DNA damage seen in the H1299 and H1975 cell lines, the latter were significantly more sensitive to 1. Our data demonstrate that all four of the cell lines tested show increased sensitivity to 1 in hypoxia and that the degree of sensitivity can, in part, be determined by the basal levels of DNA damage/replication stress. In order to use an agent such as 1 most effectively, the degree of tumor hypoxia would have to be determined prior to treatment.45−47 It is clear that nonhypoxic tumors would not respond and the more hypoxic the greater the predicted response (Figure 4d). However, the sensitivity to 1 is also determined by additional factors including the levels of DNA damage/replication arrest. We propose that 1 and derivatives would be effective against tumors with high levels of hypoxia or oncogene-mediated replication stress. In mildly hypoxic tumors, which are less susceptible to Chk1 inhibition due to low levels of replication stress for example, we predict that combining 1 with standard therapies would be effective.
Figure 6.

The sensitivity of cell lines to CH-01 correlates with levels of DNA damage and replication stress. (a) Endogenous DNA damage for each cell line was determined in the absence of additional stress by staining for the presence of 53BP1 (green) and γH2AX (red). (b) Graph represents the quantification of the percentage of cells with more than 6 53BP1 foci (black) or the presence of γH2AX foci positive staining (white) for the three cell lines. The three lung cancer cell lines A549 (c), H1299 (d), and H1975 (e) were exposed to DMSO or 25 μM CH-01 for 24 h in normoxia or hypoxia (≤0.02% O2), and colony survival assays were carried out.
Conclusions
We confirm earlier findings that inhibition of Chk1 is a valid approach to target hypoxic cancer cells. In addition, we have demonstrated that hypoxic cells show similar levels of sensitivity to inhibition of the Aurora A kinase as cells in normoxia. Here, we describe a bioreductive Chk1/Aurora A inhibitor, CH-01 (1), which selectively inhibits Chk1/Aurora A in hypoxic conditions and leads to significant loss of viability in the cancer cell lines tested. Although a proof-of-concept compound, the selective activity demonstrated by 1 suggests the potential for the bioreductive release of targeted therapies and demonstrates this approach as a promising strategy for the targeted application of cancer chemotherapeutics.
Methods
Cell Lines
RKO (colorectal), A549, H1299, and H1975 (lung) cancer cell lines were cultured in DMEM medium containing 10% FBS, penicillin (100 U/mL) and streptomycin (100 μg/mL). WI38 nontransformed human fetal lung fibroblasts were grown in DMEM medium with 15% FBS. All cell lines were originally obtained from the ATCC and routinely mycoplasma tested and found to be negative. With the exception of colony survival experiments all others were carried out with cells at 75% confluence.
Chemical Synthesis
Details of the chemical synthesis and analytical data for the compounds described are available in the Supporting Information. Gö6976 was obtained from Sigma-Aldrich and MLN8237 from Selleckchem.
Hypoxia Treatment
Hypoxia treatments were carried out in a Bactron II (Shell laboratories), In vivo 400 (Ruskinn), or Heracell mixed gas incubator (Fisher Scientific) depending on the level of hypoxia required.
Clonogenic Assay
Colonies (>50 cells) were left to form for 10 days and visualized with methylene blue stain (70% methanol in PBS, 1% methylene blue (Fisher BioReagents)).
Western Blotting
Cells were lysed in UTB (9 M urea, 75 mM Tris-HCl pH 7.5 and 0.15 M β-mercaptoethanol) and sonicated briefly. Antibodies used were Chk1-S296, Chk1-S317, Chk1-S345, HIF1α (BD Biosciences), Chk1, γH2AX (Upstate-Millipore), H3-S10, TLK-S695 (Cell Signaling), and GAPDH (Fitzgerald Industries). The Odyssey infrared system was used for imaging (LI-COR Biosciences).
Zinc Reduction of 1
To a solution of 1 (1 mg, 0.0021 mmol) in DMF (2 mL) were added aqueous ammonium chloride (20 μL, 10% w/v) and zinc powder (5 mg, 0.0765 mmol, 36 equiv). The resulting mixture was stirred at ambient temperature for 16 h. Aliquots (200 μL) were taken at designated times (where T = 0 refers to before the addition of zinc powder), and the mixture was analyzed by HPLC.
Buffer Treatment of 5
For every time point of interest, 5 μL of the T = 1 h aliquot from the zinc reduction was injected into 95 μL of potassium phosphate buffer solution (pH 7.4), and the resulting suspensions were incubated at 37 °C. At designated times the suspensions were centrifuged. The supernatant was collected, and the precipitates were dissolved in acetonitrile. Both fractions were analyzed by HPLC.
Reductase Assay
Bactosomal human NADPH-CYP reductase (Cypex, 12.7 mg/mL, 13900 nmol/min/mL) was used in combination with an NADPH-regenerating system (BD Biosciences), and the assay was carried out according to the manufacturer’s protocol (BD Biosciences application note 467) at a CH-01 concentration of 250 nM. Vials were deoxygenated by bubbling nitrogen prior to P540 addition and then transferred into a Bactron II (Shell laboratories). Samples were taken at different time points and analyzed by HPLC.
Immunofluorescence
Staining for 53BP1 (Novus Biologicals) and γH2AX foci was carried out as previously described.21 Due to the presence of 1–2 53BP1 foci in the nuclei of unstressed cells, induction of DNA damage was quantified by counting cells with more than 6 foci. Cells were visualized using a Nikon 90i microscope.
HPLC Analysis
HPLC (Waters 2695 system) comprised an RPB column (100 mm × 3.2 mm, 35 °C). Separation was achieved at a flow rate of 0.5 mL/min with a gradient of 60–95% acetonitrile in 10 mM formic acid over 6 min. Detection used a photodiode array spectrophotometer (Waters 2996), a mass spectrometer (Waters Micromass ZQ mass spectrometer), and a fluorescence spectrophotometer (Waters 474) with λex 320 nm, λem 380 nm. Injections of 10 μL were made.
Statistical Analysis
Statistical significance of differences between means of at least n = 3 experiments was determined using Student’s t test (P-values indicated accordingly in figure legend or main text). Error bars represent ± SEM.
Acknowledgments
The authors thank M. Stratford, L. Folkes, P. Wardman, and P. O’Neill for useful discussions. E.M.H., I.M.P., and C.C.-K. are funded by Cancer Research UK (C6515/A9321 awarded to E.M.H.). Additional funding was provided by the Oxford Cancer Research Centre development fund (awarded to E.M.H.). S.J.C. thanks the Department of Chemistry, University of Oxford, for research support.
Supporting Information Available
Supplementary figures, general experimental, experimental procedures, and 1H and 13C NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ University of Hull, Department of Biological Sciences, Hull HU6 7RX, U.K.
Author Contributions
§ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Halazonetis T. D.; Gorgoulis V. G.; Bartek J. (2008) An oncogene-induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Chen T.; Stephens P. A.; Middleton F. K.; Curtin N. J. (2012) Targeting the S and G2 checkpoint to treat cancer. Drug Discovery Today 17, 194–202. [DOI] [PubMed] [Google Scholar]
- Murga M.; Campaner S.; Lopez-Contreras A. J.; Toledo L. I.; Soria R.; Montana M. F.; D’Artista L.; Schleker T.; Guerra C.; Garcia E.; Barbacid M.; Hidalgo M.; Amati B.; Fernandez-Capetillo O. (2011) Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol. 18, 1331–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilad O.; Nabet B. Y.; Ragland R. L.; Schoppy D. W.; Smith K. D.; Durham A. C.; Brown E. J. (2010) Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 70, 9693–9702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole K. A.; Huggins J.; Laquaglia M.; Hulderman C. E.; Russell M. R.; Bosse K.; Diskin S. J.; Attiyeh E. F.; Sennett R.; Norris G.; Laudenslager M.; Wood A. C.; Mayes P. A.; Jagannathan J.; Winter C.; Mosse Y. P.; Maris J. M. (2011) RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. U.S.A. 108, 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoglund A.; Nilsson L. M.; Muralidharan S. V.; Hasvold L. A.; Merta P.; Rudelius M.; Nikolova V.; Keller U.; Nilsson J. A. (2011) Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 17, 7067–7079. [DOI] [PubMed] [Google Scholar]
- Ferrao P. T.; Bukczynska E. P.; Johnstone R. W.; McArthur G. A. (2011) Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 31, 1661–1672. [DOI] [PubMed] [Google Scholar]
- Cavelier C.; Didier C.; Prade N.; Mansat-De Mas V.; Manenti S.; Recher C.; Demur C.; Ducommun B. (2009) Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: potential importance for checkpoint targeting therapy. Cancer Res. 69, 8652–8661. [DOI] [PubMed] [Google Scholar]
- Pires I. M.; Olcina M. M.; Anbalagan S.; Pollard J. R.; Reaper P. M.; Charlton P. A.; McKenna W. G.; Hammond E. M. (2012) Targeting radiation-resistant hypoxic tumour cells through ATR inhibition. Br. J. Cancer 107, 291–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peasland A.; Wang L. Z.; Rowling E.; Kyle S.; Chen T.; Hopkins A.; Cliby W. A.; Sarkaria J.; Beale G.; Edmondson R. J.; Curtin N. J. (2011) Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br. J. Cancer 105, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo L. I.; Murga M.; Zur R.; Soria R.; Rodriguez A.; Martinez S.; Oyarzabal J.; Pastor J.; Bischoff J. R.; Fernandez-Capetillo O. (2011) A cell-based screen identifies ATR inhibitors with synthetic lethal properties for cancer-associated mutations. Nat. Struct. Mol. Biol. 18, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C. X.; Janetka J. W.; Piwnica-Worms H. (2011) Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 17, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaper P. M.; Griffiths M. R.; Long J. M.; Charrier J. D.; Maccormick S.; Charlton P. A.; Golec J. M.; Pollard J. R. (2011) Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 7, 428–430. [DOI] [PubMed] [Google Scholar]
- Prevo R.; Fokas E.; Reaper P. M.; Charlton P. A.; Pollard J. R.; McKenna W. G.; Muschel R. J.; Brunner T. B. (2012) The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 13, 1072–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasina A.; Hallin J.; Chen E.; Arango M. E.; Kraynov E.; Register J.; Grant S.; Ninkovic S.; Chen P.; Nichols T.; O’Connor P.; Anderes K. (2008) Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 7, 2394–2404. [DOI] [PubMed] [Google Scholar]
- Janetka J. W.; Ashwell S.; Zabludoff S.; Lyne P. (2007) Inhibitors of checkpoint kinases: from discovery to the clinic. Curr. Opin. Drug Discovery Devel 10, 473–486. [PubMed] [Google Scholar]
- Syljuasen R. G.; Sorensen C. S.; Hansen L. T.; Fugger K.; Lundin C.; Johansson F.; Helleday T.; Sehested M.; Lukas J.; Bartek J. (2005) Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol. 25, 3553–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrassa L.; Damia G. (2011) Unleashing Chk1 in cancer therapy. Cell Cycle 10, 2121–2128. [DOI] [PubMed] [Google Scholar]
- Vaupel P.; Hockel M. (2003) Tumor oxygenation and its relevance to tumor physiology and treatment. Adv. Exp. Med. Biol. 510, 45–49. [DOI] [PubMed] [Google Scholar]
- Brown J. M.; Giaccia A. J. (1998) The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 58, 1408–1416. [PubMed] [Google Scholar]
- Bencokova Z.; Kaufmann M. R.; Pires I. M.; Lecane P. S.; Giaccia A. J.; Hammond E. M. (2009) ATM activation and signaling under hypoxic conditions. Mol. Cell. Biol. 29, 526–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcina M.; Lecane P. S.; Hammond E. M. (2010) Targeting hypoxic cells through the DNA damage response. Clin. Cancer Res. 16, 5624–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires I. M.; Bencokova Z.; Milani M.; Folkes L. K.; Li J. L.; Stratford M. R.; Harris A. L.; Hammond E. M. (2010) Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 70, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson W. R.; Hay M. P. (2011) Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 11, 393–410. [DOI] [PubMed] [Google Scholar]
- Evans J. W.; Chernikova S. B.; Kachnic L. A.; Banath J. P.; Sordet O.; Delahoussaye Y. M.; Treszezamsky A.; Chon B. H.; Feng Z.; Gu Y.; Wilson W. R.; Pommier Y.; Olive P. L.; Powell S. N.; Brown J. M. (2008) Homologous recombination is the principal pathway for the repair of DNA damage induced by tirapazamine in mammalian cells. Cancer Res. 68, 257–265. [DOI] [PubMed] [Google Scholar]
- Albertella M. R.; Loadman P. M.; Jones P. H.; Phillips R. M.; Rampling R.; Burnet N.; Alcock C.; Anthoney A.; Vjaters E.; Dunk C. R.; Harris P. A.; Wong A.; Lalani A. S.; Twelves C. J. (2008) Hypoxia-selective targeting by the bioreductive prodrug AQ4N in patients with solid tumors: results of a phase I study. Clin. Cancer Res. 14, 1096–1104. [DOI] [PubMed] [Google Scholar]
- Jameson M. B.; Rischin D.; Pegram M.; Gutheil J.; Patterson A. V.; Denny W. A.; Wilson W. R. (2010) A phase I trial of PR-104, a nitrogen mustard prodrug activated by both hypoxia and aldo-keto reductase 1C3, in patients with solid tumors. Cancer Chemother. Pharmacol. 65, 791–801. [DOI] [PubMed] [Google Scholar]
- Wang J.; Foehrenbacher A.; Su J.; Patel R.; Hay M. P.; Hicks K. O.; Wilson W. R. (2012) The 2-nitroimidazole EF5 is a biomarker for oxidoreductases that activate the bioreductive prodrug CEN-209 under hypoxia. Clin. Cancer Res. 18, 1684–1695. [DOI] [PubMed] [Google Scholar]
- Rischin D.; Peters L. J.; O’Sullivan B.; Giralt J.; Fisher R.; Yuen K.; Trotti A.; Bernier J.; Bourhis J.; Ringash J.; Henke M.; Kenny L. (2010) Tirapazamine, cisplatin, and radiation versus cisplatin and radiation for advanced squamous cell carcinoma of the head and neck (TROG 02.02, HeadSTART): a phase III trial of the Trans-Tasman Radiation Oncology Group. J. Clin. Oncol. 28, 2989–2995. [DOI] [PubMed] [Google Scholar]
- Peters L. J.; O’Sullivan B.; Giralt J.; Fitzgerald T. J.; Trotti A.; Bernier J.; Bourhis J.; Yuen K.; Fisher R.; Rischin D. (2010) Critical impact of radiotherapy protocol compliance and quality in the treatment of advanced head and neck cancer: results from TROG 02.02. J. Clin. Oncol. 28, 2996–3001. [DOI] [PubMed] [Google Scholar]
- Hunter F. W.; Wang J.; Patel R.; Hsu H. L.; Hickey A. J.; Hay M. P.; Wilson W. R. (2012) Homologous recombination repair-dependent cytotoxicity of the benzotriazine di-N-oxide CEN-209: comparison with other hypoxia-activated prodrugs. Biochem. Pharmacol. 83, 574–585. [DOI] [PubMed] [Google Scholar]
- Sun J. D.; Liu Q.; Wang J.; Ahluwalia D.; Ferraro D.; Wang Y.; Duan J. X.; Ammons W. S.; Curd J. G.; Matteucci M. D.; Hart C. P. (2012) Selective tumor hypoxia targeting by hypoxia-activated prodrug TH-302 inhibits tumor growth in preclinical models of cancer. Clin. Cancer Res. 18, 758–770. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Tanabe K.; Hatta H.; Nishimoto S. (2005) Bioreduction activated prodrugs of camptothecin: molecular design, synthesis, activation mechanism and hypoxia selective cytotoxicity. Org. Biomol. Chem. 3, 1905–1910. [DOI] [PubMed] [Google Scholar]
- Granchi C.; Funaioli T.; Erler J. T.; Giaccia A. J.; Macchia M.; Minutolo F. (2009) Bioreductively activated lysyl oxidase inhibitors against hypoxic tumours. ChemMedChem 4, 1590–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu R.; Liu M. C.; Luo M. Z.; Penketh P. G.; Baumann R. P.; Shyam K.; Sartorelli A. C. (2011) 4-nitrobenzyloxycarbonyl derivatives of O(6)-benzylguanine as hypoxia-activated prodrug inhibitors of O(6)-alkylguanine-DNA alkyltransferase (AGT), which produces resistance to agents targeting the O-6 position of DNA guanine. J. Med. Chem. 54, 7720–7728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond E. M.; Dorie M. J.; Giaccia A. J. (2004) Inhibition of ATR leads to increased sensitivity to hypoxia/reoxygenation. Cancer Res. 64, 6556–6562. [DOI] [PubMed] [Google Scholar]
- Pires I. M.; Bencokova Z.; McGurk C.; Hammond E. M. (2010) Exposure to acute hypoxia induces a transient DNA damage response which includes Chk1 and TLK1. Cell Cycle 9, 2502–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foloppe N.; Fisher L. M.; Howes R.; Kierstan P.; Potter A.; Robertson A. G.; Surgenor A. E. (2005) Structure-based design of novel Chk1 inhibitors: insights into hydrogen bonding and protein-ligand affinity. J. Med. Chem. 48, 4332–4345. [DOI] [PubMed] [Google Scholar]
- Coumar M. S.; Chu C. Y.; Lin C. W.; Shiao H. Y.; Ho Y. L.; Reddy R.; Lin W. H.; Chen C. H.; Peng Y. H.; Leou J. S.; Lien T. W.; Huang C. T.; Fang M. Y.; Wu S. H.; Wu J. S.; Chittimalla S. K.; Song J. S.; Hsu J. T.; Wu S. Y.; Liao C. C.; Chao Y. S.; Hsieh H. P. (2010) Fast-forwarding hit to lead: aurora and epidermal growth factor receptor kinase inhibitor lead identification. J. Med. Chem. 53, 4980–4988. [DOI] [PubMed] [Google Scholar]
- Manchado E.; Guillamot M.; Malumbres M. (2012) Killing cells by targeting mitosis. Cell Death Differ. 19, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigenaga A.; Ogura K.; Hirakawa H.; Yamamoto J.; Ebisuno K.; Miyamoto L.; Ishizawa K.; Tsuchiya K.; Otaka A. (2012) Development of a reduction-responsive amino acid that induces peptide bond cleavage in hypoxic cells. ChemBioChem 13, 968–971. [DOI] [PubMed] [Google Scholar]
- Sorensen C. S.; Syljuasen R. G. (2011) Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 10.1093/nar/gkr697 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosio C.; Fimia G. M.; Loury R.; Kimura M.; Okano Y.; Zhou H.; Sen S.; Allis C. D.; Sassone-Corsi P. (2002) Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol. Cell. Biol. 22, 874–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan J. A.; Belotserkovskaya R.; Coates J.; Dimitrova D. S.; Polo S. E.; Bradshaw C. R.; Fraser P.; Jackson S. P. (2011) Replication stress induces 53BP1-containing OPT domains in G1 cells. J. Cell Biol. 193, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffa F. M.; Harris A. L.; West C. M.; Miller C. J. (2010) Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 102, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon E. J.; Brizel D. M.; Chi J. T.; Dewhirst M. W. (2007) The potential role of intrinsic hypoxia markers as prognostic variables in cancer. Antioxid. Redox Signaling 9, 1237–1294. [DOI] [PubMed] [Google Scholar]
- Ebbesen P.; Pettersen E. O.; Gorr T. A.; Jobst G.; Williams K.; Kieninger J.; Wenger R. H.; Pastorekova S.; Dubois L.; Lambin P.; Wouters B. G.; Van Den Beucken T.; Supuran C. T.; Poellinger L.; Ratcliffe P.; Kanopka A.; Gorlach A.; Gasmann M.; Harris A. L.; Maxwell P.; Scozzafava A. (2009) Taking advantage of tumor cell adaptations to hypoxia for developing new tumor markers and treatment strategies. J. Enzyme Inhib. Med. Chem. 24(Suppl 1), 1–39. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.