

Abstract
The small Rho GTPase Cdc42 regulates key signaling pathways required for multiple cell functions, including maintenance of shape, polarity, proliferation, invasion, migration, differentiation, and morphogenesis. As the role of Cdc42-dependent signaling in fibroblasts in vivo is unknown, we attempted to specifically delete it in these cells by crossing the Cdc42fl/fl mouse with an fibroblast-specific protein 1 (FSP1)-Cre mouse, which is thought to mediate recombination exclusively in fibroblasts. Surprisingly, the FSP1-Cre;Cdc42fl/fl mice died at 3 weeks of age due to overwhelming suppurative upper airway infections that were associated with neutrophilia and lymphopenia. Even though major aberrations in lymphoid tissue development were present in the mice, the principal cause of death was severe migration and killing abnormalities of the neutrophil population resulting in an inability to control infection. We also show that in addition to fibroblasts, FSP1-Cre deleted Cdc42 very efficiently in all leukocytes. Thus, by using this nonspecific Cre mouse, we inadvertently demonstrated the importance of Cdc42 in host protection from lethal infections and suggest a critical role for this small GTPase in innate immunity.
INTRODUCTION
Cdc42 is a member of the Rho GTPase family of proteins. Like all the Rho GTPases, it is a molecular switch that is regulated by cycling between a GTP-bound active form and a GDP-bound inactive form (1). In the GTP-bound form, Cdc42 interacts with its effectors to induce biological actions. The major classes of Cdc42 effectors are the Wiskott-Aldrich syndrome protein (WASP), p21-activated kinases (PAKs), and the partitioning-defective (PAR) protein family. WASPs are involved in regulating actin polymerization and filopodia formation by direct interactions both with profilin and with actin. PAKs alter the activity of the actin-binding protein cofilin, a key cytoskeletal protein, by regulating the serine/threonine kinase LIMK1. Cdc42 also complexes with the PAR protein family, atypical PKCs, and cadherins to regulate cell polarity and centrosome reorientation by altering phosphorylation of glycogen synthase kinase-3β (2). Thus, Cdc42 can regulate multiple critical cell functions, including cell shape, polarity, proliferation, invasion, migration, differentiation, and morphogenesis.
Cdc42 is ubiquitously expressed, and its in vitro roles in cell function have been examined in numerous different cell types, including fibroblasts. Cdc42-null fibroblasts have abnormalities in adhesion to extracellular matrix proteins, directed migration, wound healing, polarity establishment, and cell survival (3). These morphological defects were associated with alterations in PAK1, glycogen synthase kinase-3β, myosin light chain, and focal adhesion kinase phosphorylation, as well as defects in Jun N-terminal protein kinase, p70 S6K, and extracellular signal-regulated kinase 1/2 activation. Despite the well-characterized abnormalities of Cdc42-null fibroblasts in vitro, there is little known about the role of Cdc42 in fibroblasts in vivo. Indeed, despite a few studies pertaining to initial development or homeostasis of hematopoietic cells (4–7), there is a paucity of information about the consequences for overall host function in a setting where Cdc42 function is absent.
Because a constitutive lack of Cdc42 disrupts embryogenesis, we set out to define the in vivo role of Cdc42 in response to injury by deleting it using a Cre transgene driven by the fibroblast-specific protein 1 (FSP1) promoter (8, 9). FSP1, also known as S100A4, was identified as a protein whose gene was expressed in kidney fibroblasts but not epithelial cells (10). This protein was also shown to be expressed in fibroblasts in different organs, such as the lung and heart (10–12). More recently, however, it was shown that FSP1 is not expressed in either liver fibroblasts or myofibroblasts but is found in a myeloid-monocytic lineage of cells in the liver as well as bone marrow-derived and peritoneal macrophages (13). The FSP1-Cre mouse has been shown to express Cre in stromal fibroblasts of the kidney (14), heart (15, 16), prostate (8), stomach (8), mammary gland (9), and chondrocytes (17) as well as in dendritic cells (18). In this report, we present evidence of a major role for Cdc42 in host defense against infection on the basis of the unique phenotype observed when the Cdc42fl/fl mouse is crossed with the FSP1-Cre mouse.
MATERIALS AND METHODS
Reagents and mice.
Mice harboring the FSP1-Cre recombinase transgene (8) were a gift from Eric Neilson (Vanderbilt University Medical Center, Nashville, TN). Generation of the Cdc42fl/fl and ROSA26-LacZ (flox-STOP) reporter mice has been described previously (19). After each line of animals was bred onto a pure C57BL/6 background, the FSP1-Cre transgene was intercrossed onto a Cdc42fl/fl background. For experiments involving preventive antibiotic treatment, mice were placed on enrofloxacin (22.7 mg/ml; Bayer, Wuppertal, Germany), administered on a continuous basis through their drinking water. To define Cre expression in leukocytes, FSP1-Cre;ROSA26-LacZ reporter mice (19) were bred and analyzed without further manipulation. All experiments were performed according to institutional animal care guidelines after approval by the Institutional Animal Care and Use Committee of Vanderbilt University and conducted in facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care.
Gross and microscopic pathological examinations.
Animals were euthanized humanely for complete necropsy or analyses of cells and organs. At necropsy, blood for complete blood counts was collected via intracardiac puncture and placed in an EDTA collection tube. Complete blood counts were performed on a Forcyte hematology analyzer (Oxford Science, Oxford, CT). Tissues were collected and fixed for 24 h in 10% neutral buffered formalin. After fixation, the whole head was placed in 23% formic acid for 48 h for decalcification. Tissues were processed routinely, embedded in paraffin, sectioned at 5 μm, and stained with hematoxylin-eosin. Microscopic histopathology evaluations were performed by an experienced veterinary pathologist (K.L.B.) blinded to the genotypes of the animals.
Preparation and analysis of bone marrow neutrophils.
Starting at day 1, FSP1-Cre;Cdc42fl/fl mice and Cdc42fl/fl mice were placed on enrofloxacin (22.7 mg/ml) as a preventive antibiotic measure, regardless of health status at the outset. After harvesting of organs from euthanized mice at 8 weeks of age, femurs and tibias were dissected and the marrow was flushed with phosphate-buffered saline (PBS) and layered on a 3-step Percoll gradient (78%, 69%, and 52%) that was centrifuged at 1,060 × g for 30 min. Cytospin samples of the 78%-69% interface revealed more than 90% neutrophils, as assessed morphologically. These samples were then used as the polymorphonuclear leukocyte (PMN) population for the functional assays described below.
Flow cytometry.
Single-cell suspensions were prepared from bone marrow, thymus, and spleens, as described previously (20). Antibodies against CD11b (M1/70), CD4 (RM4-5), B220 (RA3-6B2), and IgM (II/41) were from BD Biosciences (La Jolla, CA). CD8 (53-6.7), anti-Gr1 (RB6-8C5), and anti-Thy1.2 (53-2.1) antibodies were from (eBioscience, San Diego, CA). Cells were analyzed with a FACSCalibur apparatus (BD Biosciences) and FlowJo software (TreeStar, Ashland, OR) after direct immunofluorescent (DIF) staining with fluorochrome-conjugated antibodies as described previously (20). To identify β-galactosidase-expressing cells, single-cell suspensions were preincubated (10 min at 37°C) in PBS containing 1% fetal bovine serum (FBS), followed by addition of fluorescein di-β-d-galactopyranoside (FDG; Marker Gene Technologies, Eugene, OR) to a final concentration of 2 nM and further incubation for 5 min at 37°C. The reaction was stopped by adding 4% paraformaldehyde and incubating on ice for 20 min, followed by DIF staining of cell surface markers.
Immunoblotting for Cdc42 expression.
Spleens or purified neutrophils from the mice were lysed with radioimmunoprecipitation assay buffer. Lysates were clarified by centrifugation, and 30 μg of total protein was electrophoresed onto a 10% SDS-polyacrylamide gel and subsequently transferred to nitrocellulose membranes. Membranes were blocked in 5% milk–Tris-buffered saline–Tween and then incubated with a Cdc42 antibody followed by the appropriate horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were identified using enhanced chemiluminescence.
Bacterial killing assay.
Bacterial killing assays were performed with purified neutrophils as described previously (21), with the following modification. Staphylococcus aureus strain Newman was grown overnight in tryptic soy broth (TSB) at 37°C. The culture was backdiluted 1/100 into fresh TSB, incubated at 37°C for 3 h, and harvested. Bacterial pellets were resuspended in PBS to an optical density at 600 nm of 0.4 (∼1 × 108 CFU/ml) and coated with complement. The complement-coated bacteria were mixed 1:1 by volume with bone marrow-derived neutrophils (resuspended in antibiotic-free Iscove's modified Dulbecco's medium [IMDM] with 10% FBS to a density of ∼0.75 × 107 cells/ml) in serum-coated 96-well flat-bottom plates. The plates were centrifuged for 8 min at 300 × g and then incubated for 2 h at 37°C with 5% CO2. The neutrophils were then lysed by the addition of a 1/10 volume of 1% Triton X-100, and serial dilutions were plated to enumerate viable bacteria.
Migration assays.
As described previously (22), 5 × 104 neutrophils purified from Cdc42fl/fl or FSP1-Cre;Cdc42fl/fl mice were diluted in serum-free medium placed on Transwells with 5-μm pores coated with fibronectin (2.5 μg/ml). Cells that migrated through the filter after 4 h were counted.
In vitro neutrophil survival and functions.
Bone marrow cells (4 × 106 cells/ml) were cultured in the presence or absence of interleukin-3 and granulocyte-macrophage colony-stimulating factor for 3 days. Viable cells were counted by Trypan blue dye exclusion, stained with anti-Gr1–phycoerythrin, anti-CD11b–fluorescein isothiocyanate (FITC), and 7-amino-actinomycin D (7-AAD) (Invitrogen, Carlsbad, CA, USA), and analyzed by fluorescence-activated cell sorting (FACS). Neutrophil survival was calculated as the percentage of 7-AAD− Gr1+ CD11b+ cell numbers relative to the input neutrophil cell number. Measurements of reactive oxygen species (ROS) production were conducted using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) and flow cytometry (23). Freshly isolated or activated marrow cells were incubated with 10 μM H2DCFDA for 15 min on ice, followed by surface marker staining and flow cytometry. The phagocytosis (endocytosis of antigen) assay was performed on fresh neutrophils using FITC-conjugated latex beads (Cayman Chemical, Ann Arbor, MI). The latex beads were diluted in PBS and added to purified neutrophils in serum-free IMDM. Following incubation for 7.5 min at 37°C, the reaction was stopped by adding cold PBS, and the results were analyzed with a FACSCalibur apparatus (BD Biosciences) and FlowJo software (TreeStar). Phagocytic maturation (acidification of the phagosome) was assayed using a pHrodo Escherichia coli Bioparticles phagocytosis kit (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions.
Statistics.
Histology, flow phenotypes, and cell numbers are representative of a minimum of six FSP1-Cre;Cdc42fl/fl and Cdc42fl/fl mice. Differences between sample sets were analyzed statistically using Student's t unpaired two-tail nonparametric test of significance (Instat; GraphPad Software).
RESULTS
FSP1-Cre;Cdc42fl/fl mice are susceptible to severe upper respiratory tract infections characterized by a neutrophil-predominant infiltrate and lymphocyte depletion.
To define the potential role of Cdc42 in kidney development and/or fibrosis, we crossed FSP1-Cre mice with Cdc42fl/fl mice. All offspring were kept together in the same cages in a nonbarrier facility until weaning. Mice in the colony were specific pathogen free by routine sentinel testing for viral pathogens at Vanderbilt Medical Center. The mice were born in normal Mendelian ratios, and the genotypes were indistinguishable. Within the first week of life, all the FSP1-Cre;Cdc42fl/fl mice showed signs of a failure to thrive and experienced 100% mortality by 4 weeks of age. The kidneys of these mice were removed, subjected to histological examination, and found to be normal (data not shown). There were also no abnormalities in the cardiovascular system, gastrointestinal tract, central nervous system, reproductive tract, lungs, endocrine organs, or muscle of these mice (data not shown). In particular, there was no evidence of abnormal leukocyte infiltration of any of these organs. However, the mice were noted to have discharge from the nares and eyes. Complete blood counts with differentials were performed, and leukocytosis with neutrophilia and lymphopenia in the FSP1-Cre;Cdc42fl/fl mice was observed (Table 1). Taken together, these data suggest that the enhanced mortality of the FSP1-Cre;Cdc42fl/fl mice was due to an underlying infective process, hematological abnormality, or both.
Table 1.
Complete blood counts of Cdc42-deficient mice
| Cell type | Mean cell count (106 cells/ml blood) ± SEM |
Mean cell % ± SEM |
||||||
|---|---|---|---|---|---|---|---|---|
| Without antibioticsa |
Antibiotic treatedb |
Without antibiotics |
Antibiotic treated |
|||||
| Cdc42fl/fl | FSP1-Cre;Cdc42fl/fl | Cdc42fl/fl | FSP1-Cre;Cdc42fl/fl | Cdc42fl/fl | FSP1-Cre;Cdc42fl/fl | Cdc42fl/fl | FSP1-Cre;Cdc42fl/fl | |
| White blood cells | 1.98 ± 0.23 | 3.13 ± 0.53c | 3.31 ± 0.26 | 4.65 ± 1.44 | ||||
| Neutrophils | 0.78 ± 0.13 | 2.13 ± 0.45c | 0.61 ± 0.07 | 3.66 ± 1.31c | 39.3 ± 5.4 | 64.9 ± 4.4d | 18.9 ± 2.0 | 70.2 ± 4.4d |
| Lymphocytes | 0.83 ± 0.13 | 0.67 ± 0.08 | 2.28 ± 0.21 | 0.81 ± 0.16d | 41.9 ± 3.9 | 23.8 ± 3.3d | 69.0 ± 3.1 | 24.1 ± 3.6d |
| Monocytes | 0.22 ± 0.04 | 0.18 ± 0.03 | 0.24 ± 0.02 | 0.11 ± 0.01d | 11.6 ± 1.7 | 6.1 ± 1.0c | 7.6 ± 1.1 | 4.0 ± 0.8c |
| Eosinophils | 0.12 ± 0.04 | 0.12 ± 0.23 | 0.15 ± 0.07 | 0.05 ± 0.02 | 5.4 ± 1.6 | 4.0 ± 1.0 | 3.9 ± 1.7 | 1.3 ± 0.6 |
| Basophils | 0.04 ± 0.01 | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.02 ± 0.01 | 1.8 ± 0.3 | 1.1 ± 0.2 | 0.55 ± 0.20 | 0.47 ± 0.28 |
Three- to 4-week-old mice; n = 9 Cdc42fl/fl mice versus 10 FSP1-Cre;Cdc42fl/fl mice.
Seven- to 9-week-old mice; n = 9 Cdc42fl/fl mice versus 9 FSP1-Cre;Cdc42fl/fl mice.
P < 0.05.
P < 0.01.
To define these abnormalities further, we performed necropsies of the FSP1-Cre;Cdc42fl/fl mice, concentrating on the organs that could potentially explain the premature mortality. Every FSP1-Cre;Cdc42fl/fl mouse examined was significantly smaller than its wild-type littermate (6 ± 2 g versus 14 ± 2.3 g) (Fig. 1A) and had a small thymus and discharge around the nose and eyes. The most striking findings were seen on microscopic examination of the head sections, which revealed copious mucopurulent exudates in the nasal passages, often occluding much of the air space and eroding the mucosa (Fig. 1C and E). This mucopurulent exudate was also present in the middle ear and Eustachian tube (Fig. 1G and I). Furthermore, gingivitis and conjunctivitis characterized by neutrophilic inflammation were also observed (Fig. 1K and M). To determine if the severe rhinitis, otitis, conjunctivitis, and gingivitis had a bacterial component, aerobic cultures were performed on seven Cdc42fl/fl mice and four FSP1-Cre;Cdc42fl/fl mice. Organisms considered normal flora, including Streptococcus acidominimus, Streptococcus hyovaginalis, Streptococcus thoraltensis, Staphylococcus xylosus, Streptococcus downei, and Streptococcus uberis, were isolated from Cdc42fl/fl and FSP1-Cre;Cdc42fl/fl mice. Pasteurella pneumotropica, a known respiratory pathogen of mice, was isolated from two of four FSP1-Cre;Cdc42fl/fl mice but not Cdc42fl/fl mice. To assess the potential for systemic infection, aerobic culturing of heart blood from FSP1-Cre;Cdc42fl/fl mice was performed and found to be negative. Taken together, these results suggest that the FSP1-Cre;Cdc42fl/fl mice were susceptible to severe suppurative upper respiratory tract infections resulting in upper airway obstruction that is characterized by a neutrophil-predominant infiltration. The significance of the organisms cultured from the Cre;Cdc42fl/fl mice is unclear. In the face of the severe inflammation and mucosal necrosis, even organisms considered to be normal flora may be contributing to morbidity.
Fig 1.
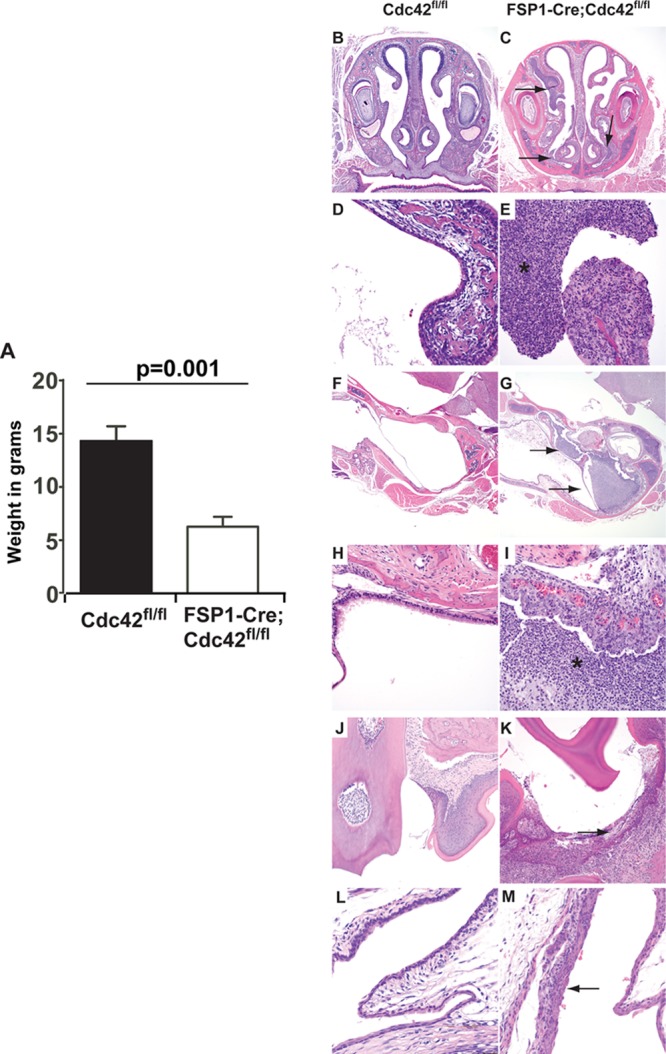
FSP1-Cre;Cdc42fl/fl mice lose weight and develop lethal suppurative infections of the upper respiratory tract. (A) FSP1-Cre;Cdc42fl/fl mice have significant weight loss by 4 weeks of age (mean weights ± SEMs are shown). (B to M) Hematoxylin-eosin staining of the upper respiratory tract of Cdc42fl/fl mice (B, D, F, and H) and FSP1-Cre;Cdc42fl/fl mice (C, E, G, and I). FSP1-Cre;Cdc42fl/fl mice have severe purulent rhinitis (C, arrows), while nasal passages in Cdc42fl/fl mice are normal (B). Higher magnification of the nasal turbinates of the FSP1-Cre;Cdc42fl/fl mice show that the mucosal surface is denuded and there is copious exudation of neutrophils and mucous, denoted by an asterisk in the nasal passage (E), while the Cdc42fl/fl mice have normal turbinates and clear nasal passages (D). FSP1-Cre;Cdc42fl/fl mice have otitis (G, arrows), while the ears are normal in Cdc42fl/fl mice (F). The higher magnification of the middle ear of the FSP1-Cre;Cdc42fl/fl mice shows that it is filled by neutrophilic exudate (I), while the middle ear of the Cdc42fl/fl mice is covered with normal cuboidal ciliated epithelium (H). FSP1-Cre;Cdc42fl/fl mice also had a mild neutrophilic gingivitis (K) and conjunctivitis (M) which was not observed in the wild-type mice (J and L). Magnifications, ×40 (B, C, F, and G), ×400 (D, E, H, and I), ×200 (J and K), and ×600 (L and M).
In addition to the severe upper respiratory infections, moderate to marked depletion of lymphocytes was noted in the thymus (Fig. 2B) and spleen (Fig. 2F). There was decreased cellular density in the thymic cortex (Fig. 2D) and marked depletion of lymphocytes in the splenic white pulp (Fig. 2H). In the spleen, periarticular T cell zones were depleted of T cells, and extramedullary hematopoiesis replaced the lymphocyte population with purely granulocytic cells (Fig. 2J). Moreover, a striking component of extramedullary hematopoiesis mainly involving the granulocytic lineages also expanded the red pulp. Granulopoiesis (Fig. 2J, arrow) also replaced the lymphocytes in the periarticular lymphoid sheath. Exuberant granulopoiesis in the bone marrow (Fig. 2L) accompanied this extramedullary hematopoiesis in the spleen. The histologic findings in the hematopoietic organs mirrored the abnormalities (lymphopenia and neutrophilia) observed on the complete blood count.
Fig 2.
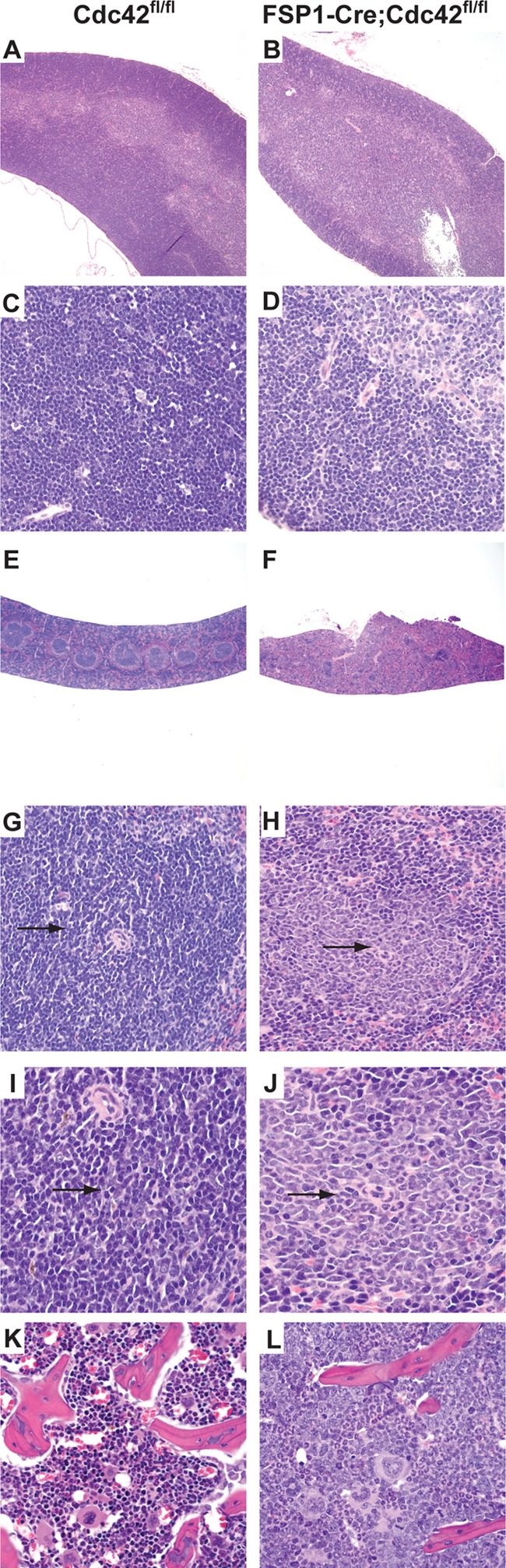
FSP1-Cre;Cdc42fl/fl mice developed lymphocyte depletion and granulocytosis. (A to D) Hematoxylin-eosin staining of the thymus of control Cdc42fl/fl mice (A, C) and FSP1-Cre;Cdc42fl/fl mice (B, D). There is a marked decrease in cellularity and depletion of lymphocytes in the thymus of the FSP1-Cre;Cdc42fl/fl mice (D, asterisk) relative to Cdc42fl/fl mice (C, asterisk). (E to J) Hematoxylin-eosin staining of the spleens of Cdc42fl/fl mice (E, G, I) and FSP1-Cre;Cdc42fl/fl mice (F, H, J). The spleen showed marked depletion of lymphocytes and prominent proliferation of granulocytes in the white pulp (lymphoid tissue) of the spleen of FSP1-Cre;Cdc42fl/fl mice (H, arrow) relative to the normal spleen of Cdc42fl/fl mice (G, arrow). Granulopoiesis (arrow) replaces the lymphocytes in the periarticular lymphoid sheath in the FSP1-Cre;Cdc42fl/fl mice (J) but not the Cdc42fl/fl mice (I). There is a marked increase in granulocytosis in the bone marrow of the FSP1-Cre;Cdc42fl/fl mice (L) relative to the Cdc42fl/fl mice (K). Magnifications, ×40 (A, B, E, and F), ×400 (C, D, G, H, K, and L), and ×1,000 (I and J).
Fatal upper respiratory tract infections in FSP1-Cre;Cdc42fl/fl mice respond to antibiotic treatment.
Since mice are unable to breathe through their mouths and are obliged to maintain airflow through their nares, we reasoned that the early mortality of FSP1-Cre;Cdc42fl/fl mice may have been due to uncontrolled infections of the upper respiratory tract. To test this hypothesis, mice were placed on enrofloxacin delivered in the drinking water. FSP1-Cre;Cdc42fl/fl mice treated with antibiotics were rescued from early mortality, and they lived until sacrifice at 8 weeks of age. The severity of the rhinitis (Fig. 3A and B) was dramatically decreased, and the otitis (Fig. 3C and D) and the conjunctivitis and gingivitis (data not shown) were completely resolved in all examined mice upon antibiotic treatment; however, the leukocytosis with neutrophilia and lymphopenia was unchanged (Table 1). Depletion of lymphocytes and prominent extramedullary granulopoiesis remained in the spleen (Fig. 3E and F); increased granulopoiesis was still observed in the bone marrow (Fig. 3G), and severe lymphoid depletion was still detected in the thymus (Fig. 3H). Thus, antibiotic therapy of the FSP1-Cre;Cdc42fl/fl mice decreased the suppurative infections of the upper respiratory tract but did not alter their severe lymphoid depletion and increased granulopoeisis. These data suggest that loss of Cdc42 results in defects in the ability to control bacterial pathogens and immune cell development.
Fig 3.

Suppurative infections of the upper respiratory tract of FSP1-Cre;Cdc42fl/fl mice are prevented by antibiotic therapy. (A to D) Hematoxylin-eosin staining of FSP1-Cre;Cdc42fl/fl mice shows the absence of rhinitis (A, B) and otitis (C, D) following antibiotic administration. Depletion of lymphocytes and extramedullary granulopoiesis remain prominent in the spleen (E, F); granuolopoiesis is observed in the bone marrow (G), and severe lymphoid depletion is observed in the thymus (H). Magnifications, ×40 (A, C, and E) and ×400 (B, D, and F to H).
FSP1-Cre is expressed in all leukocyte lineages in mice.
The unexpected phenotype observed in the FSP1-Cre;Cdc42fl/fl mice, which lacked overt fibroblast defects and which had features resembling many features seen when Cdc42 was deleted during hematopoietic cell development (4–7), prompted us to define whether FSP1-Cre expression occurred in hematopoietic cells. Accordingly, we delineated cell lineages in which FSP1-Cre is active by crossing FSP1-Cre mice with ROSA26-LacZ reporter mice, where expression of β-galactosidase occurs only after Cre recombinase excises a loxP-flanked stop codon upstream from the lacZ gene. After direct immunofluorescent staining of cell suspensions with lineage-specific monoclonal antibodies combined with the use of FDG, a cell-permeant fluorogenic substrate of β-galactosidase, bone marrow and lymphoid organs were analyzed by flow cytometry. The results of these studies indicated that Cre excision was widespread among cells in these organs (Fig. 4A). Gating on lineage-specific markers disclosed that FSP-Cre led to ample β-galactosidase activity in a variety of the most prevalent lymphoid and myeloid lineage cells, i.e., CD4 and CD8 T lymphocytes, B220-marked B lymphocytes, Gr1+ neutrophils, and the various myeloid cells marked by combinations of CD11b or CD11c expression (Fig. 4B). The development of fluorescence from the FDG substrate was specific for mice that bore both an FSP1-Cre allele and a ROSA26-LacZ tracer allele, indicating that it was not due to an endogenous enzymatic activity capable of cleaving the β-galactoside. Consistent with this, immunoblots showed that the Cdc42 protein was absent from unfractionated spleen cells and from purified neutrophils of the FSP1-Cre;Cdc42fl/fl mice (Fig. 4C). Together, these results indicate that Cdc42 is deleted in most or all leukocytes.
Fig 4.

FSP1-Cre transgenic mice express Cre in hematopoietic lineage cells. (A) ROSA26-LacZ reporter mice were crossed with FSP1-Cre transgenic mice. Cre expression results in the removal of a loxP-flanked DNA element, which induces expression of lacZ. Cells were isolated from bone marrow (BM), thymus, and spleen, stained with FDG, and analyzed by flow cytometry. Shown are representative FACS profiles of three independent experiments. Shaded histogram, Cre-negative control; line histogram, FSP1-Cre-positive cells. The percentages of FDG-positive events are indicated. (B) Cells were isolated from the spleens of ROSA26-LacZ reporter mice having the FSP1-Cre transgene or not and incubated with FDG, followed by staining for surface markers. Shown are FACS histograms in the viable gates of each surface marker-positive population in one experiment representative of three experiments. (C) Western blot analysis of Cdc42 in splenocytes and purified neutrophils from Cdc42fl/fl and FSP1-Cre;Cdc42fl/fl mice. β-Actin was immunoblotted as a loading control.
Distorted development of granulocytes versus lymphocytes.
The bacterial infections, severe abnormalities of lymphoid tissues, and loss of the Cdc42 protein in leukocytes of the FSP1-Cre;Cdc42fl/fl mice prompted us to analyze their leukocyte phenotype and development in more detail. Using mice maintained on antibiotics to allow the conditionally Cdc42-deficient mice to survive, we determined that total cellularity was substantially decreased in the thymus but this was normal in the spleen and marrow of the FSP1-Cre;Cdc42fl/fl mice (Fig. 5A). Flow cytometry revealed a dramatic increase in mature granulocyte populations in peripheral blood and lymphoid organs, as detected by both parameters of size and granularity (forward scatter [FSC] versus side scatter [SSC]; Fig. 5B) and surface staining for Gr1+ CD11b+ cells (Fig. 5C and D). Strikingly, almost half of the cells in spleens of the conditional knockout mice were of the myeloid lineage with characteristics of PMNs. Conversely, populations of T and B lymphocytes were decreased in the spleen, with an even greater decrease in the numbers of T and B cell precursors observed in the thymus and bone marrow (Fig. 5E and F). The dramatically increased granulocyte/neutrophil population in bone marrow and spleen in the FSP1-Cre;Cdc42fl/fl mice was also confirmed using CD11b+ Ly6G+ Ly6Clo surface markers and Wright-Giemsa staining (Fig. 5G; see Fig. S1 in the supplemental material). These data confirmed the accumulation of neutrophils found in the histology of the FSP1-Cre;Cdc42fl/fl mice (Fig. 2). It was of interest to note that in addition to Ly6G+ neutrophils, CD11b+ Ly6G− Ly6Chi cells were also increased in the Cdc42-deficient mice (Fig. 5G; see Fig. S1A in the supplemental material). Taken together, these results indicate that FSP1-Cre was expressed in and depleted Cdc42 from the hematopoietic lineages, thereby suggesting that the decrease in lymphocytes and the increase in neutrophils are due to an essential role of this small GTPase in establishing appropriate populations of leukocytes. Moreover, because Cdc42 appears to restrain PMN numbers yet promote the lymphoid cell populations, the data indicate that lack of this protein during hematopoiesis results in a bias toward granulocyte development over lymphocyte development.
Fig 5.

Leukocyte development in FSP1-Cre;Cdc42fl/fl mice is abnormal. Profiling of hematopoietic compartment in mice maintained on antibiotics. FSP1-Cre;Cdc42fl/fl and Cdc42fl/fl mice were maintained with antibiotic-containing water and lymphoid organs were harvested at 8 ± 1 weeks of age and analyzed by flow cytometry, as described in the legend to Fig. 4. (A) Cellularity of bone marrow, thymus, and spleen. Shown are the mean cell numbers ± SEMs from each lymphoid organ of five Cdc42fl/fl mice and five FSP1-Cre;Cdc42fl/fl mice (7 to 9 weeks old). (B to D) Abnormal granulocyte (neutrophil) population in FSP1-Cre;Cdc42fl/fl mice. (B) Profiles of FSC and SSC of cells in the indicated organs from one representative pair of mice of the five analyzed; (C) flow cytometric profiles for Gr1 and CD11b in the indicated FSC × SSC gates. Numbers represent the frequencies (in percent) of FSChi SSChi cells (B) and Gr1+ CD11b+ cells (C). (D) Mean cell number ± SEM of mature granulocytes in bone marrow and spleen and mean percentage of those cells in blood. (E, F) Impaired lymphocyte development in FSP1-Cre;Cdc42fl/fl mice, despite antibiotic therapy. T- and B-lineage cell numbers from lymphoid organs were calculated using the cellularity of each lymphoid organ and the frequencies of T- and B-lineage subsets. Shown are the mean ± SEM values from the five Cdc42fl/fl mice and five FSP1-Cre;Cdc42fl/fl mice (7 to 9 weeks old), along with the results of tests of statistical significance. (G) Increased CD11b+ Ly6G+ Ly6Clo and CD11b+ Ly6G+ Ly6Chi populations in bone marrow and spleen of FSP1-Cre;Cdc42fl/fl mice. Mean cell numbers ± SEMs were calculated from the flow cytometry data shown in Fig. S1 in the supplemental material (five Cdc42fl/fl mice and five FSP1-Cre;Cdc42fl/fl mice). DN, CD4− CD8− double-negative population; DP, CD4+ CD8+ double-positive population.
Functional analyses of Cdc42-deficient neutrophils.
The paradox of an increase in PMN number along with increased susceptibility to bacterial infection of the respiratory tract which could be reversed with preventive antibiotic treatment suggested a defect in antimicrobial activity and prompted us to analyze the functionality of FSP1-Cre;Cdc42fl/fl neutrophils as well as their survival. We initially defined the microbicidal activity ability of neutrophils against Staphylococcus aureus, an organism known to cause robust inflammatory responses highlighted by increased recruitment of neutrophils. The capacity of Gr1+ CD11b+ cells from FSP1-Cre;Cdc42fl/fl mice to control the growth of Staphylococcus aureus in vitro was found to be lower than that of the same cells isolated from wild type (Fig. 6A). As the ability of PMNs to control bacterial infection depends on their ability to migrate and Cdc42 is known to be a key regulator of neutrophil migration, it was not surprising to find that Cdc42-deficient neutrophil migration on fibronectin was severely impaired (Fig. 6B). In contrast to these abnormalities, there was at most a modest decrease in formyl-Met-Leu-Phe-induced ROS production, and phagosomal acidification by Cdc42-deficient PMNs appeared to be normal (Fig. 6C and D). Despite this, Cdc42-deficient neutrophils exhibited impaired endocytosis of fluorochrome-conjugated latex beads (Fig. 6E), suggesting a requirement of Cdc42 in the early stage of phagocytosis. Since neutrophils are postreplicative but short-lived, a possibility for their massive increase in the periphery in FSP1-Cre;Cdc42fl/fl mice included increased survival or overproduction. However, our ex vivo survival assay results provided evidence that the absence of Cdc42 impaired PMN survival (Fig. 6F). Together, these results indicate that although there is substantial overproduction of, accumulation of, and tissue infiltration by neutrophils in the absence of Cdc42 driven by FSP1-Cre, despite an apparently normal capacity to generate ROS, these PMNs are less able to migrate on fibronectin as well as uptake and kill bacteria.
Fig 6.

Cdc42 is required for normal killing activity, migration, and survival of neutrophils. (A) Reduced antimicrobial activity of Cdc42-deficient neutrophils. Purified neutrophils were incubated with S. aureus precoated with complement, after which bacterial viability was determined by plating of serial dilutions on solid medium. Shown are the percentages of bacterial growth after incubation with neutrophils relative to the number of bacteria incubated in the absence of neutrophils. Data represent the mean ± SEM of six Cdc42fl/fl mice and six FSP1-Cre;Cdc42fl/fl mice. (B) Impaired migration of Cdc42-deficient neutrophils. Purified neutrophils were plated in the upper chamber with a 5-μm-pore-size polycarbonate membrane coated with fibronectin and stained with crystal violet after 4 h of incubation. Cells in 10 independent high-powered fields were counted and averaged. (C) Cdc42 is dispensable for ROS production of neutrophils. Bone marrow cells were stimulated with N-formyl-methionyl-leucyl-phenylalanine (fMLP), phorbol myristate acetate (PMA), or vehicle for 30 min and incubated with H2DCFDA and antibodies against specific surface markers. Shown are representative histograms of Gr1+ CD11b+ gated populations. Three independent experiments were performed. The changes in the mean fluorescence intensity (ΔMFI) were calculated by subtracting the mean fluorescence intensity of unstained controls (shaded) from the mean fluorescence intensity of DCF (line). (D) Cdc42 is dispensable for phagosomal acidification of neutrophils. Acidification of blood neutrophils was measured using a pHrodo E. coli Bioparticles phagocytosis kit. Shown are representative histograms of two independent experiments. (E) Impaired phagocytic endocytosis of Cdc42-deficient neutrophils. Purified neutrophils were incubated with FITC-conjugated latex beads, and endocytosis was measured by flow cytometry. Shown are representative histograms of viable granulocyte populations (shaded histogram, neutrophil alone; line histogram, neutrophils incubated with the beads). The mean frequency of phagocytic cells (percentage of FITC-positive cells) and phagocytic activity (ΔMFI = mean fluorescence intensity of experimental sample − mean fluorescence intensity of neutrophils alone) for five Cdc42fl/fl mice and five FSP1-Cre;Cdc42fl/fl mice are presented. (F) Bone marrow cells were cultured, and cell survival was determined at days 1 to 3. Percent survival was calculated as the percentage of viable neutrophil numbers determined by counting viable cells and FACS analysis of live neutrophils (7-AAD-negative CD11b+ Gr1+ cells) at each day relative to the initial cell numbers. Shown is a representative result of two independent experiments.
DISCUSSION
The ubiquitously expressed molecular switch Cdc42 is required for the normal development and survival of mice (3, 24). In this study, we originally set out to define the unexplored role of Cdc42-dependent signaling in fibroblasts and following injury. We found that this FSP1-Cre transgene deleted Cdc42 in all leukocytes, in addition to fibroblast populations. Furthermore, early lethality was observed for all FSP1-Cre;Cdc42fl/fl mice and could be attributed to increased susceptibility to severe suppurative infections. Involvement of the upper respiratory tract and extensive purulent accumulation therein appeared to be rapidly fatal for these obligate nose breathers. After rescue using systemic antibacterial treatment, the FSP1-Cre;Cdc42fl/fl mice also demonstrated defects in neutrophil migration and bacterial killing, along with abnormalities in lymphoid tissue development. Collectively, these findings demonstrate the requirement for Cdc42 in host protection from lethal infections and suggest a critical role for this small GTPase in innate immunity.
Prior work, largely with in vitro models, has reported that chemotaxis can depend on Cdc42, including in neutrophils (25–28). However, the ultimate impact of blocking Cdc42 function on the susceptibility to infection of an otherwise intact animal has not been determined. Our results provide evidence that deleting Cdc42 in neutrophils resulted in defects in bacterial killing, migration, phagocytic endocytosis, and survival; however, there were no deficiencies in ROS production or phagosomal acidification. These findings are consistent with those of previous work demonstrating a migration phenotype in Cdc42−/− neutrophils in vitro (29). In that study, Cdc42 played a critical role in the maintenance of neutrophil polarity by modulating activation of the myosin light chain pathway by integrin αMβ2. Both that study and our data confirmed previous in vitro findings demonstrating a key role for Cdc42 in neutrophil function (25–28). Collectively, the clinical presentation of the FSP1-Cre;Cdc42fl/fl mice as well as their improved survival with enrofloxacin treatment suggests that although there may be yet further contributing mechanisms, neutrophil defects were the principal reason for the lethal phenotype. While our data support a role for neutrophils in the bacterially driven mortality, they also raise the intriguing possibility that cells other than neutrophils also participate in the high susceptibility to infection in the model of Cdc42 deficiency.
The predominant pathological features of the FSP1-cre;Cdc42fl/fl mice were a leukocytosis with a neutrophil predominance and lethal infection of the upper respiratory passages with a massive neutrophil infiltrate. The inflammation of the upper airways was so severe that it caused asphyxiation in the mice, as they are obligate nasal breathers. There was no evidence of such severe inflammation in any other organs within the mice. These mice had some of the same characteristics found in human leukocyte adhesion deficiency (LAD) syndromes, which are clinically defined by recurrent bacterial infections primarily localized to skin and mucosal surfaces, mild to moderate basal neutrophilia, and marked granulocytosis during acute infection. LAD is thought to be primarily due to the inability of leukocytes to migrate and exit the vasculature, resulting in a lack of granulocytes at the site of infections. The 3 LAD syndromes found in humans are (i) LAD I, caused by integrin β2 deletions; (ii) LAD II, resulting from abnormalities in fucosylated selectin ligands required for neutrophil rolling; and (iii) LAD III, produced by deficiencies in kindlin 3, a protein required for integrin activation (30). Our mice had the neutrophilia and bacterial infections; however, they also had a severe neutrophil infiltrate in the upper airways. In addition, the phenotype of the FSP1-Cre;Cdc42fl/fl mice was far more severe than that of the nonfatal mouse models of LAD I (31, 32) or LAD II (33) deficiency. These data suggest that under physiological conditions in vivo, Cdc42 has a fundamental role in cellular functions, in addition to the β2 integrin- or the selectin-based functions. The kindlin 3-null mice die within a week; however, this is due to severe bleeding, as kindlin 3 is required for the activation of both β2 and β3 integrins (34, 35).
In other work seeking to define the role of Cdc42 in hematological cells, this allele was deleted in hematopoietic stem cells using the interferon-induced Mx1-Cre transgene (6, 7). These studies identified that deletion of Cdc42 at the stem cell stage, but not later in B lymphocyte development, resulted in abnormalities of the B lymphoid lineage (6, 7) that were largely recapitulated with FSP1-Cre-driven deletion. Nonetheless, only a severe lack of circulating natural antibodies would likely give rise to the observed antibiotic-sensitive early mortality of FSP1-Cre;Cdc42fl/fl mice; however, serum Ig levels were normal in these mice. In addition, our findings with T-lineage cells were similar to those recently reported by others (4). However, the presence of normal frequencies of FoxP3-expressing CD4 T cells, the nature of the cellular infiltrate, and the impact of antibiotics all represent evidence against a primary T lymphocyte-driven etiology of the mortality. Similar to the FSP1-Cre;Cdc42fl/fl mice, the Mx1-Cre;Cdc42fl/fl mice died within 3 weeks of Cre induction; however, the cause of death was different. The Mx1-Cre;Cdc42fl/fl mice developed a fatal myeloproliferative disorder manifested by significant leukocytosis with neutrophilia in the peripheral blood, myeloid hyperproliferation of the bone marrow and spleen, and myeloid cell infiltration of other organs, including lungs and liver (7). In contrast, the FSP1-Cre;Cdc42fl/fl mice had a more moderate leukocytosis and neutrophilia and died of suppurative infections. A possible explanation for these differences is that the Mx1-Cre;Cdc42fl/fl mice but not the FSP1-cre;Cdc42fl/fl mice were maintained in a sterile pathogen-free facility. Consistent with this possibility, the FSP1-Cre;Cdc42fl/fl mice survived for at least 8 weeks when placed on antibiotics; however, even under these circumstances they never developed a myeloproliferative disorder. An alternative explanation is that the respective Cre transgenes delete Cdc42 in different myeloid progenitor cell types.
In conclusion, we show that deleting Cdc42 from mice using FSP1-Cre results in lethal abnormalities of immune function that yield a completely penetrant and fatal susceptibility to natural infection. This surprising phenotype emphasizes the nonspecificity of the FspI-Cre which we demonstrate is clearly active in all leukocytes, in addition to fibroblast and dendritic cell lineages, as previously shown (8, 9, 15–18). In addition, we establish the absolute requirement for Cdc42 in host defenses against infections, suggesting that therapeutically targeting this vital protein could have fatal consequences.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants DK083187 (to R.Z.), DK075594 (to R.Z.), DK069221 (to R.Z.), HL106812 (to M.B. and K.L.), and R56 AI091771 (to E.P.S.); O'Brien Center grant DK79341-01 (to R.Z.); and an American Heart Association Established Investigator Award (to R.Z.). R.Z. has a merit award (1I01BX002196-01) from the U.S. Department of Veterans Affairs. T.E.K.-F. was supported by postdoctoral fellowships from the American Heart Association and NIH (F32 AI100480).
We thank Eric Neilson for the FSP1-Cre mice.
Footnotes
Published ahead of print 20 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.01114-12.
REFERENCES
- 1. Melendez J, Grogg M, Zheng Y. 2011. Signaling role of Cdc42 in regulating mammalian physiology. J. Biol. Chem. 286:2375–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chen C, Wirth A, Ponimaskin E. 2012. Cdc42: an important regulator of neuronal morphology. Int. J. Biochem. Cell Biol. 44:447–451 [DOI] [PubMed] [Google Scholar]
- 3. Yang L, Wang L, Zheng Y. 2006. Gene targeting of Cdc42 and Cdc42GAP affirms the critical involvement of Cdc42 in filopodia induction, directed migration, and proliferation in primary mouse embryonic fibroblasts. Mol. Biol. Cell 17:4675–4685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guo F, Zhang S, Tripathi P, Mattner J, Phelan J, Sproles A, Mo J, Wills-Karp M, Grimes HL, Hildeman D, Zheng Y. 2011. Distinct roles of Cdc42 in thymopoiesis and effector and memory T cell differentiation. PLoS One 6:e18002. 10.1371/journal.pone.0018002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang L, Yang L, Filippi MD, Williams DA, Zheng Y. 2006. Genetic deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, adhesion, and engraftment. Blood 107:98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L, Wang L, Geiger H, Cancelas JA, Mo J, Zheng Y. 2007. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc. Natl. Acad. Sci. U. S. A. 104:5091–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yang L, Wang L, Kalfa TA, Cancelas JA, Shang X, Pushkaran S, Mo J, Williams DA, Zheng Y. 2007. Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood 110:3853–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL. 2004. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303:848–851 [DOI] [PubMed] [Google Scholar]
- 9. Cheng N, Bhowmick NA, Chytil A, Gorksa AE, Brown KA, Muraoka R, Arteaga CL, Neilson EG, Hayward SW, Moses HL. 2005. Loss of TGF-beta type II receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of TGF-alpha-, MSP- and HGF-mediated signaling networks. Oncogene 24:5053–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. 1995. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 130:393–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lawson WE, Polosukhin VV, Zoia O, Stathopoulos GT, Han W, Plieth D, Loyd JE, Neilson EG, Blackwell TS. 2005. Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 171:899–907 [DOI] [PubMed] [Google Scholar]
- 12. Schneider M, Kostin S, Strom CC, Aplin M, Lyngbaek S, Theilade J, Grigorian M, Andersen CB, Lukanidin E, Lerche Hansen J, Sheikh SP. 2007. S100A4 is upregulated in injured myocardium and promotes growth and survival of cardiac myocytes. Cardiovasc. Res. 75:40–50 [DOI] [PubMed] [Google Scholar]
- 13. Osterreicher CH, Penz-Osterreicher M, Grivennikov SI, Guma M, Koltsova EK, Datz C, Sasik R, Hardiman G, Karin M, Brenner DA. 2011. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. U. S. A. 108:308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Inoue T, Takenaka T, Hayashi M, Monkawa T, Yoshino J, Shimoda K, Neilson EG, Suzuki H, Okada H. 2010. Fibroblast expression of an IkappaB dominant-negative transgene attenuates renal fibrosis. J. Am. Soc. Nephrol. 21:2047–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN. 2012. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 485:599–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. 2007. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13:952–961 [DOI] [PubMed] [Google Scholar]
- 17. Teng Y, Kanasaki K, Bardeesy N, Sugimoto H, Kalluri R. 2011. Deletion of Smad4 in fibroblasts leads to defective chondrocyte maturation and cartilage production in a TGFbeta type II receptor independent manner. Biochem. Biophys. Res. Commun. 407:633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boomershine CS, Chamberlain A, Kendall P, Afshar-Sharif AR, Huang H, Washington MK, Lawson WE, Thomas JW, Blackwell TS, Bhowmick NA. 2009. Autoimmune pancreatitis results from loss of TGFbeta signalling in S100A4-positive dendritic cells. Gut 58:1267–1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Soriano P. 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21:70–71 [DOI] [PubMed] [Google Scholar]
- 20. Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M. 2010. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32:743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kehl-Fie TE, Chitayat S, Hood MI, Damo S, Restrepo N, Garcia C, Munro KA, Chazin WJ, Skaar EP. 2011. Nutrient metal sequestration by calprotectin inhibits bacterial superoxide defense, enhancing neutrophil killing of Staphylococcus aureus. Cell Host Microbe 10:158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen D, Roberts R, Pohl M, Nigam S, Kreidberg J, Wang Z, Heino J, Ivaska J, Coffa S, Harris RC, Pozzi A, Zent R. 2004. Differential expression of collagen- and laminin-binding integrins mediates ureteric bud and inner medullary collecting duct cell tubulogenesis. Am. J. Physiol. Renal Physiol. 287:F602–F611 [DOI] [PubMed] [Google Scholar]
- 23. Eruslanov E, Kusmartsev S. 2010. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods Mol. Biol. 594:57–72 [DOI] [PubMed] [Google Scholar]
- 24. Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, Geffers R, Rottner K, Brakebusch C. 2005. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol. Biol. Cell 16:4473–4484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lerm M, Brodin VP, Ruishalme I, Stendahl O, Sarndahl E. 2007. Inactivation of Cdc42 is necessary for depolymerization of phagosomal F-actin and subsequent phagosomal maturation. J. Immunol. 178:7357–7365 [DOI] [PubMed] [Google Scholar]
- 26. Zhong B, Jiang K, Gilvary DL, Epling-Burnette PK, Ritchey C, Liu J, Jackson RJ, Hong-Geller E, Wei S. 2003. Human neutrophils utilize a Rac/Cdc42-dependent MAPK pathway to direct intracellular granule mobilization toward ingested microbial pathogens. Blood 101:3240–3248 [DOI] [PubMed] [Google Scholar]
- 27. Szczur K, Xu H, Atkinson S, Zheng Y, Filippi MD. 2006. Rho GTPase CDC42 regulates directionality and random movement via distinct MAPK pathways in neutrophils. Blood 108:4205–4213 [DOI] [PubMed] [Google Scholar]
- 28. Van Keymeulen A, Wong K, Knight ZA, Govaerts C, Hahn KM, Shokat KM, Bourne HR. 2006. To stabilize neutrophil polarity, PIP3 and Cdc42 augment RhoA activity at the back as well as signals at the front. J. Cell Biol. 174:437–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Szczur K, Zheng Y, Filippi MD. 2009. The small Rho GTPase Cdc42 regulates neutrophil polarity via CD11b integrin signaling. Blood 114:4527–4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanna S, Etzioni A. 2012. Leukocyte adhesion deficiencies. Ann. N. Y. Acad. Sci. 1250:50–55 [DOI] [PubMed] [Google Scholar]
- 31. Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, Smith CW, Montgomery CA, Rich S, Beaudet AL. 1998. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 188:119–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson RW, Ballantyne CM, Smith CW, Montgomery C, Bradley A, O'Brien WE, Beaudet AL. 1993. Gene targeting yields a CD18-mutant mouse for study of inflammation. J. Immunol. 151:1571–1578 [PubMed] [Google Scholar]
- 33. Hellbusch CC, Sperandio M, Frommhold D, Yakubenia S, Wild MK, Popovici D, Vestweber D, Grone HJ, von Figura K, Lubke T, Korner C. 2007. Golgi GDP-fucose transporter-deficient mice mimic congenital disorder of glycosylation IIc/leukocyte adhesion deficiency II. J. Biol. Chem. 282:10762–10772 [DOI] [PubMed] [Google Scholar]
- 34. Moser M, Bauer M, Schmid S, Ruppert R, Schmidt S, Sixt M, Wang HV, Sperandio M, Fassler R. 2009. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat. Med. 15:300–305 [DOI] [PubMed] [Google Scholar]
- 35. Moser M, Nieswandt B, Ussar S, Pozgajova M, Fassler R. 2008. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 14:325–330 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.