

Abstract
Francisella tularensis, the bacterial cause of tularemia, infects the liver and replicates in hepatocytes in vivo and in vitro. However, the factors that govern adaptation of F. tularensis to the intrahepatocytic niche have not been identified. Using cDNA microarrays, we determined the transcriptional profile of the live vaccine strain (LVS) of F. tularensis grown in the FL83B murine hepatocytic cell line compared to that of F. tularensis cultured in broth. The fslC gene of the fsl operon was the most highly upregulated. Deletion of fslC eliminated the ability of the LVS to produce siderophore, which is involved in uptake of ferric iron, but it did not impair its growth in hepatocytes, A549 epithelial cells, or macrophages. Therefore, we sought an alternative means by which F. tularensis might obtain iron. Deletion of feoB, which encodes a putative ferrous iron transporter, retarded replication of the LVS in iron-restricted media, reduced its growth in hepatocytic and epithelial cells, and impaired its acquisition of iron. Survival of mice infected intradermally with a lethal dose of the LVS was slightly improved by deletion of fslC but was not altered by loss of feoB. However, the ΔfeoB mutant showed diminished ability to colonize the lungs, liver, and spleen of mice that received sublethal inocula. Thus, FeoB represents a previously unidentified mechanism for uptake of iron by F. tularensis. Moreover, failure to produce a mutant strain lacking both feoB and fslC suggests that FeoB and the proteins of the fsl operon are the only major means by which F. tularensis acquires iron.
INTRODUCTION
Francisella tularensis is a Gram-negative, facultative intracellular coccobacillus and the causative agent of tularemia (1). Tularemia is a zoonotic disease, and the natural hosts include small mammals and arthropods. Of the identified subspecies, F. tularensis subsp. tularensis (type A) and subsp. holarctica (type B) are important causes of human disease. F. tularensis subsp. tularensis exists mainly in North America, whereas subsp. holarctica exists on all continents of the Northern Hemisphere (1, 2). Inhalation of as few as 10 CFU of the highly virulent F. tularensis subsp. tularensis can cause severe disease in humans (3). Due to the high mortality associated with infection by F. tularensis subsp. tularensis, this pathogen was once developed as a biological weapon and is still a potential threat to public health if it were to be released intentionally (4). A live vaccine strain (LVS) of F. tularensis subsp. holarctica was developed in the former Soviet Union, with subsequent modification in the United States. This strain exhibits attenuated virulence in humans but causes lethal disease in mice. Thus, the LVS is a widely employed tool in the study of the pathogenesis of tularemia (5).
F. tularensis has developed mechanisms to replicate within mammalian cells, including macrophages and a variety of nonphagocytic cells (6–14). As for almost all bacteria, iron is essential for F. tularensis (15, 16). Amounts of free iron within host cells are believed to be relatively low (16), so F. tularensis must possess mechanisms for efficient uptake of this nutrient when replicating intracellularly. Growth of F. tularensis in macrophages requires genes of the Francisella pathogenicity island (17). Some of these genes are also upregulated when F. tularensis LVS grows in iron-restricted medium (16, 18), raising the possibility that they aid the bacterium in adapting to the low concentrations of iron with host cells. Moreover, intracellular LVS organisms stimulate acquisition of iron by murine macrophages via the host cell transferrin receptor 1, and they induce the macrophages to express ferrireductase (Steap3), iron membrane transporter Dmt1, and iron regulatory proteins IRP1 and IRP2 (13). Despite the need of F. tularensis for iron, levels must be carefully controlled by the bacterium in its mammalian hosts, where H2O2 is produced by immune cells. Iron drives the production of the highly toxic hydroxyl radical from H2O2 via the Fenton reaction. F. tularensis subsp. tularensis strains contain relatively low levels of iron compared to subsp. holarctica strains, a property that is linked to better survival when H2O2 is added to their medium. This resistance to killing by H2O2 may also contribute to the generally greater virulence of F. tularensis subsp. tularensis strains (19).
F. tularensis is known to take up iron by a siderophore pathway. Siderophores are small iron-binding molecules used as shuttles to acquire ferric iron (Fe3+) from the environment (20). All of the F. tularensis subspecies encode an fsl operon, which produces a siderophore that is similar in structure to rhizoferrin (21). The fsl operon is composed of six genes (fslA to fslF) that are involved in the synthesis of the siderophore and its receptor. A gene encoding a repressor, Fur, and the repressor's binding site, the Fur box, are located upstream of the fsl operon (18, 21–23). When iron is abundant, Fur, together with ferrous iron (Fe2+), represses the expression of the operon by binding to the Fur box. Under conditions of restricted iron, Fur dissociates from the Fur box. Consequently, expression of the operon and the siderophore-Fe3+ uptake pathway is turned on (18, 21).
In many bacteria, the ferrous iron transport (Feo) system serves as an alternative means for acquisition of iron (24). Some Gram-negative bacteria, such as Escherichia coli, Campylobacter jejuni, Yersinia pestis, and Helicobacter pylori, use the Feo system to obtain Fe2+ (25–28). Typically, this system consists of three proteins, FeoA, FeoB, and FeoC, and the genes encoding them are usually in an operon. However, in some organisms, FeoA, FeoC, or both are absent. The functions of FeoA and FeoC have not been determined. As the core of the system, FeoB forms a channel in the bacterial inner membrane via its C terminus, whereas the N terminus is a G protein-like domain in the cytosol that is believed to regulate the uptake of ferrous iron (29). The genes feoA and feoB have been identified in F. tularensis (19, 21). Inactivation of feoB by insertion of a Tn5-based transposon attenuates the growth of the mutant in lungs in a murine model of respiratory tularemia (30), but the biological function of FeoB in this bacterium has not been studied in detail.
Regardless of the route of inoculation, the liver is one of the important targets for infection by F. tularensis (6, 10, 12, 31–33). Furthermore, F. tularensis invades and replicates to high numbers in hepatocytes (6, 10, 12, 14, 31, 34), which constitute 70% of the cell number and 80% of the volume of the liver (35). In this study, we demonstrate for the first time that FeoB participates in uptake of iron by F. tularensis. Loss of FeoB resulted in reduced growth of the LVS in cells of cultured hepatocytic and epithelial lines, although replication in macrophages was unaffected. Moreover, the FeoB-deficient strain was significantly impaired in its ability to colonize the liver, lungs, and spleen of infected mice.
MATERIALS AND METHODS
Bacterial strains and culture media.
F. tularensis LVS (American Type Culture Collection [ATCC] 29684; Manassas, VA) was a generous gift from Karen L. Elkins, Center for Biologics Evaluation and Research, Food and Drug Administration, Rockville, MD. Frozen stocks of bacteria were prepared as previously described (36). For each in vitro experiment, frozen bacteria were thawed and grown on chocolate II agar (Becton, Dickinson and Co. [BD], Sparks, MD) at 37°C in 5% CO2 for 2 to 3 days. Single colonies were inoculated into the appropriate medium and cultured overnight at 37°C with shaking at 100 rpm. E. coli DH5α (Invitrogen, Carlsbad, CA) was cultured in LB medium (Sigma-Aldrich Chemical Co., St. Louis, MO) supplemented with 100 μg/ml of ampicillin or 50 μg/ml of kanamycin where appropriate. F. tularensis strains with plasmids were cultured with either 5 μg/ml of kanamycin or 200 μg/ml of hygromycin B in Mueller-Hinton (MH) II broth (BD) supplemented with 2% IsoVitaleX enrichment (BD), 5.6 mM d-glucose, 625 μM CaCl2, 530 μM MgCl2, and 335 μM ferric pyrophosphate. All antibiotics were from Sigma-Aldrich.
Mammalian cells and culture media.
FL83B (CRL-2390; ATCC) is a hepatocytic cell line derived from the normal liver of a fetal mouse (37). FL83B cells were cultured in F-12K medium (ATCC) supplemented with 10% fetal bovine serum (FBS; HyClone, Logan, UT). The AML12 cell line (CRL-2254; ATCC) was derived from hepatocytes of a mouse transgenic for human transforming growth factor α (38). The AML12 cells were grown in Dulbecco's modified Eagle medium (DMEM)–F-12 (ATCC) supplemented with 5 μg/ml of insulin, 5 μg/ml of transferrin, 5 ng/ml of sodium selenite, 40 ng/ml of dexamethasone, and 10% FBS. HepG2/C3A (CRL-10741; ATCC) is a clonal derivative of the human HepG2 hepatocytic cell line and was grown in Eagle's minimal essential medium (ATCC) containing 10% FBS. HH4 is a human fetal hepatocytic cell line provided by Nelson Fausto (University of Washington School of Medicine, Seattle, WA) and was cultured in Williams' Medium E (Invitrogen) with supplements as described previously (39). A549 (CCL-185; ATCC) is a human lung epithelial cell line derived from carcinomatous pulmonary tissue. A549 cells were cultured in DMEM (Life Technologies, Grand Island, NY) containing 10% FBS. All supplements for the various media were from Sigma-Aldrich unless otherwise indicated.
Human macrophages were derived from monocytes isolated from peripheral blood of healthy adult donors using the human monocyte isolation kit II (Miltenyi Biotec, Auburn, CA) as described by Bolger et al. (7). To prepare human monocyte-derived macrophages, purified monocytes were cultured for 5 days in RPMI 1640 medium (Invitrogen) supplemented with 10% heat-inactivated (56°C for 30 min) FBS and 50 ng/ml of macrophage colony-stimulating factor (R&D Systems, Inc., Minneapolis, MN). Collection of human blood was approved by the Stony Brook University Committee on Research Involving Human Subjects.
Intracellular growth of bacteria.
To infect host cells, bacteria grown in supplemented MH II broth were centrifuged at 4,000 × g for 10 min, and the pellet was resuspended in the appropriate cell culture medium to the desired multiplicity of infection (MOI). The number of bacteria was estimated by measuring the optical density at 600 nm (OD600), and the precise number was determined by retrospective plating on chocolate II agar. Infection protocols were optimized for each type of host cell as follows: hepatocytes, MOI of 1,000 for 2 h; epithelial cells, MOI of 250 for 3 h; and macrophages, MOI of 50 for 2 h. After addition of the bacteria, tissue culture plates were centrifuged at 240 × g for 5 min and then incubated for the indicated times at 37°C to allow uptake of the bacteria. After incubation, the cultures were washed and treated with 5 μg/ml of gentamicin (Invitrogen) for 1 h to kill extracellular organisms. At this point, some cultures were washed and lysed with 10 mg/ml of saponin (Sigma-Aldrich) in Dulbecco's phosphate-buffered saline (PBS) for 15 min at 37°C to measure uptake. The lysates were serially diluted and plated on chocolate II agar, and the number of colonies was counted after 2 days of incubation at 37°C. To measure replication, infected hepatocytes or epithelial cells were subsequently incubated for various times in the presence of gentamicin, and macrophages were cultured for an additional 13 h in antibiotic-free medium. Lysates then were prepared to enumerate CFU.
For visualization of intracellular F. tularensis by immunofluorescence, infected cells were washed, fixed with 2.5% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA) in PBS, permeabilized with 0.5% Triton X-100 (Sigma-Aldrich) in PBS for 5 min at 37°C, blocked with 3% bovine serum albumin (Sigma-Aldrich) in PBS for 5 min at 37°C, and stained with rabbit antiserum to F. tularensis LVS and goat anti-rabbit immunoglobulin conjugated to fluorescein isothiocyanate (both from BD).
cDNA microarray analysis.
To analyze the transcriptional profile of F. tularensis within hepatocytes, cDNA microarrays were used. The F. tularensis microarrays, covering 2,073 and 1,804 open reading frames of the LVS and type A Schu S4 strain genomes, respectively, were provided by the Pathogen Functional Genomics Resource Center at the J. Craig Venter Institute, Rockville, MD, which was sponsored by the National Institute of Allergy and Infectious Diseases. FL83B cells were grown to confluence on 150-mm dishes and incubated with the LVS at an initial MOI of 150 for 24 h. Cultures then were washed, and incubation was continued for another 24 h in the presence of 5 μg/ml of gentamicin. Bacteria were released from the hepatocytes with 10 mg/ml of saponin, 19% (vol/vol) ethanol, and 1% (vol/vol) phenol in water, along with vigorous pipetting and vortexing of the suspension. Hepatocytic debris was removed by centrifugation at 1,000 × g at 4°C for 5 min. Bacteria were pelleted by centrifugation at 10,000 × g at 4°C for 20 min. Total bacterial RNA was isolated using the RNeasy Midi kit (Qiagen, Valencia, CA). LVS organisms grown in MH II broth overnight at 37°C and in the presence of 5% CO2 were processed identically for comparison. cDNA was synthesized and microarrays were processed and analyzed as described by Bakshi et al. (40). Four experiments were performed with dye swaps applied between each experiment to eliminate any possible dye bias.
To verify the microarray data, the same RNA samples were subjected to real-time reverse transcription-PCR (RT-PCR) to assess levels of expression of fslA (FTL_1832) and fslC (FTL_1834). The total RNA was reverse transcribed into cDNA using Qiagen's QuantiTech reverse transcription kit with random hexamers (Qiagen) as the RT primers instead of the primer mix in the kit. Assays were performed in the 7500 real-time PCR system (Applied Biosystems, Carlsbad, CA) using SYBR green as the detector (QuantiTech primer assays; Qiagen). Primers for fslA were primers 1 and 2, and those for fslC were primers 3 and 4 (see Table S1 in the supplemental material). The expression of the 16S rRNA gene was used as an endogenous control (primers 5 and 6). Relative quantification of gene expression was determined according to the method of Pfaffl (41).
Construction of gene deletion mutants and complemented strains.
To produce an in-frame deletion of fslC, two fragments within the gene were amplified using the Expand high-fidelity PCR system (Roche Diagnostics, Indianapolis, IN). Primers for the upstream fragment were primers 7 and 8. Primers for the downstream fragment were primers 9 and 10. Both fragments were purified from agarose gels using the QIAquick gel extraction kit (Qiagen), inserted into the pGEM-T Easy vector (Promega, Madison, WI), and transformed into E. coli DH5α. The fragments were isolated by digesting the plasmids from E. coli DH5α with SalI and NdeI, and then the fragments were ligated at the SalI site of the suicide plasmid pMP590 (42). Primers 11 and 12 on each side of the SalI site on pMP590 were used to verify the correct insertion of up- and downstream fragments. Deletion of fslC from the LVS chromosome was achieved by allelic exchange using the pMP590 vector as described by LoVullo et al. (42). Primers 13 and 14 within the deleted fragment were employed to confirm the success of the procedure. RT-PCR with primers 15 and 16, specific for fslB (FTL_1833), and primers 17 and 18, specific for fslD (FTL_1835), was performed to demonstrate that the in-frame deletion of fslC did not affect the expression of these adjacent genes. In-frame deletion of feoB (FTL_0133) was carried out similarly. The upstream fragment was cloned using primers 19 and 20, and the downstream fragment was cloned using primers 21 and 22. The up- and downstream fragments were linked via an NdeI sequence and were inserted into pMP590 between BamHI and SalI sites. Primers 23 and 24 were used to confirm deletion of feoB in the LVS chromosome. Expression of fslA and fslC in the ΔfeoB mutant grown in MH II broth was compared to that in the LVS by real-time RT-PCR as described above, and levels of the mRNAs were similar between the two strains.
A complementing plasmid carrying fslC, with the shuttle vector pMP633 as the backbone, was constructed and introduced into the ΔfslC strain by electroporation (42). The fslC gene was cloned using primers 25 and 26, which contained NdeI or MluI restriction sites. To optimize expression of fslC, a 518-nucleotide fragment covering part of the fslA gene and its upstream sequence was cloned as the promoter region using primers 27 and 28. The two fragments were then ligated and inserted at the MluI site of pMP633. The correct orientation of insertion was verified using primers 29 and 30 on each side of the MluI site on pMP633. This ΔfslC complemented strain was used for the experiments shown in Fig. 2A and B. Complementation of the ΔfeoB mutant strain via this method was unsuccessful.
Fig 2.
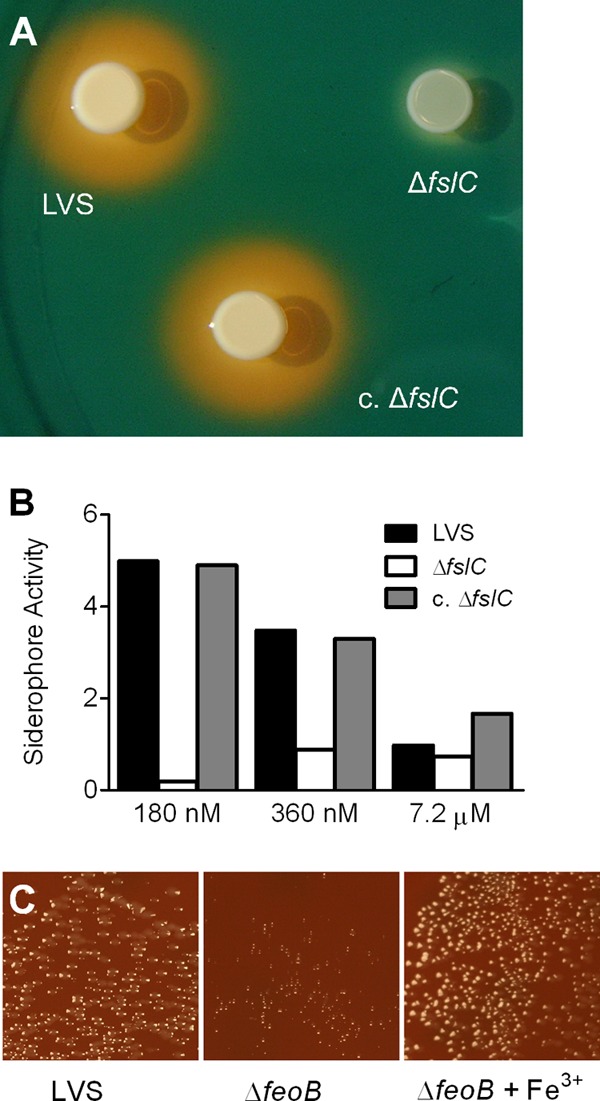
ΔfslC mutant fails to produce siderophore, and growth of the ΔfeoB mutant is diminished on chocolate II agar. (A) Five μl of broth cultures containing the wild-type LVS, the ΔfslC mutant, or the complemented ΔfslC (c. ΔfslC) strain was spotted on CAS plates and incubated for 60 h. Production of siderophore is indicated by orange halos surrounding the bacteria. (B) The wild-type LVS, ΔfslC mutant, or complemented ΔfslC (c. ΔfslC) strain was inoculated into Che-CDM with the indicated concentrations of FeSO4. Siderophore activity in the conditioned media was measured after 120 h and normalized to the OD600. (C) Broth cultures of the LVS or ΔfeoB mutant were plated onto chocolate II agar or chocolate II agar supplemented with ferric pyrophosphate (+Fe3+) and incubated for 48 h.
Complementation of the ΔfeoB mutant strain was achieved by integrating the gene into the chromosome at the site of blaB as described by LoVullo et al. (43). The feoB gene and its promoter were amplified using primers 31 and 32. The resulting fragment was cloned into the pMP815 vector (43) between two blaB flanking sequences. This plasmid then was introduced into the ΔfeoB mutant by electroporation, and blaB in the chromosome was replaced with feoB by allelic exchange. We also used this method with primers 33 and 34 to create a new complemented ΔfslC strain, which produced siderophore (data not shown). The complemented strains produced by chromosomal insertion were used in the experiments shown in Fig. 3 to 8. The pMP590, pMP633, and pMP815 plasmids were generous gifts from Martin S. Pavelka, Jr., University of Rochester Medical Center, Rochester, NY.
Fig 3.

ΔfeoB mutant shows impaired growth in medium containing low levels of iron. Growth of the wild-type LVS, the ΔfeoB and ΔfslC mutants, and their corresponding complemented strains (c. ΔfeoB and c. ΔfslC) was assessed in MH II broth supplemented with ferric pyrophosphate (MH+) (A), MH II broth without addition of ferric pyrophosphate (MH−) (B), Che-CDM containing 7.2 μM FeSO4 (C), or Che-CDM containing 360 nM FeSO4 (D). Growth of the bacterial strains was quantified by measuring the OD600 at various times. All experiments were repeated two more times with similar results.
Fig 8.
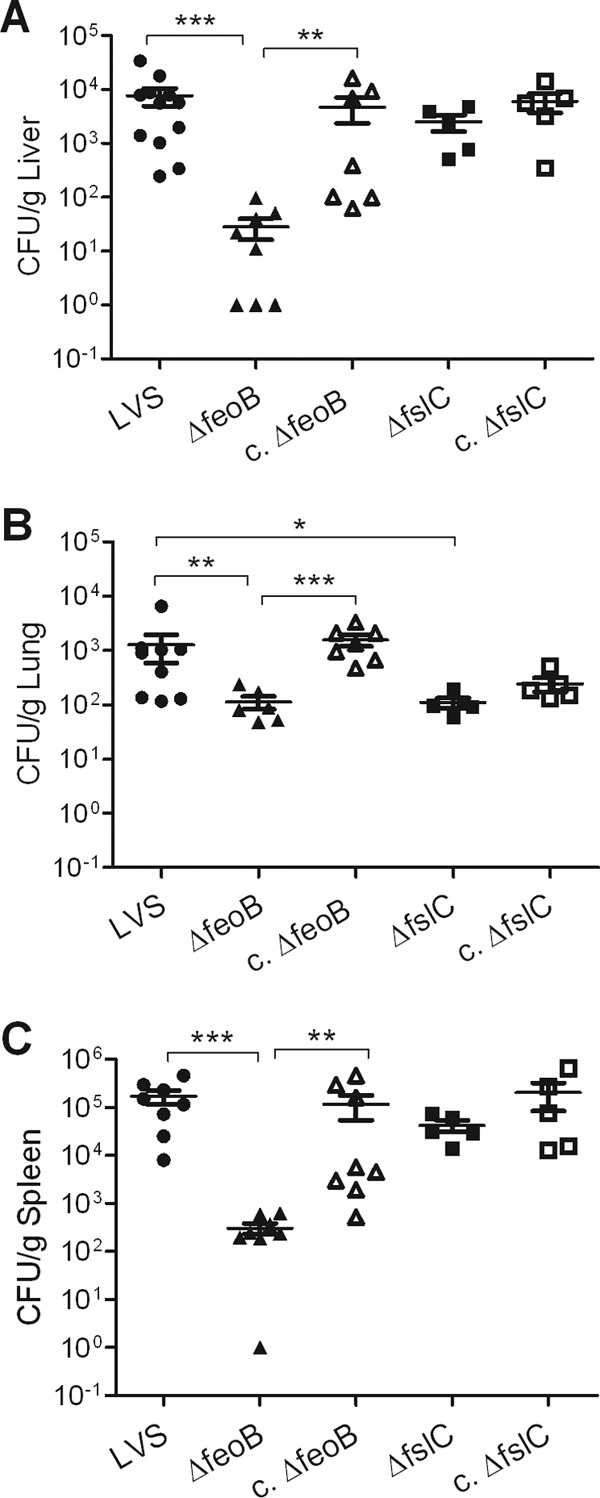
ΔfeoB mutant is defective for colonization of the liver, lungs, and spleen of infected mice. C3H/HeN mice were inoculated intradermally with a sublethal dose (∼3 × 105 CFU) of the wild-type LVS, the ΔfeoB or ΔfslC mutant, or their complemented strains (c. ΔfslC and c. ΔfeoB). Organs were harvested 3 days after infection, and the CFU/g of liver (A), lungs (B), and spleen (C) were determined. The data are combined from three independent experiments. A total of 8 mice per group in two experiments was used in studies of the ΔfeoB and c. ΔfeoB strains; samples from nine organs were lost to contamination. A total of 5 mice per group was used in one experiment with the ΔfslC and c. ΔfslC strains. The bars denote medians ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Growth of bacteria in iron-replete and iron-restricted media.
The MH II broth in which we routinely cultured F. tularensis (designated here MH+) was rich in iron due to supplementation with IsoVitaleX and 335 μM ferric pyrophosphate. We compared the growth of the LVS and mutant strains in MH+ and in MH II broth supplemented with IsoVitalex but not ferric pyrophosphate (designated MH−). To this end, bacterial strains were cultured in MH+ overnight to approximately the same OD600 and washed in PBS. MH+ or MH− then was inoculated with equal numbers of each strain, as confirmed by retrospective plating, and replication with time was monitored by measuring the OD600.
Growth of the bacterial strains also was evaluated using Chamberlain's defined medium (CDM) (44, 45). To prepare Chelex-100-treated CDM (Che-CDM) with known levels of iron, CDM lacking FeSO4 and MgSO4 was treated twice with 1% (wt/vol) Chelex-100 (sodium form; Bio-Rad Laboratories, Hercules, CA) overnight with stirring, and the beads were removed by filtration (46). The medium then was supplemented with essential divalent cations (550 μM MgSO4, 1.5 μM ZnSO4, 0.2 μM CuCl2, 1 μM MnCl2, and 5 μM CaCl2). Che-CDM was prepared with highly purified water and stored in plastic bottles to avoid any contamination with iron. Che-CDM supplemented with 7.2 μM FeSO4 is considered to be replete with iron; that with 720 nM or less is considered to have restricted iron (21). Bacteria of each strain were scraped from chocolate II agar plates and resuspended in Che-CDM with 7.2 μM FeSO4 to the same OD600. Equal volumes were inoculated into Che-CDM with 7.2 μM FeSO4, grown overnight to approximately the same OD600, and washed three times with PBS. The OD600 of suspensions of each strain were adjusted to the same level, and 500 μl was inoculated into Che-CDM supplemented with various amounts of a freshly prepared solution of FeSO4. Bacterial growth then was assessed by determining the OD600 of the cultures at different times.
Measurement of siderophore activity.
Colorimetric chrome azurol S (CAS; Sigma-Aldrich) assays were adapted to a 96-well microtiter plate format, as well as an agar plate format, as previously described (47). If present, siderophores will remove ferric iron from the CAS-Fe3+ complex, causing a change in color from blue to orange that is measurable at OD630. For some experiments, the specific siderophore activity was obtained by normalizing the OD630 to the bacterial densities (OD600). The agar plate format incorporated the CAS reagent into CDM agar. If bacteria grown on CAS plates produce a siderophore, orange halos will form around the colonies.
Uptake of iron by F. tularensis.
The ability of F. tularensis strains to assimilate iron was determined by measuring the total amount of intracellular iron. Bacteria were scraped from chocolate II agar plates, resuspended in Che-CDM to an OD600 of 0.2, and incubated overnight at 37°C with shaking at 100 rpm. The next day, the bacteria were washed, resuspended to an OD600 of 0.2 in Che-CDM supplemented with 7.2 μM FeSO4, and again incubated at 37°C with shaking. At various times, the OD600 of each culture was measured, and three aliquots of equal volumes from each culture were collected. The bacteria were pelleted by centrifugation and washed, and the amount of intracellular iron was determined using a colorimetric ferrozine assay as described previously (48).
Infection of mice with F. tularensis.
To measure the ability of F. tularensis to disseminate to and/or grow in organs of inoculated mice, the wild-type LVS or mutant strains were grown overnight in MH+ broth to exponential phase. Groups of 3 to 5 C3H/HeN mice (Taconic, Hudson, NY) from 6 to 8 weeks old were infected intradermally with sublethal inocula (3 × 105 CFU in 100 μl of PBS). We chose to use intradermal infections for these studies, since they yield more consistent results than intranasal inoculation. The mice were euthanized on day 3 postinfection, and their lungs, livers, and spleens were harvested and weighed. Organs were homogenized in PBS in sterile Whirl-Pak bags (Nasco, Fort Atkinson, WI), and serial dilutions of the homogenates were plated on chocolate II agar to determine the CFU per g of tissue. To test the ability of the LVS and mutant strains to cause fatal disease, groups of 5 mice were infected intradermally with 2 × 107 CFU. In our experience, this is the smallest inoculum of the LVS that reproducibly causes death of wild-type animals. The mice then were monitored, and the time of death was recorded. The infectious doses for all experiments were confirmed by retrospective plating. These studies were approved by Stony Brook University's Institutional Animal Care and Use Committee.
Statistics.
The results from organ burden assays were analyzed using the Kruskal-Wallis test for nonparametric data, followed by the Dunn's multiple-comparison posttest. The log-rank test was used to analyze survival of infected mice. Statistical significance of all other data was determined using an unpaired analysis of variance and the Tukey-Kramer multiple-comparison posttest (Prism version 5.00; GraphPad Software, San Diego, CA). With Bonferroni's correction, a difference between survival curves was considered significant if the P value was less than 0.008. For all other studies, the criterion for significance was a P value of less than 0.05.
Microarray data accession number.
All microarray data described in this study have been deposited in the National Center for Biotechnology Information's Gene Expression Omnibus (49) and are accessible through GEO series accession number GSE43731 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43731).
RESULTS
Francisella tularensis LVS grows in hepatocytes.
It has been reported that F. tularensis replicates in murine hepatocytes in vivo (6, 10, 12, 31, 34), as well as in cultured rat hepatocytes (14) and the human HepG2 hepatocytic cell line (9). We found that the LVS also grew to high numbers in several other well-established hepatocytic cell lines. These lines were incubated with the LVS at an initial MOI of 150 for 24 h. Extracellular bacteria then were washed away, and incubation was continued for another 24 h in medium with gentamicin to kill any remaining extracellular organisms. Visualization of intracellular bacteria in murine FL83B hepatocytes by fluorescence microscopy revealed that cells became infected during the first 24 h of coincubation, but relatively small amounts of intracellular bacteria were observed (Fig. 1A). However, the number of intracellular bacteria increased greatly after an additional 24 h of culture (Fig. 1B). Human HH4 hepatocytes also became infected after 24 h of incubation with the LVS (Fig. 1C), and we observed substantial growth of intracellular bacteria following another day of culture (Fig. 1D). Replication was quantitated using CFU assays. After a 2-h period of infection at an MOI of 1,000, the number of viable intracellular bacteria in FL83B cells increased about three logs in the subsequent 28 h (Fig. 1E). The LVS also infected and grew well in murine AML12 and human HepG2/C3A hepatocytic lines (data not shown). These results demonstrate that murine and human hepatocytic cell lines generally are capable of supporting extensive replication of the LVS.
Fig 1.
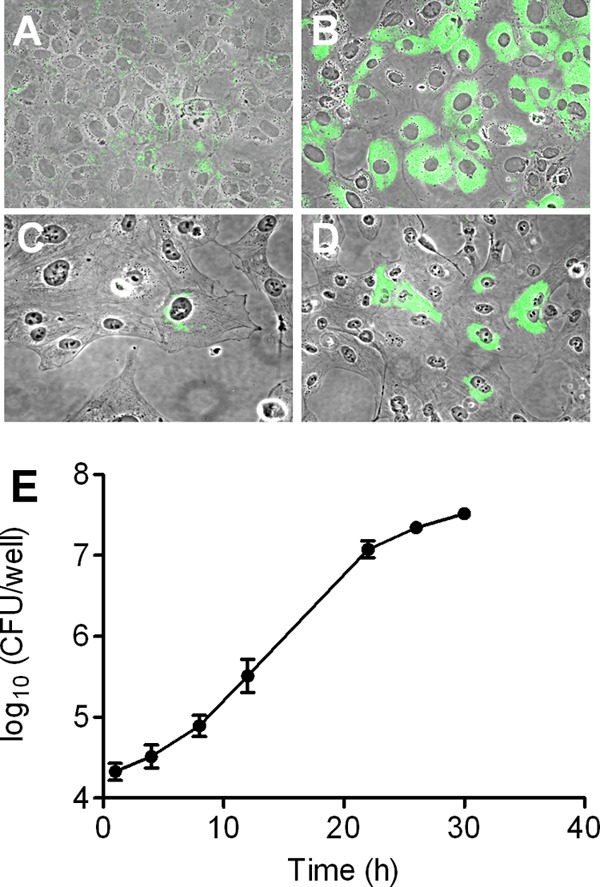
Hepatocytes support extensive intracellular growth of F. tularensis. Murine FL83B (A and B) or human HH4 (C and D) hepatocytes were incubated with the LVS at an initial MOI of 150 for 24 h, and the extracellular bacteria were removed. The cells then were fixed for visualization of bacteria with an antibody to F. tularensis either immediately (A and C) or after an additional 24-h incubation (B and D). The bacteria appear green in these merged phase-contrast and immunofluorescent images (original magnification, ×400). (E) FL83B hepatocytic cells were incubated with the LVS at an MOI of 1,000 for 2 h, extracellular bacteria were removed, and the cells were cultured in medium with gentamicin for the indicated times. Hepatocytes were lysed, and the amounts of viable bacteria were determined by a CFU assay. Data are the means ± standard deviations (SD) from three replicate samples. This experiment was repeated twice more with similar results.
The fslC gene is highly expressed by F. tularensis growing in hepatocytes and contributes to uptake of iron.
The global gene expression profile of the LVS growing in hepatocytes was assessed to gain insight into how the bacterium adapts to this intracellular niche. Murine FL83B hepatocytes were incubated with the LVS at an MOI of 150 for 24 h, washed, and cultured in medium with gentamicin for an additional 24 h. The intracellular bacteria then were released by selective lysis of the hepatocytes. Total RNA was isolated from the intracellular bacteria or bacteria grown in MH+, and the expression patterns were compared using cDNA microarrays. A summary of these data is provided in the supplemental material. Four genes of the fsl operon, fslA, fslB, fslC, and fslD, were the most highly upregulated in the intracellular organisms, ranging from 6.7-fold for fslB to 13.1-fold for fslC (see Table S2 in the supplemental material). The upregulation of fslA and fslC was verified by real-time RT-PCR, with fold changes in mRNA levels of 33.3 ± 12.7 and 20.2 ± 5.8, respectively. Similarly, expression of the fsl operon is upregulated when the type A F. tularensis Schu S4 strain is grown in primary human macrophages (50).
FslA of F. tularensis has been shown to be involved in production of a siderophore (18, 21). Since it is encoded by a gene in the same operon, FslC might have a similar biological function. To test this premise, fslC was inactivated by in-frame deletion. Cultivation of bacteria on CAS plates showed that the wild-type LVS produced a siderophore, but the ΔfslC mutant did not. Complementation by expression of fslC in trans restored the production of siderophore to the same level as that in the wild-type LVS (Fig. 2A), whereas the empty vector did not rescue the phenotype (data not shown). Similar results were observed when liquid media conditioned by the wild-type LVS, the ΔfslC mutant, or the complemented ΔfslC bacteria were subjected to a CAS assay. Che-CDM conditioned by the ΔfslC mutant showed little siderophore activity, but both the wild-type LVS and the complemented ΔfslC strain produced large amounts of siderophore in Che-CDM with limited iron (180 or 360 nM). As would be expected for an iron-regulated molecule, the level of siderophore decreased when these strains were cultured in Che-CDM with a high concentration (7.2 μM) of iron (Fig. 2B).
FeoB is necessary for efficient acquisition of iron by the LVS.
Iron is an essential nutrient for the LVS (15, 16). We observed that the ΔfslC mutant grew similarly to wild-type bacteria in MH+ (Fig. 3A), raising the possibility that F. tularensis possesses an additional mechanism for acquisition of iron. A gene encoding a putative ferrous iron transporter, feoB (FTL_0133), was identified in the genome of the LVS (19, 21). In-frame deletion of feoB significantly slowed the growth of bacteria on chocolate II agar, as shown by substantially smaller-sized colonies compared to those of the wild-type LVS (Fig. 2C). Supplementation of the agar with ferric pyrophosphate restored the normal replication of the ΔfeoB mutant (Fig. 2C). However, supplementation did not alter growth of the wild-type LVS (data not shown), suggesting that the small colony size of the ΔfeoB strain was due to its inability to acquire iron efficiently. Eleven attempts to generate F. tularensis lacking both fslC and feoB failed. This failure argues that bacteria that lack the two genes are not viable and strongly implies that both FslC and FeoB are involved in acquisition of iron.
Growth of the ΔfeoB mutant is inhibited in iron-restricted broth.
The wild-type LVS, ΔfeoB mutant, and ΔfslC mutant all replicated well in either MH II broth supplemented with IsoVitalex and ferric pyrophosphate (Fig. 3A) or MH II broth containing only IsoVitalex (Fig. 3B). Both of these media are relatively rich in iron. To test the capacity of the strains to grow in an environment where iron is limited, we measured their replication in Che-CDM with defined concentrations of iron. Although ferrous iron was added to the medium, oxidation results in a mixture of the ferrous and ferric forms. The wild-type LVS, the mutant strains, and their complemented counterparts all grew well in medium replete with iron (7.2 μM) (Fig. 3C). In the presence of limiting amounts of iron (360 nM), the ΔfslC mutant grew similarly to wild-type organisms (Fig. 3D). In contrast, the ΔfeoB strain displayed almost no growth during the first 20 h of culture but, with time, reached a density comparable to that of wild-type bacteria. The lag in growth of the ΔfeoB mutant was eliminated by complementation, demonstrating that the phenotype is specific to loss of feoB (Fig. 3D).
We considered the possibility that increased growth of the ΔfeoB mutant after 20 h in medium with low iron results from accumulation of siderophore. To test this premise, replication of the bacteria and secretion of siderophore were measured in the same cultures. Indeed, growth and production of siderophore increased in parallel, whether the ΔfeoB mutant was cultured in medium that was iron replete (Fig. 4A and B) or iron restricted (Fig. 4C and D). Together, these data indicate that FeoB is a major mechanism for uptake of iron by F. tularensis, but in vitro its loss can be overcome by secretion of siderophore.
Fig 4.

Production of siderophore by F. tularensis lacking FeoB is delayed in iron-restricted medium. The wild-type LVS or ΔfeoB mutant was inoculated into Che-CDM containing 7.2 μM (A and B) or 360 nM (C and D) FeSO4. At the indicated times the OD600 of each culture was measured (A and C), and the total siderophore activity in the conditioned medium was assayed (B and D). Data represent one experiment, which was repeated with similar results.
Loss of FeoB diminishes uptake of iron by F. tularensis.
To determine directly the contribution of FslC and FeoB to assimilation of iron, the wild-type LVS, the mutants, and their complemented strains were starved for this nutrient by growing them overnight in Che-CDM. Equal numbers of bacteria then were inoculated into Che-CDM containing 7.2 μM FeSO4, and replication and amounts of intracellular iron, as determined by a ferrozine assay (48), were measured in parallel. Growth of all strains was similar throughout the course of the experiment (Fig. 5A). However, normalization of the amount of intracellular iron in each culture to the number of bacteria revealed that individual ΔfeoB organisms contained significantly less iron than did wild-type or ΔfslC organisms (P < 0.001). Whereas amounts in the wild-type LVS and the ΔfslC bacteria increased similarly from 0 to 11 h and declined thereafter, the level in the ΔfeoB organisms did not rise at all. Notably, the phenotype of the ΔfeoB mutant was restored to that of the wild-type strain by complementation (Fig. 5B). Thus, loss of feoB reduces the ability of F. tularensis to take up iron from the environment.
Fig 5.
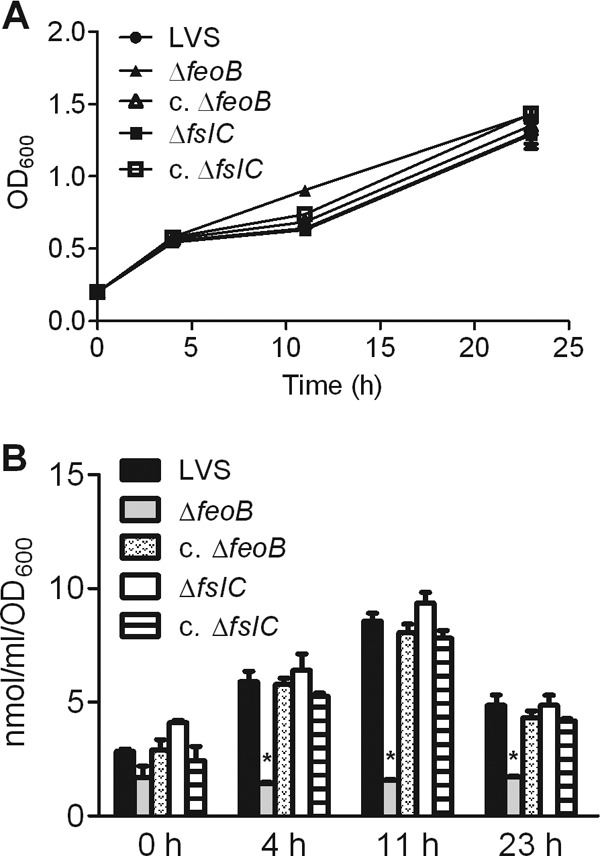
ΔfeoB mutant is deficient in its ability to take up extracellular iron. The wild-type LVS, ΔfslC and ΔfeoB mutants, and their complemented strains (c. ΔfslC and c. ΔfeoB) were grown overnight in Che-CDM. The strains were then inoculated into CDM supplemented with 7.2 μM FeSO4. At the indicated times, the growth of the bacteria was determined by measuring the OD600 (A), and the total intracellular iron of each strain was evaluated using a colorimetric ferrozine assay (B). Bars in panel B represent the means ± SD from 3 replicate samples. *, P < 0.001 compared to all other groups at the same time point. This study was repeated for the wild-type LVS and mutant strains, yielding similar results.
Loss of FeoB limits the growth of F. tularensis in cultured hepatocytes and epithelial cells.
To address whether FslC and FeoB are important for growth in host cells, murine FL83B hepatocytes, human A549 lung epithelial cells, or human monocyte-derived macrophages were incubated with the wild-type LVS, ΔfslC mutant, ΔfeoB mutant, or complemented strains. Uptake of all strains by the hepatocytes, epithelial cells, or macrophages was similar (Fig. 6). Whereas growth of the ΔfslC mutant in hepatocytes (Fig. 6A) and epithelial cells (Fig. 6B) was comparable to that of the wild-type LVS, intracellular replication of the ΔfeoB mutant in these cell types was negligible. In both cases, complementation rescued the ability of the ΔfeoB mutant to replicate. In contrast, all strains grew equivalently in the macrophages (Fig. 6C). These results suggest that FeoB is necessary for efficient replication of F. tularensis in some, but not all, host cells.
Fig 6.
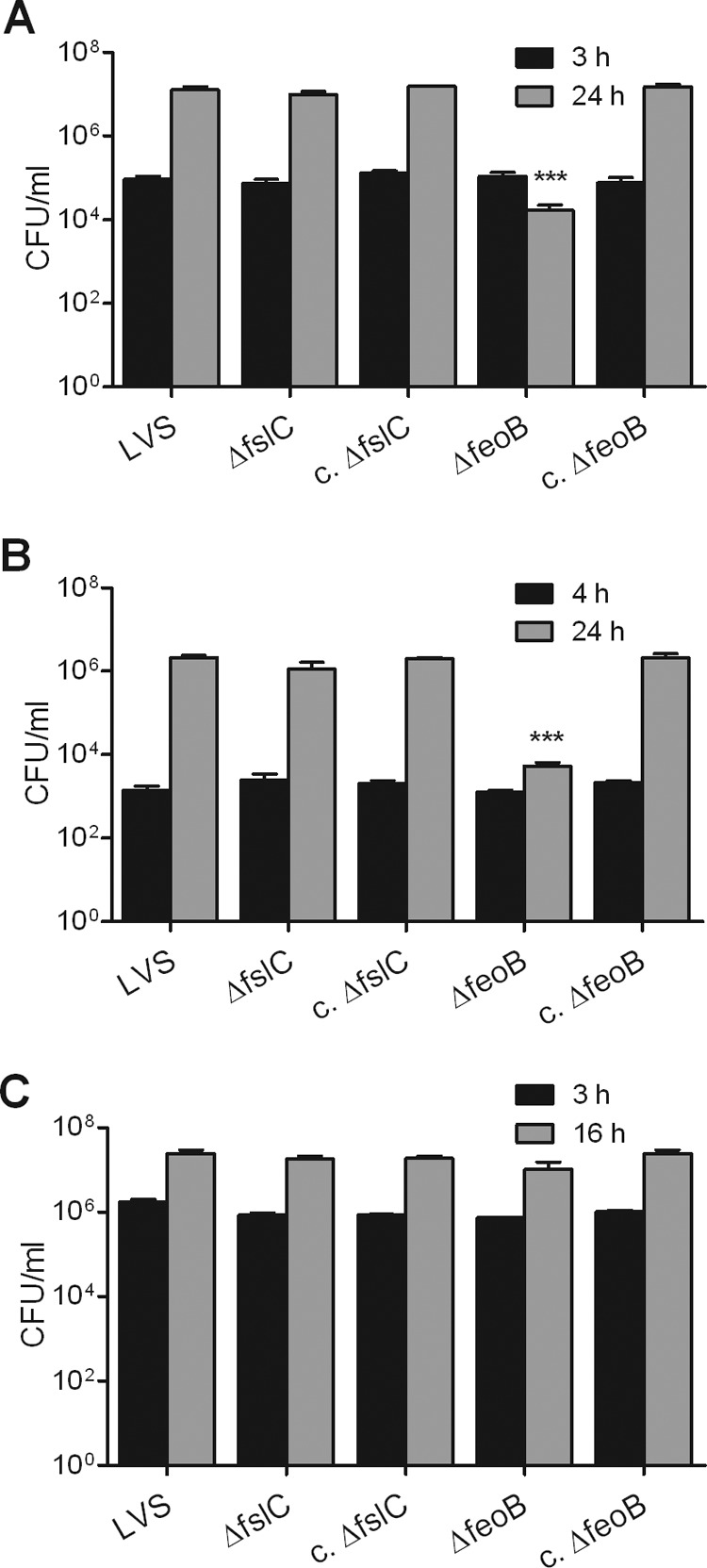
ΔfeoB mutant shows impaired replication in hepatocytes and epithelial cells but not in macrophages. Murine FL83B hepatocytes (A), human A549 lung epithelial cells (B), or human monocyte-derived macrophages (C) were infected with the wild-type LVS, ΔfeoB and ΔfslC mutants, or their corresponding complemented strains (c. ΔfslC and c. ΔfeoB) as described in Materials and Methods. They were then treated with gentamicin for 1 h to kill extracellular bacteria. Some samples were lysed immediately after treatment with antibiotic to measure uptake using a CFU assay. Other samples were further incubated for a total of 24 h (A and B) or 16 h (C) to permit replication. Bars represent the means ± SD from 3 replicate samples. ***, P < 0.001 compared to replication of all other strains. For each type of host cell, there was no significant difference in uptake of the various bacterial strains. The experiments shown in panels A and B were repeated one or two more times, respectively, yielding similar results. Similar growth of the wild-type LVS and ΔfeoB mutant in human macrophages, as shown in panel C, was confirmed in three additional experiments.
The ΔfeoB mutant retains its capacity to cause lethal disease in mice but has a diminished ability to infect key organs.
We assessed the contribution of fslC and feoB to the virulence of F. tularensis by infecting C3H/HeN mice intradermally with a lethal amount of the LVS or comparable numbers of the mutant bacteria. The mice then were monitored for time of death. All mice that received either the wild-type LVS or ΔfeoB mutant succumbed by day 7 postinfection, whereas 30% of mice inoculated with the ΔfslC strain survived until the experiment was ended at day 20 postinfection (Fig. 7). When pairwise comparisons were made, only the survival curves for the ΔfslC mutant and the LVS differed significantly (P = 0.0009). However, complementation did not restore the virulence of the ΔfslC strain (Fig. 7). To investigate whether the mutants have a detectable phenotype when administered at a lower dose, bacterial burdens were measured in target organs after mice were inoculated intradermally with a sublethal amount of the mutants, their complemented counterparts, or the wild-type LVS. Three days after infection, livers and spleens contained comparable numbers of the ΔfslC mutant and wild-type organisms (Fig. 8A and C). In the lung, there was a modest decrease in ΔfslC organisms compared to wild-type bacteria (P < 0.05), but there was no significant difference between the ΔfslC mutant and its complemented strain (Fig. 8B). In striking contrast, the ΔfeoB mutant showed substantially reduced burdens in all three organs (P < 0.01), a defect that was reversed by complementation (Fig. 8). These results indicate that the loss of feoB does not prevent F. tularensis from causing lethal disease in mice. Nonetheless, the ΔfeoB mutant strain has a markedly diminished ability to establish systemic infection after intradermal inoculation of a sublethal dose.
Fig 7.
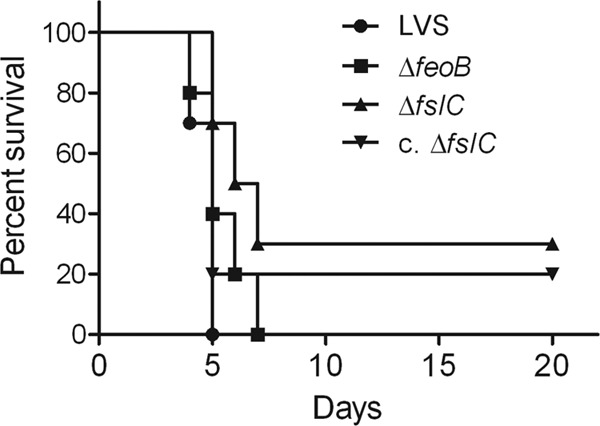
Loss of FeoB does not prevent F. tularensis LVS from causing lethal disease in mice. C3H/HeN mice were infected intradermally with ∼2.0 × 107 bacteria of the wild-type LVS, the ΔfeoB or ΔfslC strain, or the complemented ΔfslC strain (c. ΔfslC). Mice were monitored for 20 days, and the time of death was recorded. Data are combined from two experiments, each of which included 5 mice per group. The wild-type LVS and the ΔfslC and ΔfeoB mutants were tested in the first experiment; the second experiment studied the wild-type LVS, the ΔfslC strain, and the complemented ΔfslC strain. Pairwise comparison revealed a significant difference only between the wild-type LVS and ΔfslC groups (P = 0.0009).
DISCUSSION
In this study, we demonstrated that hepatocytic cells of murine and human origin generally supported extensive intracellular growth of F. tularensis LVS, and we analyzed the gene expression of the bacteria within hepatocytes. Four genes of the fsl operon were the most highly upregulated. Deletion of fslC eliminated production of siderophore, which is used for importation of ferric iron. However, the ΔfslC strain grew similarly to the wild-type LVS in iron-rich broth, hepatocytes, epithelial cells, and macrophages, strongly suggesting that the LVS possesses other means for acquisition of iron. Our results assign such a role to FeoB.
The involvement of the fsl operon in acquisition of iron by Francisella has been investigated previously. The genes of this operon are the most highly upregulated when F. tularensis LVS grows in iron-restricted conditions (18). FslA is required for production of siderophore in the LVS (21) and the highly virulent F. tularensis Schu S4 strain (46). Francisella novicida rarely causes human disease but retains pathogenicity in mice (51). In F. novicida, the genes fslA, fslB, and fslC all have been shown to be required for optimal production of siderophore (22), and our data demonstrate that the FslC protein of the LVS is similarly needed (Fig. 2). FslE has been identified as an outer membrane protein (52). Unlike that of other F. novicida fsl mutants, the growth of an ΔfslE strain cannot be restored by addition of exogenous siderophore (22). Similarly, a ΔfslE mutant of the Schu S4 strain makes, but cannot use, the siderophore, indicating that FslE is a siderophore receptor (23). However, fslE is dispensable for utilization of siderophore in the LVS, since this strain possesses a paralogous fusion gene, fupA/B (FTL_0439), that plays the predominant role in uptake of siderophore-bound iron (53).
We found that the growth of the LVS ΔfslC mutant was similar to that of wild-type bacteria under a variety of conditions, suggesting that the LVS can obtain iron by mechanisms that do not involve a siderophore. The Feo transport system is used by many other bacteria for acquisition of ferrous iron. Of 250 completely sequenced bacterial genomes, 46% possess genes of the Feo system (29). Mutation of feo genes in E. coli results in defective import of ferrous iron, lower levels of intracellular iron, and impaired ability to colonize the mouse intestine (25, 54). The structure of the transmembrane domain of FeoB and the mechanism by which this protein transports iron are unknown. Genes encoding FeoA and FeoB were identified in the LVS (19, 21), but their biological functions had not been determined. We therefore carried out in-frame deletion of feoB to explore its potential role in acquisition of iron. Similar growth of the wild-type LVS, the ΔfslC mutant, and the ΔfeoB mutant in media replete with iron revealed that neither FslC nor FeoB by itself is essential for acquisition of iron. The loss of FeoB, however, affected growth in defined medium containing restricted iron (Fig. 3D and 4C). Although replication of the ΔfeoB mutant was greatly delayed when iron was limited, a burst of growth occurred eventually, and the density of the culture ultimately reached that of the wild-type LVS. This burst correlated with an increase in siderophore activity in the medium (Fig. 4C and D). The ΔfeoB organisms also contained lower levels of iron than both the wild-type LVS and the ΔfslC strain (Fig. 5B). Collectively, these results establish a role for FeoB in acquisition of iron by F. tularensis. Despite repeated attempts, we could not delete fslC and feoB simultaneously. This failure suggests that siderophore and FeoB are the major mechanisms by which F. tularensis takes up iron.
It has been well documented that F. tularensis invades hepatocytes in vivo. Inoculation of mice with the LVS by the intravenous or intradermal route results in infection of the liver, where the bacteria grow in both Kuppfer cells and hepatocytes (6, 10). In the absence of gamma interferon, replication of the LVS in hepatocytes in vivo is extensive (34), and a highly virulent type A strain shows enhanced growth in hepatocytes compared to the LVS in vitro and in vivo (12, 14). Our observations in cultured hepatocytic lines confirm that this cell type offers a hospitable niche for F. tularensis. The mechanisms by which hepatocytes foster growth of F. tularensis are not known. However, our results and those of others indicate that the siderophore-ferric iron system is not essential for intracellular replication. Growth of an LVS ΔfslA mutant and a Schu S4 ΔfslE mutant in murine J774 macrophage-like cells is the same as that of the wild-type parental bacteria (18, 55). Similarly, growth of the ΔfslC mutant in hepatocytes, epithelial cells, and macrophages was similar to that of the wild-type LVS (Fig. 6). To avoid toxicity, levels of free iron in hosts are tightly regulated. Extracellular iron is bound to proteins in the ferric form. Free ferrous iron is needed for various physiological and pathological processes within host cells, but any excess is oxidized and either exported or stored bound to ferritin (56). Our data suggest that intracellular F. tularensis can acquire this ferrous iron via FeoB, since replication of the ΔfeoB mutant was greatly reduced in both hepatocytic and epithelial cells (Fig. 6A and B). Notably, replication of the ΔfeoB mutant was similar to that of the wild-type LVS in human macrophages (Fig. 6C). This result raises the possibility that the amount of available iron differs in various types of cells and that loss of FeoB impairs the ability of intracellular F. tularensis to acquire iron when amounts are limited.
We evaluated the impact of deletion of feoB or fslC on the virulence of F. tularensis in mice. The ΔfeoB mutant and the wild-type LVS were equally capable of causing fatal disease, at least by the intradermal route and using the dose tested. Fewer mice died when infected with a comparable inoculum of the ΔfslC mutant, but complementation did not alter this outcome (Fig. 7). Similarly, complementation did not reverse the diminished bacterial loads in the lungs of mice infected with a sublethal amount of the ΔfslC mutant (Fig. 8B). The complemented ΔfslC strain secreted siderophore in vitro, but it is possible that complementation was not effective in the more stringent in vivo environment. Regardless, we cannot conclude with certainty that the diminished virulence of the ΔfslC strain was specific to loss of the gene. Notably, ablation of fslA was previously reported to have no effect on the ability of F. tularensis Schu S4 to kill mice infected intradermally (46). Although deletion of feoB produced no phenotype in the survival assay, we observed that loss of this gene resulted in significantly reduced bacterial burdens in the lungs, liver, and spleen of mice infected with a sublethal dose. Moreover, these reductions were reversed by complementation (Fig. 8). Our observation is in agreement with the previous finding that an feoB::Tn5 mutant of the LVS is defective for replication in the lungs of mice after intranasal inoculation (30). Collectively, these results indicate that FeoB contributes to virulence of F. tularensis in mice, but the consequences of disrupting its function become less important when large numbers of bacteria are administered.
In other Gram-negative bacteria, FeoB is located in the inner membrane (29). This observation raises the question of how iron crosses the outer membrane and enters the periplasm of F. tularensis for subsequent transport via FeoB. In the Schu S4 strain, this function is carried out by FupA, an outer membrane protein that is required for high-affinity uptake of ferrous iron (55). As mentioned, the LVS does not have a distinct fupA gene but rather an fupA/B hybrid gene. Whether transport of ferrous iron across the outer membrane of the LVS is carried out by FupA/B or a different molecule remains to be determined. If fupA and feoB are members of a sequential pathway, then mutants in either gene should have the same phenotype. However, Schu S4 ΔfupA strains are attenuated in survival studies in mice (46, 55), whereas our LVS ΔfeoB mutant retained wild-type virulence in this assay (Fig. 7). This seeming disparity may reflect differences in the ways by which the LVS and Schu S4 strain acquire iron, or it may show that FupA has functions in addition to uptake of iron. In support of the latter possibility, the FupA homologue of F. novicida has been implicated in both immune evasion and maintenance of integrity of the bacterial outer membrane (57).
F. tularensis likely requires more than one mechanism to obtain iron due to the many different environments in which it grows. F. tularensis has been isolated from water and moist soil. The bacterium also has a broad range of hosts; it infects over 250 animal species, including mammals, birds, fish, invertebrates, and amoebae (51, 58). In mice, viable F. tularensis is found both intracellularly and extracellularly in the blood throughout the course of infection (59). We determined here that FslC and FeoB both contribute to the acquisition of iron, but their functions are not completely redundant. FslC is required for production of a siderophore, which likely provides the bulk of iron when the bacterium grows in niches where ferric iron predominates. FeoB appears more important for replication of the bacterium within cells, at least in the case of hepatocytes and epithelial cells. FeoB also contributes to growth of F. tularensis before sufficient amounts of siderophore accumulate. Thus, multiple mechanisms for acquisition of iron appear to benefit F. tularensis by allowing it to flourish in diverse environments.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by Public Health Service grant P01 AI055621 from the National Institute of Allergy and Infectious Diseases (NIAID). C.A.T.-C. was supported by grant T32 AI007539 from the NIAID.
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIAID or the National Institutes of Health.
We are grateful to Jaime Italo Bhalla, Indralatha Jayatilaka, Varya Kirillov, and Gloria Monsalve for expert technical assistance. We thank Hong Wang for processing of cDNA microarrays, Nelson Fausto for providing HH4 human hepatocytic cells, and Martin S. Pavelka, Jr., for providing plasmids for genetic manipulation of F. tularensis.
Footnotes
Published ahead of print 28 May 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00170-13.
REFERENCES
- 1. Tärnvik A, Berglund L. 2003. Tularaemia. Eur. Respir. J. 21:361–373 [DOI] [PubMed] [Google Scholar]
- 2. Ellis J, Oyston PCF, Green M, Titball RW. 2002. Tularemia. Clin. Microbiol. Rev. 15:631–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saslaw S, Eigelsbach HT, Prior JA, Wilson HE, Carhart S. 1961. Tularemia vaccine study. II. Respiratory challenge. Arch. Intern. Med. 107:702–714 [DOI] [PubMed] [Google Scholar]
- 4. Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O'Toole T, Parker G, Perl TM, Russell PK, Tonat K. 2001. Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773 [DOI] [PubMed] [Google Scholar]
- 5. Fortier AH, Slayter MV, Ziemba R, Meltzer MS, Nacy CA. 1991. Live vaccine strain of Francisella tularensis: infection and immunity in mice. Infect. Immun. 59:2922–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Conlan JW, North RJ. 1992. Early pathogenesis of infection in the liver with the facultative intracellular bacteria Listeria monocytogenes, Francisella tularensis, and Salmonella typhimurium involves lysis of infected hepatocytes by leukocytes. Infect. Immun. 60:5164–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bolger CE, Forestal CA, Italo JK, Benach JL, Furie MB. 2005. The live vaccine strain of Francisella tularensis replicates in human and murine macrophages but induces only the human cells to secrete proinflammatory cytokines. J. Leukoc. Biol. 77:893–897 [DOI] [PubMed] [Google Scholar]
- 8. Bosio CM, Dow SW. 2005. Francisella tularensis induces aberrant activation of pulmonary dendritic cells. J. Immunol. 175:6792–6801 [DOI] [PubMed] [Google Scholar]
- 9. Qin A, Mann BJ. 2006. Identification of transposon insertion mutants of Francisella tularensis tularensis strain Schu S4 deficient in intracellular replication in the hepatic cell line HepG2. BMC Microbiol. 6:69. 10.1186/1471-2180-6-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rasmussen JW, Cello J, Gil H, Forestal CA, Furie MB, Thanassi DG, Benach JL. 2006. Mac-1+ cells are the predominant subset in the early hepatic lesions of mice infected with Francisella tularensis. Infect. Immun. 74:6590–6598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hall JD, Craven RR, Fuller JR, Pickles RJ, Kawula TH. 2007. Francisella tularensis replicates within alveolar type II epithelial cells in vitro and in vivo following inhalation. Infect. Immun. 75:1034–1039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wickstrum JR, Bokhari SM, Fischer JL, Pinson DM, Yeh HW, Horvat RT, Parmely MJ. 2009. Francisella tularensis induces extensive caspase-3 activation and apoptotic cell death in the tissues of infected mice. Infect. Immun. 77:4827–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pan X, Tamilselvam B, Hansen EJ, Daefler S. 2010. Modulation of iron homeostasis in macrophages by bacterial intracellular pathogens. BMC Microbiol. 10:64. 10.1186/1471-2180-10-64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ray HJ, Chu P, Wu TH, Lyons CR, Murthy AK, Guentzel MN, Klose KE, Arulanandam BP. 2010. The Fischer 344 rat reflects human susceptibility to Francisella pulmonary challenge and provides a new platform for virulence and protection studies. PLoS One 5:e9952. 10.1371/journal.pone.0009952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tresselt HB, Ward MK. 1964. Blood-free medium for the rapid growth of Pasteurella tularensis. Appl. Microbiol. 12:504–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lenco J, Hubalek M, Larsson P, Fucikova A, Brychta M, Macela A, Stulik J. 2007. Proteomics analysis of the Francisella tularensis LVS response to iron restriction: induction of the F. tularensis pathogenicity island proteins IglABC. FEMS Microbiol. Lett. 269:11–21 [DOI] [PubMed] [Google Scholar]
- 17. Nano FE, Schmerk C. 2007. The Francisella pathogenicity island. Ann. N. Y. Acad. Sci. 1105:122–137 [DOI] [PubMed] [Google Scholar]
- 18. Deng K, Blick RJ, Liu W, Hansen EJ. 2006. Identification of Francisella tularensis genes affected by iron limitation. Infect. Immun. 74:4224–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lindgren H, Honn M, Salomonsson E, Kuoppa K, Forsberg A, Sjöstedt A. 2011. Iron content differs between Francisella tularensis subspecies tularensis and subspecies holarctica strains and correlates to their susceptibility to H2O2-induced killing. Infect. Immun. 79:1218–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Andrews SC, Robinson AK, Rodriguez-Quinones F. 2003. Bacterial iron homeostasis. FEMS Microbiol. Rev. 27:215–237 [DOI] [PubMed] [Google Scholar]
- 21. Sullivan JT, Jeffery EF, Shannon JD, Ramakrishnan G. 2006. Characterization of the siderophore of Francisella tularensis and role of fslA in siderophore production. J. Bacteriol. 188:3785–3795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kiss K, Liu W, Huntley JF, Norgard MV, Hansen EJ. 2008. Characterization of fig operon mutants of Francisella novicida U112. FEMS Microbiol. Lett. 285:270–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ramakrishnan G, Meeker A, Dragulev B. 2008. fslE is necessary for siderophore-mediated iron acquisition in Francisella tularensis Schu S4. J. Bacteriol. 190:5353–5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hantke K. 2003. Is the bacterial ferrous iron transporter FeoB a living fossil? Trends Microbiol. 11:192–195 [DOI] [PubMed] [Google Scholar]
- 25. Kammler M, Schon C, Hantke K. 1993. Characterization of the ferrous iron uptake system of Escherichia coli. J. Bacteriol. 175:6212–6219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Velayudhan J, Hughes NJ, McColm AA, Bagshaw J, Clayton CL, Andrews SC, Kelly DJ. 2000. Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol. Microbiol. 37:274–286 [DOI] [PubMed] [Google Scholar]
- 27. Naikare H, Palyada K, Panciera R, Marlow D, Stintzi A. 2006. Major role for FeoB in Campylobacter jejuni ferrous iron acquisition, gut colonization, and intracellular survival. Infect. Immun. 74:5433–5444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Perry RD, Mier I, Jr, Fetherston JD. 2007. Roles of the Yfe and Feo transporters of Yersinia pestis in iron uptake and intracellular growth. Biometals 20:699–703 [DOI] [PubMed] [Google Scholar]
- 29. Cartron ML, Maddocks S, Gillingham P, Craven CJ, Andrews SC. 2006. Feo-transport of ferrous iron into bacteria. Biometals 19:143–157 [DOI] [PubMed] [Google Scholar]
- 30. Su J, Yang J, Zhao D, Kawula TH, Banas JA, Zhang JR. 2007. Genome-wide identification of Francisella tularensis virulence determinants. Infect. Immun. 75:3089–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Conlan JW, Chen W, Shen H, Webb A, Kuo Lee R. 2003. Experimental tularemia in mice challenged by aerosol or intradermally with virulent strains of Francisella tularensis: bacteriologic and histopathologic studies. Microb. Pathog. 34:239–248 [DOI] [PubMed] [Google Scholar]
- 32. Wickstrum JR, Hong KJ, Bokhari S, Reed N, McWilliams N, Horvat RT, Parmely MJ. 2007. Coactivating signals for the hepatic lymphocyte gamma interferon response to Francisella tularensis. Infect. Immun. 75:1335–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Collazo CM, Meierovics AI, De PR, Wu TH, Lyons CR, Elkins KL. 2009. T cells from lungs and livers of Francisella tularensis-immune mice control the growth of intracellular bacteria. Infect. Immun. 77:2010–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bokhari SM, Kim KJ, Pinson DM, Slusser J, Yeh HW, Parmely MJ. 2008. NK cells and gamma interferon coordinate the formation and function of hepatic granulomas in mice infected with the Francisella tularensis live vaccine strain. Infect. Immun. 76:1379–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gao B, Jeong WI, Tian Z. 2008. Liver: an organ with predominant innate immunity. Hepatology 47:729–736 [DOI] [PubMed] [Google Scholar]
- 36. Noah CE, Malik M, Bublitz DC, Camenares D, Sellati TJ, Benach JL, Furie MB. 2010. GroEL and lipopolysaccharide from Francisella tularensis live vaccine strain synergistically activate human macrophages. Infect. Immun. 78:1797–1806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breslow JL, Sloan HR, Ferrans VJ, Anderson JL, Levy RI. 1973. Characterization of the mouse liver cell line FL83B. Exp. Cell Res. 78:441–453 [DOI] [PubMed] [Google Scholar]
- 38. Wu JC, Merlino G, Fausto N. 1994. Establishment and characterization of differentiated, nontransformed hepatocyte cell lines derived from mice transgenic for transforming growth factor alpha. Proc. Natl. Acad. Sci. U. S. A. 91:674–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yan W, Lee H, Deutsch EW, Lazaro CA, Tang W, Chen E, Fausto N, Katze MG, Aebersold R. 2004. A dataset of human liver proteins identified by protein profiling via isotope-coded affinity tag (ICAT) and tandem mass spectrometry. Mol. Cell. Proteomics 3:1039–1041 [DOI] [PubMed] [Google Scholar]
- 40. Bakshi CS, Malik M, Mahawar M, Kirimanjeswara GS, Hazlett KR, Palmer LE, Furie MB, Singh R, Melendez JA, Sellati TJ, Metzger DW. 2008. An improved vaccine for prevention of respiratory tularemia caused by Francisella tularensis SchuS4 strain. Vaccine 26:5276–5288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pfaffl MW. 2001. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. LoVullo ED, Sherrill LA, Perez LL, Pavelka MS., Jr 2006. Genetic tools for highly pathogenic Francisella tularensis subsp. tularensis. Microbiology 152:3425–3435 [DOI] [PubMed] [Google Scholar]
- 43. LoVullo ED, Molins-Schneekloth CR, Schweizer HP, Pavelka MS., Jr 2009. Single-copy chromosomal integration systems for Francisella tularensis. Microbiology 155:1152–1163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chamberlain RE. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl. Microbiol. 13:232–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cherwonogrodzky JW, Knodel MH, Spence MR. 1994. Increased encapsulation and virulence of Francisella tularensis live vaccine strain (LVS) by subculturing on synthetic medium. Vaccine 12:773–775 [DOI] [PubMed] [Google Scholar]
- 46. Lindgren H, Honn M, Golovlev I, Kadzhaev K, Conlan W, Sjostedt A. 2009. The 58-kilodalton major virulence factor of Francisella tularensis is required for efficient utilization of iron. Infect. Immun. 77:4429–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Schwyn B, Neilands JB. 1987. Universal chemical assay for the detection and determination of siderophores. Anal. Biochem. 160:47–56 [DOI] [PubMed] [Google Scholar]
- 48. Riemer J, Hoepken HH, Czerwinska H, Robinson SR, Dringen R. 2004. Colorimetric ferrozine-based assay for the quantitation of iron in cultured cells. Anal. Biochem. 331:370–375 [DOI] [PubMed] [Google Scholar]
- 49. Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30:207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, Brouwer D, Nair V, Fischer ER, Wicke L, Curda AJ, Kupko JJ, Martens IIIC, Crane DD, Bosio CM, Porcella SF, Celli J. 2009. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol. 11:1128–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Foley JE, Nieto NC. 2010. Tularemia. Vet. Microbiol. 140:332–338 [DOI] [PubMed] [Google Scholar]
- 52. Huntley JF, Conley PG, Hagman KE, Norgard MV. 2007. Characterization of Francisella tularensis outer membrane proteins. J. Bacteriol. 189:561–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sen B, Meeker A, Ramakrishnan G. 2010. The fslE homolog, FTL_0439 (fupA/B), mediates siderophore-dependent iron uptake in Francisella tularensis LVS. Infect. Immun. 78:4276–4285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stojiljkovic I, Cobeljic M, Hantke K. 1993. Escherichia coli K-12 ferrous iron uptake mutants are impaired in their ability to colonize the mouse intestine. FEMS Microbiol. Lett. 108:111–115 [DOI] [PubMed] [Google Scholar]
- 55. Ramakrishnan G, Sen B, Johnson R. 2012. Paralogous outer membrane proteins mediate uptake of different forms of iron and synergistically govern virulence in Francisella tularensis tularensis. J. Biol. Chem. 287:25191–25202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doherty CP. 2007. Host-pathogen interactions: the role of iron. J. Nutr. 137:1341–1344 [DOI] [PubMed] [Google Scholar]
- 57. Nallaparaju KC, Yu JJ, Rodriguez SA, Zogaj X, Manam S, Guentzel MN, Seshu J, Murthy AK, Chambers JP, Klose KE, Arulanandam BP. 2011. Evasion of IFN-gamma signaling by Francisella novicida is dependent upon Francisella outer membrane protein C. PLoS One 6:e18201. 10.1371/journal.pone.0018201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sjöstedt A. 2007. Tularemia: history, epidemiology, pathogen physiology, and clinical manifestations. Ann. N. Y. Acad. Sci. 1105:1–29 [DOI] [PubMed] [Google Scholar]
- 59. Forestal CA, Malik M, Catlett SV, Savitt AG, Benach JL, Sellati TJ, Furie MB. 2007. Francisella tularensis has a significant extracellular phase in infected mice. J. Infect. Dis. 196:134–137 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.