

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) is the etiologic agent of primary effusion lymphoma (PEL), multicentric Castleman's disease (MCD), and the inflammation-driven neoplasm Kaposi's sarcoma (KS). A triad of processes, including abnormal proliferation of endothelial cells, aberrant angiogenesis, and chronic inflammation, characterize KS lesions. STAT3 is a key transcription factor governing these processes, and deregulation of STAT3 activity is linked to a wide range of cancers, including PEL and KS. Using primary human endothelial cells (ECs), I demonstrate that KSHV infection modulated STAT3 activation in two ways: (i) KSHV induced uncoupling of canonical tyrosine (Y) and serine (S) phosphorylation events while (ii) concomitantly inducing the phosphorylation and inactivation of TRIM28 (also known as KAP-1 or TIF-1β), a newly identified negative regulator of STAT3 activity. KSHV infection of primary ECs induced chronic STAT3 activation characterized by a shift from the canonical dual P-STAT3 Y705 S727 form to a mono P-STAT3 S727 form. Expression of the latent protein kaposin B promoted the unique phosphorylation of STAT3 at S727, in the absence of Y705, activated the host kinase mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 (MK2), and stimulated increased expression of STAT3-dependent genes, including CCL5, in ECs. TRIM28-mediated repression of STAT3 is relieved by phosphorylation of S473, and in vitro kinase assays identified TRIM28 S473 as a bona fide target of MK2. Together, these data suggest that kaposin B significantly contributes to the chronic inflammatory environment that is a hallmark of KS by unique activation of the proto-oncogene STAT3, coupled with MK2-mediated inactivation of the STAT3 transcriptional repressor TRIM28.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is a human gammaherpesvirus 2 etiologically linked to several AIDS-associated malignancies, including Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD) (1–3). KS is a unique angiogenic tumor that closely resembles a chronic inflammatory state with lesions rich in inflammatory infiltrates (4, 5). This inflammatory state is required for maintenance of the lesion, providing autocrine and paracrine factors essential for driving KSHV-mediated oncogenesis. In KS, the predominant tumor cell is the KSHV-infected, proliferating “spindle cell.” Spindle cells are thought to be endothelial in origin and secrete pathogenetically important proinflammatory cytokines and growth factors (6–9) necessary to maintain endothelial cell (EC) proliferation and extended life span. The exact mechanism of KSHV-induced tumorigenesis remains to be elucidated, but is thought to depend upon multiple viral gene products that collaborate to promote inflammation, EC proliferation, and neoangiogenesis. Most spindle cells support latent KSHV infection, and latent viral gene products are thought to directly contribute to tumorigenesis. One central latent protein shown to contribute to the inflammatory milieu within lesions is kaposin B. Kaposin B upregulates inflammatory cytokine levels linked to KS pathogenesis through mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 (MK2)-mediated stabilization of normally labile proinflammatory mRNAs (10), thereby promoting inflammation and maintenance of lesions.
Inflammation drives KSHV-induced pathogenesis, and a key transcription factor involved in the inflammatory response is the proto-oncoprotein signal transducer and activator of transcription 3 (STAT3). STAT3 plays a critical role in selectively inducing and maintaining a procarcinogenic inflammatory microenvironment, contributing to both induction and maintenance of cancer progression (11–17). STAT3 is constitutively active in a significant proportion of human solid tumors, including breast, brain, colon, lung, prostate and hematological malignancies, such as lymphomas and leukemias (18–23). STAT3 regulates a number of pathways important in tumorigenesis, including cell cycle progression, apoptosis, tumor angiogenesis, invasion and metastasis, and tumor cell evasion of the immune system (24–28). Accumulating evidence points to an important role for STAT3 activation in KSHV pathogenesis. STAT3 is constitutively activated in some KSHV-positive PEL cell lines, and expression of a dominant-negative form of STAT3 induces apoptosis in PEL cells (29). Latently infected telomerase reverse transcriptase (TERT)-immortalized endothelial cells display persistent activation of STAT3 (30); however, the role of STAT3 in primary endothelial cells, the major in vivo target of KSHV, is unclear. Despite the importance of STAT3 in inflammation and tumorigenesis, our understanding of the mechanisms governing STAT3 transcriptional activation in primary endothelial cells during KSHV infection remains incomplete.
In the canonical STAT3 activation pathway, interleukin-6 (IL-6) family cytokines mediate transient activation through cytokine receptors that activate receptor-associated Janus kinases (JAKs) to phosphorylate STAT3 on tyrosine 705 (P-STAT3 Y705), leading to the formation of dimers through reciprocal phosphotyrosine-SH2 domain interactions (31, 32). P-STAT3 Y705 dimers translocate to the nucleus and are able to bind specific interferon gamma activation sequence (GAS) elements and induce gene expression. A second phosphorylation event in the STAT3 transactivation domain on serine 727 (S727) is thought to be required for maximal transcription activity (33). Interestingly, unlike other STAT proteins, STAT3 nuclear import occurs in the absence of cytokine-mediated tyrosine phosphorylation and dimerization (34–36). In this case, monophosphorylation of STAT3 on S727 can occur. Current work illustrates that non-tyrosine-phosphorylated STAT3 resides in the nucleus, driving the expression of a unique subset of STAT3-responsive genes (36). Increasing evidence suggests an important role for monomeric nuclear P-S727 STAT3 in promoting the expression of a distinct subset of STAT3-responsive genes that are thought to contribute to chronic inflammation and tumorigenesis (37).
Given the important role STAT3 plays in multiple cellular functions and the implications of deregulated activation, STAT3 activity must be tightly controlled. The tripartite motif-containing protein 28 (TRIM28, also known as TIF-1β or KAP-1) is a potent negative regulator of STAT3 activation. TRIM28 is a universal corepressor of the Kruppel-associated box zinc-finger protein superfamily of transcriptional repressors (38, 86, 87) and binds directly to the central coiled-coil and DNA-binding domains of STAT3, facilitating the recruitment of proteins involved in gene silencing to inhibit STAT3-mediated transcription (39). TRIM28 recruits and coordinates the assembly of several chromatin-remodeling proteins, including histone deacetylase (HDAC) multiprotein complexes, histone methyltransferases, and heterochromatin protein 1 (HP1), through the plant homeodomain (PhD), bromodomain, and PXVXL motif, respectively (40, 41). Phosphorylation of TRIM28 by protein kinase Cδ (PKCδ) on the serine residue at position 473 inhibits repressor activity by disrupting TRIM28-mediated transcriptional silencing complexes, leading to enhanced STAT3 gene expression (39, 42). Inactivation of TRIM28, coupled with aberrant activation of STAT3, likely contributes to tumorigenesis via enhanced inflammation.
Here, I describe a new mechanism by which the latent/lytic KSHV protein kaposin B aberrantly activated STAT3 in primary human endothelial cells. I demonstrate kaposin B induced uncoupling of STAT3 serine phosphorylation from tyrosine phosphorylation, concomitant with phosphorylation and derepression of the STAT3-negative regulator, TRIM28. Kaposin B expression in primary ECs induced monophosphorylation of the STAT3 transactivation domain at serine 727, in the absence of tyrosine 705 phosphorylation. Furthermore, I have identified TRIM28, as a previously unknown target of MK2 and provide evidence that TRIM28 is a bona fide target of MK2. My results demonstrate kaposin B-induced phosphorylation of STAT3 at S727 and the concomitant phosphorylation of TRIM28 at S473 by MK2 results in aberrant STAT3 activation, inducing a subset of STAT3-activated genes, including CCL5. I propose that kaposin B promotes Kaposi's sarcoma tumorigenesis by enforcing constitutive and aberrant activation of the proto-oncogene STAT3.
MATERIALS AND METHODS
Cells and KSHV infections.
Primary human umbilical vein endothelial cells (HUVECs) were purchased from Lonza or isolated from the large vein of human umbilical cords after treatment with 0.5 mg of collagenase (Cooper Biomedical, Mississauga, Ontario, Canada) per ml in 0.01 M phosphate-buffered saline (PBS) (pH 7.4) as previously described (43). Cultures were expanded in EBM-2 medium (Lonza) supplemented with a bullet kit containing 2% fetal bovine serum, vascular endothelial growth factor, basic fibroblast growth factor, insulin-like growth factor 3, epidermal growth factor, hydrocortisone, ascorbic acid, gentamicin, amphotericin B, and heparin (EGM-2), in 6-well tissue culture plates coated with 0.1% (wt/vol) gelatin (in phosphate-buffered saline [PBS]) and used between passages 2 and 6 for experiments. Wild-type and MK2/3−/− mouse embryonic fibroblasts (MEFs) were a kind gift from M. Gaestel and were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS). KSHV was obtained from cultures of BCBL-1 cells (a kind gift from C. McCormick) that harbor latent KSHV (44, 45). Lytic reactivation of 5 × 105 BCBL-1 cells/ml was induced by addition of 0.3 mM valproic acid (Sigma) to cultures for 7 days. On day 7, cell-free virus-containing supernatant was harvested from cultures and concentrated by centrifugation at 15,000 × g for 2 h. The viral pellet was resuspended in EBM-2 medium, aliquoted, and frozen at −80°C. For infection studies, endothelial cells were plated out on gelatin-coated 6-well plates with or without glass coverslips at 1 ×105 cells/well. The next day, monolayers were inoculated with KSHV supplemented with 8 μg/ml Polybrene (Sigma) for 2 h at 37°C followed by spinoculation at 2,000 rpm for 2 h at 30°C, after which the inoculum was removed and cells were incubated at 37°C in 5% CO2 in fresh EBM-2 medium for the indicated times. For UV inactivation, KSHV was exposed to 5 × 1,200 μJ UV in a UV Stratalinker (30). For some experiments, at 48 h postinfection cells were mock treated or treated with 20 ng/ml recombinant human IL-6 (rhIL-6) (R&D Systems) for 30 min at 37°C. For other experiments, MK2 was inhibited by addition of 10 μM MK2 inhibitor III 4 h postinfection (p.i.). At 48 h p.i., cells were serum starved for 3 h in the presence of fresh inhibitors. At this time, some cells were treated with 10 μM the MK2 inhibitor rottlerin.
Chemicals, antibodies, and reagents.
The following reagents were purchased from commercial vendors: recombinant human IL-6 (rhIL-6), Peprotech, Rocky Hill, NJ; rabbit anti-phospho-STAT3 Y705 (D3A7), rabbit anti-phospho-STAT3 S727, rabbit anti-STAT3, rabbit anti-MK2, rabbit anti-phospho-MK2 Thr222, rabbit anti-phospho-MK2 Thr334, and mouse anti-β-actin, Cell Signaling; mouse anti-TRIM28, Transduction Laboratories; rabbit anti-phospho-TRIM28 S473, Biolegend, San Diego, CA; rat anti-LANA, Advanced Biotechnologies; the RNeasy kit, Qiagen, Valencia, CA; quantitative PCR (qPCR) primers, Invitrogen; a chemical inhibitor of MK2 (MK2 inhibitor III), Calbiochem; and rottlerin, EMD Millipore. Mouse anti-K8.1, rabbit anti-LANA, and rabbit anti-kaposin B were kind gifts from Don Ganem.
Retroviral transductions. (i) Retroviral plasmids.
pBMN-kapB-IP was generated by BamHI-EcoRI digestion of pCR3.1-kapB (10), releasing the 636-bp kaposin B open reading frame (ORF) (derived from a pulmonary KS isolate). To prepare the recipient pBMN-IP vector (a kind gift from G. Nolan, Stanford University), XhoI digestion was performed, followed by a Klenow fill-in reaction, and a BamHI-EcoRI digest. The kaposin B fragment was subsequently ligated into the BamHI/EcoRI-digested pBMN-IP vector, permitting the expression of kaposin B and the puromycin resistance gene from a single bicistronic mRNA. To create pBMN-MK2EE-IP, pcDNA3-Flag-MK2EE (a kind gift from Paul Anderson and described in reference 46) was amplified with primers G42 and G43 from pcDNA3mycMK2T205E,T317E (47). The amplicons were digested with BamHI and XhoI and inserted into BamHI-XhoI-digested pBMN-IP vector.
(ii) Retrovirus preparation and inoculation.
Retrovirus stocks were produced by calcium phosphate-mediated transfection of the Phoenix amphotropic packing cell line (a kind gift from G. Nolan, Stanford University) using the Profection kit (Promega) according to the instructions of the manufacturer. Virus-containing supernatants were collected 48 h after transfection. Supernatants were frozen at −80°C for use later. HUVECs were plated out on gelatin-coated 6-well plates with or without glass coverslips at 8 ×104 cells/well. The next day, cells were spinoculated with the indicated retroviruses at 2,000 rpm for 2 h at 30°C in the presence of 5 μg/ml Polybrene. At 24 h postransduction, 1 μg/ml puromycin was added to cells for 2 days to eliminate nontransduced cells. After selection, cells received 24 h of rest in fresh EBM-2 medium containing no puromycin before being used in experiments. When retroviral stocks gave >95% transduction efficiency, no selection was carried out and the cells were used for experiments between 54 and 56 h posttransduction (p.t.).
Immunofluorescence microscopy.
HUVECs were seeded in 6- or 12-well dishes containing gelatin-coated glass coverslips and infected with KSHV or transduced with the indicated retroviruses as described above. At 48 h postinfection or 5 days postransduction, cells were washed in PBS and fixed in 4% paraformaldehyde for 10 min at room temperature. The cells were then washed and permeabilized in 0.1% Triton X-100 for 5 min at room temperature. After three PBS washes, coverslips were blocked in 1% human AB serum in 5% bovine serum albumin (BSA)–PBS for 30 min at room temperature. KSHV-infected cells were stained using either rabbit anti-LANA or rat anti-LANA(1:500) to determine the extent of KSHV infection as the latent gene LANA forms visible nuclear puncta (48). Kaposin B expression in both infected and transduced cells was detected by staining with rabbit polyclonal anti-kaposin B (1:2,000) in blocking buffer that was adsorbed to cells for 45 min at 4°C. Rabbit anti-STAT3, anti-phospho-STAT3 Y705, and anti-phospho-STAT3 S727 (1:1,000) primary antibodies were incubated overnight at 4°C. Mouse anti-TRIM28 (1:1,000) and phospho-TRIM28 Ser473 (1:1,000) were incubated for 20 to 45 min at 4°C. After primary antibody binding, cell monolayers were washed and incubated for 45 min with species-specific secondary antibodies conjugated with Alexa Fluor 488 and Alexa Fluor 555 (1:2,000; Molecular Probes) at 4°C followed by incubation with ToPro3 nuclear stain for 5 min and mounted on glass slides using Prolong Gold antifade reagent (Invitrogen). Cells were visualized and photographed under a Zeiss LSM510 scanning argon (488 nm) and neon (555 or 633 nm) laser confocal microscope, using a 40× or 63× oil immersion objective. Images were collected with similar exposure times and identical gains. For some experiments, HUVECs at 48 h postinfection or 24 h postrest were either mock treated or treated with 20 ng/ml recombinant human IL-6 for 30 min prior to fixation.
Image processing.
All images taken using the confocal microscope were acquired using the program LSM510 (Zeiss). All image processing was performed using Photoshop CS3 (Adobe).
Western blotting.
HUVECs were transduced with control or recombinant retroviruses expressing either kaposin B or MK2EE as described. At 2 to 5 days postransduction, the cells were washed with PBS and lysed with Laemmli's sample buffer. For the experiments using KSHV, HUVECs were infected with KSHV or UV-inactivated KSHV or were left uninfected. At various times p.i., cells were washed with PBS and harvested in Laemmli's sample buffer. Twenty-five to 50 μg of protein was resolved by SDS-PAGE and transferred to a nitrocellulose membrane, blocked in 2.5% BSA or 5% milk in Tris-buffered saline (TBS)–0.1% Tween 20 for 1 h at room temperature before incubation with the primary antibody overnight at 4°C in 5% BSA in TBS–0.1% Tween 20. Protein targets were detected with the appropriate horseradish peroxidase-conjugated immunoglobulin secondary antibodies (1:5,000; Cell Signaling) for 1 h at room temperature and were visualized by chemiluminescence with ECL Plus (GE Healthcare). Rabbit anti-kaposin B and rabbit anti-phospho-TRIM28 S473 were used at 1:5,000. All other antibodies were used at 1:1,000. Actin was used a loading control (1:5,000). Signals were detected using the Kodak Image Station 4000 mm Pro.
In vitro kinase assay.
Recombinant full-length active MK2 purified from Escherichia coli was incubated with the indicated glutathione S-transferase (GST) fusion proteins (GST alone, GST-heat shock protein 27 [Hsp27], GST-TRIM28, or GST-TRIM28 S473A) bound to glutathione-Sepharose beads in the presence of ATP for 1 h at 37°C. Protein samples were immunoblotted with antibodies specific for P-Hsp27 S78 (1:1,000) or P-TRIM28 S473 (1:10,000). Parallel SDS-PAGE gels were stained with Coomassie brilliant blue to stain for total proteins.
ELISA.
RANTES and IL-6 were analyzed via an in-house enzyme-linked immunosorbent assay (ELISA). The ELISA involved capture antibodies (RANTES antibody P-230-E and IL-6 monoclonal antibody M-620-E; Endogen, Woburn, MA). Nonspecific binding was blocked by using 1% bovine serum albumin (BSA) in Na2HPO4 (pH 8.3 to 8.5) for 1 h at 37°C. The matched biotinylated detection antibodies were BAF 278 for RANTES (R&D Systems) and M-621-B (Endogen) for IL-6. A commercial ELISA amplification system (Life Technologies) was used for detection. The sensitivity of both the RANTES and IL-6 assays was 7.8 pg/ml.
qRT-PCR.
Total RNA was extracted using the RNeasy minikit (Qiagen) according to the manufacturers' instructions. RNA concentration was determined by spectrophotometer, and cDNA was synthesized from 0.5 to 1 μg total RNA using the QuantiTect reverse transcription kit (Qiagen). Quantitative PCR (qPCR) was performed using the Mx3000P multiplex quantitative PCR system (Stratagene) and a QuantiFast SYBR green PCR kit (Qiagen) and analyzed with MxPro (Stratagene). Standard curves were generated for each primer set and used to extrapolate relative sample values. Primer sequences are available upon request. Results were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) and expressed as fold change from baseline (uninfected or vector-transduced cells). Experiments were performed two to three separate times, and all samples were run in duplicate.
Statistics.
Statistical analyses were performed using GraphPad Prism software. Data are represented as means ± standard errors of the means (SEM) of at least 3 separate experiments. The paired t test was used for comparison between two groups. Significance was defined as P < 0.05.
RESULTS
KSHV infection of primary ECs activates STAT3.
STAT3 is hyperphosphorylated on tyrosine 705 (Y705) and chronically activated in a diverse group of primary tumors and in many cancer cell lines. Punjabi and colleagues demonstrated that latent KSHV infection of TERT-immortalized microvascular endothelial (TIME) cells and primary human dermal microvascular endothelial cells (HMVEC-d) resulted in persistent Y705 phosphorylation of STAT3 and increased STAT3 activity (30). Data are limited regarding KSHV infection in primary human endothelial cells (ECs) and the activation status of STAT3, which is relevant to KSHV tumorigenesis. To address the effect of KSHV on STAT3 activity, I infected low-passage-number primary human umbilical vein endothelial cells (HUVECs) that display no STAT3 phosphorylation. Immunofluorescence microscopy at 48 h postinfection (p.i.) demonstrated that KSHV-infected HUVECs, but not mock-infected cells, display phosphorylation of STAT3 at Y705 and serine 727 (S727) (Fig. 1A). One hundred percent of HUVECs were positive for KSHV, as assessed by LANA staining.
Fig 1.

KSHV infection of primary endothelial cells (ECs) activates STAT3. (A) Mock-infected (uninfected) and KSHV-infected HUVECs were stained at 48 h p.i. with rabbit anti-LANA, rabbit anti-P-STAT3 Y705, rabbit anti-P-STAT3 S727, or anti-STAT3 antibodies and visualized with goat anti-rabbit Alexa 488-conjugated secondary antibody. LANA staining demonstrated establishment of latency in 100% of cells. Magnification, 630×; scale bar, 10 μm. (B) Phase-contrast images of HUVECs at various times p.i. showing characteristic spindling during KSHV infection. The final image is from immunofluorescent microsopy showing LANA-positive cells at 18 days (18d) p.i. Data are representative of 3 independent experiments. (C) HUVECs infected with live KSHV or UV-inactivated KSHV were harvested at 1, 2, 4, 6, 11, and 18 days p.i., and 20 μg of lysates was immunoblotted with antibodies specific for LANA, K8.1, kaposin B, P-STAT3 Y705, P-STAT3 S727, and STAT3. (D) Fifty micrograms of lysate was immunoblotted to demonstrate kaposin B expression (arrow) by 2 days p.i. ns band, nonspecific band. (E) Relative levels of P-STAT3, normalized to STAT3, at various times p.i. show distinct patterns of phosphorylation over time. Densitometric analysis of signal was determined using the Kodak Molecular Imaging software package 4.5.1. Uninfected controls were set to 1 for comparison. Analysis demonstrates an uncoupling of P-STAT3 Y705 and S727 levels as infection progresses. β-Actin served as a loading control. Data are representative of 2 independent experiments. (F) HUVECs were harvested at 2 days and 11 days p.i. and lysed, and RT-qPCR was carried out to determine the levels of expression of STAT3-dependent target genes. Analysis of transcripts suggest that changes in phosphorylation of STAT3 at Y705 and S727 impact gene expression, as evidenced by the findings that CCL5 transcript levels increased over the course of infection, IL-6 levels were relatively constant, but STAT3 levels decreased. Data are expressed as fold increase relative to uninfected ECs that were set to 1 for comparison. Data are expressed as means ± standard deviations (SD) of two separate experiments with duplicate samples. Cell-free supernatants were harvested from cultures of infected HUVECs at various days p.i. with CCL5 (G) and IL-6 (H) protein levels measured by ELISA. The data demonstrate a steady increase in CCL5 protein levels over the course of infection, while IL-6 levels dramatically decrease by 11 and 18 days p.i., when P-Y705 levels are also decreased. Data are expressed as means ± SD and are representative of two separate experiments with duplicate samples.
Given the importance of aberrant chronic STAT3 activation in tumorigenesis, I wanted to determine if KSHV infection results in chronic activation of STAT3 in primary ECs. To address this, I performed an extended-time-course study over 18 days to examine the activation status of STAT3. HUVECs infected with KSHV showed characteristic spindling over time, and fluorescence microscopy demonstrated nuclear LANA expression in 18-day cultures (Fig. 1B). Immunoblotting for expression of KSHV proteins LANA (encoded by a latent gene), kaposin B (encoded by a latent or lytic gene), and K8.1 (encoded by a late lytic gene) demonstrated that HUVECs expressed LANA by 1 day p.i. and expression was maintained up to 18 days p.i. Kaposin B expression was detectable by 2 days p.i. (Fig. 1C and D), with increasing levels seen over the course of infection, while K8.1 expression was evident by 4 days p.i. (Fig. 1C). Analysis of the phosphorylation status of STAT3 during this time course clearly demonstrated that both tyrosine and serine residues were phosphorylated in KSHV-infected cells by 1 day p.i. Low-level P-STAT3 S727 was observed in mock- and UV-inactivated KSHV control cultures, but it was dramatically upregulated during the course of live KSHV infection and persisted throughout the culture period, while Y705 phosphorylation levels decreased over time. Densitometric analysis of P-STAT3 S727 and Y705 levels, normalized to total STAT3, demonstrated a striking difference in the tyrosine and serine phosphorylation patterns during the course of infection. Y705 phosphorylation levels were relatively stable from 1 day until 6 days p.i., when levels dramatically increased, and were followed by a sharp decrease at 11 and 18 days p.i. In contrast, levels of S727 phosphorylation steadily increased over the course of infection, with a peak at 6 days followed by little decrease at 11 and 18 days p.i. (Fig. 1E). Furthermore, RT-qPCR analysis of STAT3-dependent transcripts at 2 and 11days p.i. demonstrated a clear change in transcript levels over the course of infection, with CL5 levels steadily increasing, IL-6 levels maintained, and STAT3 levels decreased. These data complement my phosphorylation data and suggest that changes in STAT3 phosphorylation status during KSHV infection directly impact STAT3 transcription. In addition, I analyzed CCL5 and IL-6 protein levels in cell-free supernatants from infected HUVEC cultures at various days p.i. to confirm gene expression data as cytokine and chemokine transcripts are subject to significant posttranscriptional and posttranslational modification. I demonstrate an overall steady increase in CCL5 levels over time, even at day 18, when P-Y705 levels are low but P-S727 levels are still high. Furthermore, IL-6 shows a pattern that demonstrated IL-6 levels are elevated when both Y705 and S727 residues are robustly phosphorylated and low when P-Y705 levels are low, but P-S727 levels are high. My data clearly show KSHV infection of low-passage-number primary ECs resulted in persistent STAT3 activation. Furthermore, the kinetics of phosphorylation of STAT3 S727 appeared to be distinct from Y705. In canonical cytokine-mediated STAT3 activation, phosphorylation of Y705 occurs and is followed by S727 phosphorylation, resulting in a STAT3 dimer that is dually phosphorylated and able to efficiently drive gene expression. My data illustrate KSHV infection of primary ECs resulted in a unique shift from a dually phosphorylated form of STAT3 to an S727 monophosphorylated form at later times in infection, and these changes were associated with corresponding changes in STAT3-dependent gene expression. These data indicate that phosphorylation of these two residues is uncoupled during KSHV infection and that within KSHV-infected ECs, two populations of P-STAT3 exist: the canonical, dually phosphorylated form of STAT3, where both Y705 and S727 residues are phosphorylated, as seen in early days p.i., and another that is characterized by mono-P-STAT3 S727.
Kaposin B expression promotes monophosphorylation of STAT3 at S727.
My data demonstrated KSHV-infected primary ECs displayed enhanced P-STAT3 S727 levels by 2 days p.i., coinciding with expression of kaposin B, suggesting a role for kaposin B in this phosphorylation event. To address the contribution of kaposin B to STAT3 activation in KSHV-infected cells, HUVECs were transduced with control retroviruses or retroviruses encoding kaposin B, and STAT3 activation was analyzed. HUVECs expressing kaposin B protein harbored increased nucleus-localized P-STAT3 S727 in the absence of significant Tyr705 phosphorylation (Fig. 2A and B). Densitometric analysis demonstrated a 1.4-fold increase in P-STAT3 S727 levels relative to total STAT3 in kaposin B-expressing cells. Statistical analysis of P-STAT3 S727 levels, relative to total STAT3, demonstrated that kaposin B-expressing cells had significantly enhanced levels of P-STAT3 S727 (P = 0.044, n = 5) (Fig. 2C). Together, my data suggest that kaposin B expression in ECs resulted in unique activation of STAT3 that likely contributes to the chronic inflammatory state observed in KS lesions.
Fig 2.
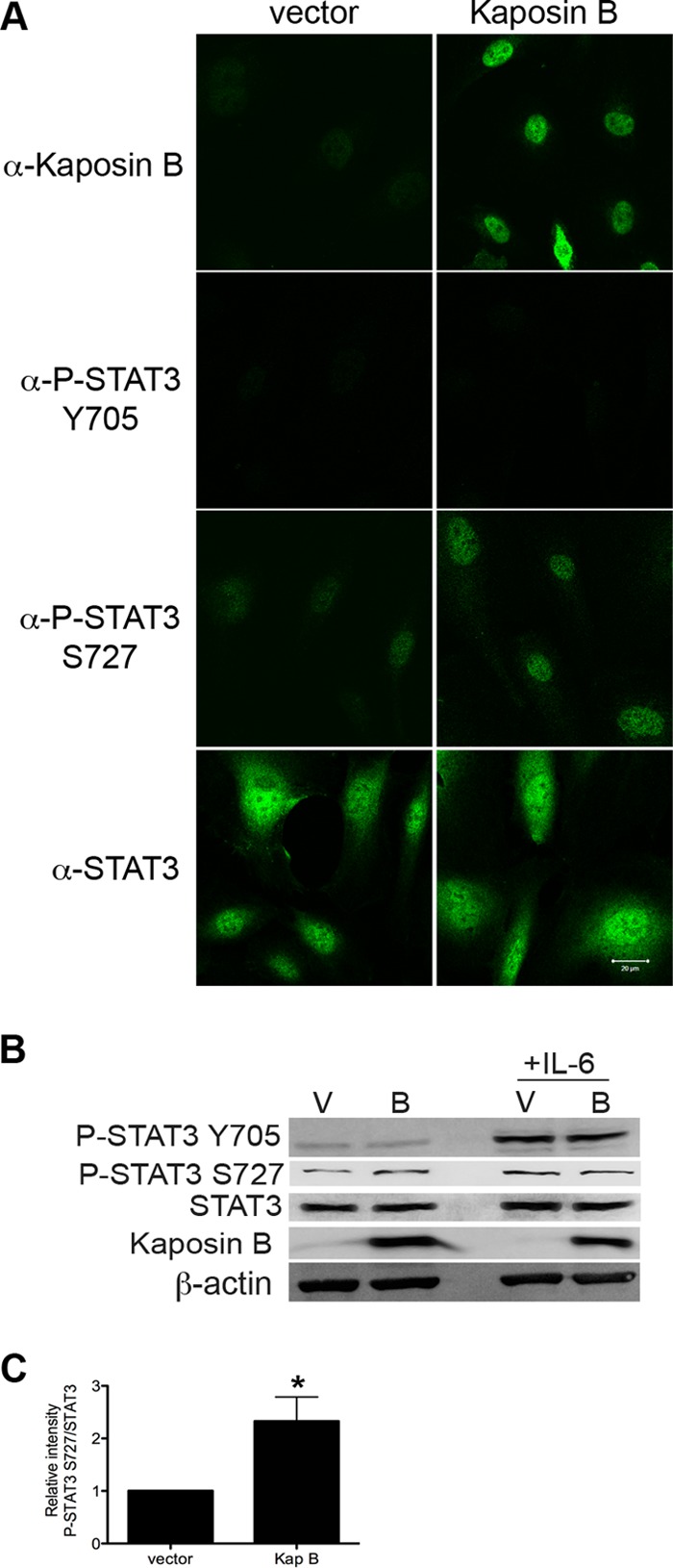
Kaposin B expression promotes monophosphorylation of STAT3 at S727. (A) HUVECs were transduced with empty retrovirus vector or retrovirus encoding kaposin B. At 5 days p.t., cultures were stained with antibodies specific for kaposin B, P-STAT3 Y705, P-STAT3 S727, or STAT3 and visualized with goat anti-rabbit Alexa 488-conjugated secondary antibody. Magnification, 630×; scale bar, 20 μm. Kaposin B-expressing cells show increased levels of the P-STAT3 S727 in the absence of phosphorylation at Y705. Vector-transduced cells were negative for phosphorylated STAT3. (B) HUVECs were transduced with empty retroviral vector (lanes V) or vector encoding kaposin B (lanes B). Protein samples were immunoblotted with rabbit anti-kaposin B, anti-P-STAT3 Y705, anti-P-STAT3 S727, and anti-STAT3 antibodies. β-Actin served as a loading control. Some cultures were treated with recombinant human IL-6 for 30 min as a positive control for STAT3 activation. (C) Kaposin B-expressing cells show a significant increase in relative P-STAT3 S727 levels, normalized to total STAT3. *, P = 0.044 (n = 5). Densitometric analysis of signal was determined performed using the Kodak Molecular Imaging software package 4.5.1. Data are representative of 5 independent experiments.
Phosphorylation of TRIM28 at S473 in KSHV-infected ECs.
TRIM28 is a central transcriptional repressor of the STAT3 proto-oncogene, and knockdown of TRIM28 increases the level of P-STAT3 S727 in epithelial cells (49). TRIM28 repressive activity is relieved by phosphorylation on residue serine 473 adjacent to the HP1-binding domain that is essential for mediating recruitment of silencing complexes (39, 42). Given the importance of TRIM28 in regulating STAT3 activity, I investigated the phosphorylation status of TRIM28 at S473 in KSHV-infected ECs. Forty-eight hours p.i., latently infected HUVECs displayed robust phosphorylation of TRIM28 at S473, while adjacent uninfected bystander cells did not stain positive (Fig. 3A). Confocal microscopy revealed P-TRIM28 S473 in a distinctive punctate pattern that corresponded strikingly to LANA puncta (Fig. 3B). Phosphorylation of TRIM28 at S473 in KSHV-infected HUVECs was confirmed in lysates (Fig. 3C). These data provide compelling evidence of in vivo phosphorylation of TRIM28 at S473, an event known to relieve repressive activity on STAT3, during KSHV infection.
Fig 3.
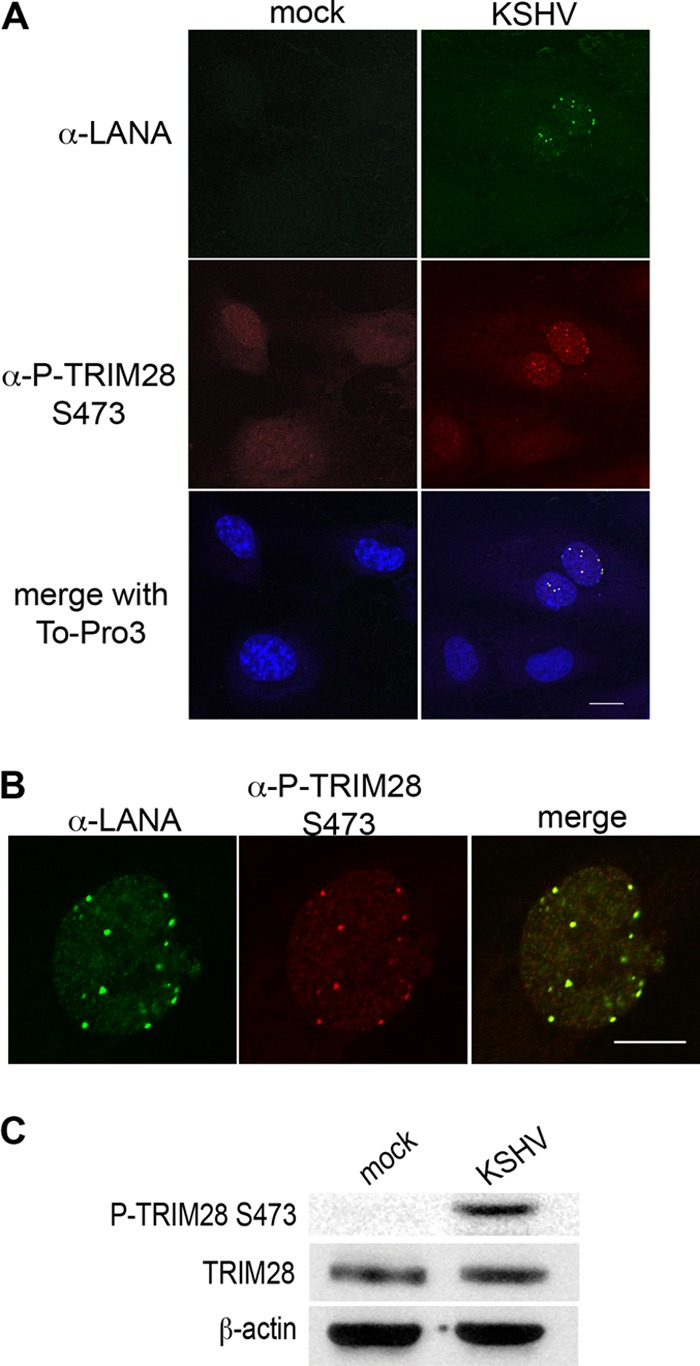
Latent KSHV infection of primary endothelial cells stimulates TRIM28 S473 phosphorylation. (A) HUVECs were infected with KSHV and at 48 h p.i. were processed for immunofluorescence microscopy as described, stained with rabbit anti-P-TRIM28 S473 and rat anti-LANA antibodies, and detected with goat anti-rabbit Alexa 488- and donkey anti-rat Alexa 555-conjugated antibodies. ToPro3 was used to visualize nuclei. Magnification, 630×; scale bar, 20 μm. Green, LANA; red, P-TRIM28 S473; blue, nuclei. (B) At 48 h, KSHV latently infected HUVECs stained as in panel A demonstrated nuclear colocalization of P-TRIM28 S473 and LANA puncta. Magnification, 1,260×; scale bar, 10 μm. (C) At 48 h, KSHV latently infected HUVEC lysates were immunoblotted with antibodies specific for P-TRIM28 S473, TRIM28, and β-actin (as a loading control). Microscopy and Western blot analysis demonstrate phosphorylation of TRIM28 at Ser 473 in KSHV latently infected endothelial cells.
KSHV induces an MK2-dependent phosphorylation of TRIM28.
Previous work using the chemical inhibitor rottlerin suggested that the MAPK pathway was responsible for phosphorylation of TRIM28 at S473 (42). A schematic of the TRIM28 protein illustrates the various domains including position S473 (Fig. 4A), and a survey of the TRIM28 sequence surrounding S473 revealed a previously unidentified candidate MK2 consensus phosphorylation site (Fig. 4B). In vitro kinase assays using a purified, Escherichia coli-derived fusion protein of glutathione S-transferase (GST) and TRIM28, but not GST alone, demonstrated that TRIM28 was efficiently phosphorylated by purified recombinant active MK2, and this phosphorylation event was prevented by mutation of serine 473 to alanine (Fig. 4C). Furthermore, MK2-mediated phosphorylation of TRIM28 at S473 was efficiently inhibited in the presence of a commercially available selective inhibitor of MK2, MK2 inhibitor III, and rottlerin (Fig. 4D) but not the relatively poor MK2 inhibitor compound 1 (50). Further evidence for the role of MK2-mediated phosphorylation of TRIM28 was demonstrated by analysis of the phosphorylation state of TRIM28 at S473 in mouse embryonic fibroblasts (MEFs) lacking MK2 and MK3 (MK2 homologue) (Fig. 4E). MK2/MK3−/− MEFs treated with hydrogen peroxide (a known activator of MAP kinases) demonstrated a 40% decrease in the amount of phosphorylated TRIM28 S473, compared to wild-type control MEFs. Taken together, these studies identify TRIM28 as a previously unknown and valid target of the host cell kinase MK2 and support a role for MK2 in vivo in the regulation of TRIM28 activity.
Fig 4.

TRIM28 is a bona fide target of MK2. (A) Schematic representation of TRIM28 illustrating the KRAB interaction domains and the HP1 binding domain. The serine residue at position 473 adjacent to the HP1 binding domain is indicated (*). (B) Partial protein sequence alignment highlighting a putative MK2 phosphorylation site in TRIM28, compared to the consensus and known MK2 target proteins. (C) Recombinant full-length active MK2 purified from E. coli was incubated with the indicated GST fusion proteins (GST alone, GST-Hsp27, GST-TRIM28, or GST-TRIM28 S473A) bound to glutathione Sepharose beads in the presence of ATP for 1 h at 37°C. Protein samples were immunoblotted with antibodies specific for phospho-serine 78 (P-S78) Hsp27 or phospho-serine 473 (P-S473). Parallel SDS-PAGE gels were stained with Coomassie brilliant blue to stain for total proteins, indicating equal loading of GST fusion proteins in this assay. (D) In vitro MK2 kinase assays were performed as described above, with the inclusion of 1 to 10 μM compound I, MK2 inhibitor III, or rottlerin (rott.). These experiments indicate that phosphorylation of either Hsp27 or TRIM28 is sensitive to a chemical inhibitor of MK2. In these assays, the well-known MK2 substrate heat shock protein 27 (Hsp27) served as a positive control. IB, immunoblot. (E) Wild-type (WT) or MK2/MK3−/− MEFs were treated with 1 mM hydrogen peroxide (H2O2), a known activator of MK2, for 40 min or left untreated (ns [not stimulated]). Cell lysates were immunoblotted with P-TRIM28 S473 antibody. β-Actin served as a loading control. Chemiluminescence signal was visualized using a Kodak Image Station 4000MM PRO (Carestream). Densitometric analysis of signal was determined using the Kodak Molecular Imaging software package 4.5.1, and results were expressed as a ratio of P-TRIM28 S473 to β-actin relative to WT levels.
KSHV activates a plethora of cell signaling pathways, including the prosurvival phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR and the MAPK pathways (8, 51–54). The latent/lytic KSHV protein kaposin B is known to activate MK2 in epithelial cells (10). However, the activation status of MK2 in KSHV-infected primary ECs is not known. Analysis of the phosphorylation state of MK2 in vitro during KSHV infection of primary ECs showed significant phosphorylation and activation of MK2 at threonine 222 by 2 days p.i. (Fig. 5A) (P ≤ 0.05, n = 3). Furthermore, robust phosphorylation and nuclear localization of MK2 at residue threonine 334 were observed in LANA-expressing KSHV-infected ECs, but not in nearby uninfected bystander cells (Fig. 5B) strongly suggesting MK2 activation was due to viral gene expression and not the result of paracrine factors. Together, these data support a role for cell autonomous, KSHV-induced, MK2 modulation of TRIM28 activity in primary endothelial cells via phosphorylation of S473.
Fig 5.
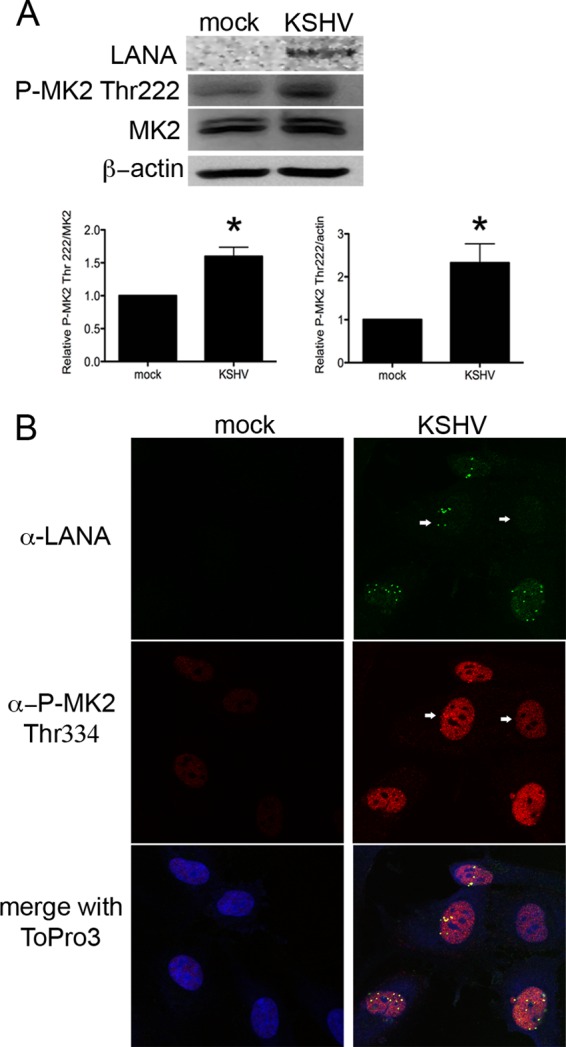
KSHV infection of primary endothelial cells triggers MK2 activation. (A) HUVECs were infected with KSHV, harvested at 2 days p.i., and immunoblotted with rabbit anti-LANA, rabbit anti-phospho-MK2 Thr222, or MK2 antibodies. β-Actin was used a loading control. Relative levels of phospho-MK2 were significantly increased in KSHV-infected ECs, compared to mock controls. *, P ≤ 0.05 (n = 3). Densitometric analysis of signal was determined using the Kodak Molecular Imaging software package 4.5.1 (B) HUVECs plated on glass coverslips were infected with KSHV. Forty-eight hours p.i., cells were stained with rat anti-LANA and rabbit anti-phospho-MK2 Thr334 antibodies. Primary antibodies were visualized with goat anti-rat-Alexa 488- and goat anti-rabbit-Alexa 555-conjugated antibodies. ToPro3 staining was used to identify nuclei. Green, LANA; red, P-MK2 Thr334; blue, nuclei. LANA staining demonstrated establishment of latency in some cells. Magnification, 630×; scale bar, 10 μm.
Kaposin B expression in ECs induces TRIM28 phosphorylation at S473.
One of the viral proteins involved in KSHV-mediated MK2 activation is kaposin B. Kaposin B binds and activates MK2, resulting in the phosphorylation of MK2 target proteins, including Hsp27, in epithelial cells (10). To confirm the role of kaposin B in MK2-mediated S473 phosphorylation of TRIM28 in primary ECs, cells were transduced with control retroviruses or retroviruses encoding kaposin B or a constitutively active form of MK2, MK2EE. Immunofluorescence microscopy clearly demonstrated ECs expressing the constitutively active form of MK2, or kaposin B, harbored significantly elevated levels of nuclear P-TRIM28 S473 (Fig. 6A). This was confirmed in total cell lysates from HUVECs transduced to express kaposin B or treated with hydrogen peroxide, a known activator of MK2 (Fig. 6B). Taken together, these experiments show for the first time that TRIM28 is a substrate of MK2 and strongly suggest that kaposin B activation of MK2 during KSHV infection contributes to the observed TRIM28 S473 phosphorylation in infected ECs.
Fig 6.
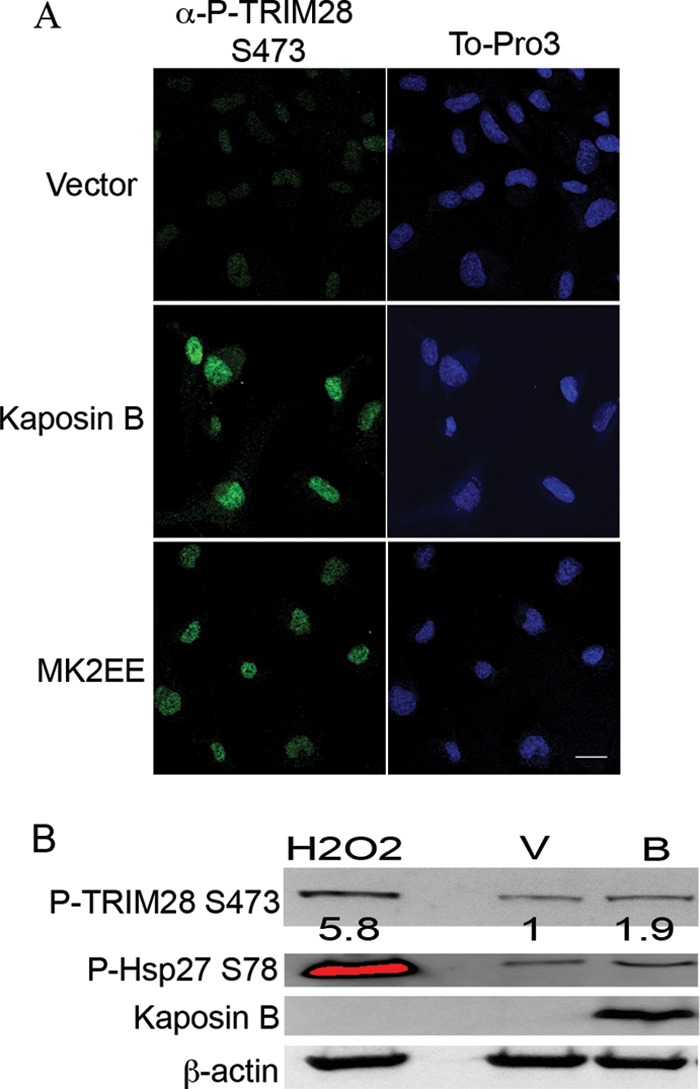
The latent/lytic kaposin B protein induced TRIM28 S473 phosphorylation in ECs. (A) HUVECs were transduced with empty retrovirus vector or retrovirus encoding kaposin B or MK2EE (a constitutively active form of MK2). Cultures were stained with the rabbit anti-P-TRIM28 S473 and visualized with goat anti-rabbit Alexa 488. ToPro3 staining was used to visualize nuclei. Magnification, 630×, scale bar, 20 μm. Kaposin B- and MK2EE-expressing endothelial cells harbor phospho-TRIM28 S473 compared to vector control cells. (B) HUVECs transduced with empty retroviral vector or vector encoding kaposin B and immunoblotted with rabbit anti-P-TRIM28 S473, rabbit anti-P-Hsp27 S78, and rabbit anti-kaposin B antibodies. HUVECs treated with hydrogen peroxide for 30 min prior to harvest serve as a positive control for MK2 activation. β-Actin serves as a loading control. Densitometric analysis was carried out, and the results are expressed as a ratio of P-TRIM28 S473 to actin. Data are representative of two independent experiments.
Kaposin B expression promotes selective STAT3 gene expression.
My data demonstrate that KSHV infection and kaposin B expression in primary ECs result in aberrant and unique activation of STAT3. To assess the downstream consequence of KSHV- and kaposin B-induced STAT3 S727 phosphorylation, coupled with phosphorylation and inactivation of TRIM28, I examined a subset of STAT3-responsive genes by RT-qPCR in HUVECs. A distinct subset of the STAT3 target genes, including the genes coding for SOCS3, CCL5, and IL-6, were found to be transcriptionally active at 48 h p.i. in latent KSHV-infected primary ECs (Fig. 7A). In one representative experiment, the levels of CCL5, IL-6, and SOCS3 mRNA were increased 19-fold, 17-fold, and 37-fold, respectively, compared to those in uninfected cells. Reverse transcription-qPCR on kaposin B-expressing primary ECs at 2 to 5 days posttransduction demonstrated a 14-fold increase in CCL5 mRNA and a 1.5–2 fold increase in SOCS3, IL-6, STAT3, and transforming growth factor β (TGF-β) mRNA levels (Fig. 7B), demonstrating that the P-STAT3 Ser727 monophosphorylated form of STAT3 induced by kaposin B was transcriptionally active. Furthermore, cells expressing the constitutively active form of MK2, MK2EE, demonstrated similar increases in the SOCS3, STAT3, and TGF-β genes. In contrast, IL-6 gene expression was augmented by MK2EE, while CCL5 expression was induced but to a significantly lower degree than that observed in kaposin B-expressing cells. Chemical inhibition of MK2 in HUVECs using the commercially available MK2 inhibitor III and rottlerin in the context of KSHV infection resulted in an ∼40% decrease in CCL5 levels compared to the levels in vehicle-treated cells (Fig. 7C). Together, these data indicate that activation of STAT3-responsive genes in kaposin B-expressing endothelial cells was due, at least in part, to activation of MK2. Taken together, my data suggest that the latent/lytic gene product kaposin B contributes to the aberrant chronic activation of STAT3 observed during KSHV infection, likely both through enforcement of the monophosphorylation of STAT3 at serine 727 and through the phosphorylation and inactivation of the negative regulator TRIM28. Together, these events result in deregulated STAT3 activity and elevated CCL5 expression. These data support a role for kaposin B directly contributing to the inflammatory milieu present within KS lesions.
Fig 7.

The KSHV kaposin B protein promotes monophosphorylation of STAT3 to drive selective STAT3 gene expression. HUVECs were infected with KSHV (A) or transduced with control retroviruses or retroviruses encoding kaposin B or MK2EE (B). At 48 h p.i. or 2 to 5 days postransduction, cells were lysed and RT-qPCR was carried out to determine the levels of expression of STAT3-dependent target genes. Data are expressed as fold increase relative to uninfected or vector-transduced ECs that were set to 1 for comparison. Each reaction was done in duplicate, and each point represents the mean ± SD of data from 2 or 3 independent experiments. (C) HUVECs were infected with KSHV, and 4 h p.i., cells were treated with 10 μM MK2 inhibitor III or DMSO (vehicle). At 48 h p.i., cells were serum starved for 3 h, fresh MK2 inhibitor was added, and 10 μM rottlerin was added to additional untreated wells. After 3 h of treatment, cells were lysed and RT-qPCR was carried out to determine the effect of MK2 inhibition on CCL5 expression. Data were normalized to GAPDH and expressed as the percentage of CCL5 expression of vehicle-treated controls. Data are expressed as means ± SD from two separate experiments carried out in duplicate.
DISCUSSION
KSHV is the etiologic agent of Kaposi's sarcoma, a multifocal (present in the skin and linings of the mouth, nose, and eye, as well as in the lungs, liver, stomach, intestines, and lymph nodes) highly vascularized neoplasm characterized by chronic inflammation, abnormal proliferation, and neoangiogenesis (55, 56). A central transcription factor governing activation of these pathways is STAT3, although its role in KSHV-induced tumorigenesis is not well understood. I now show that latent KSHV infection of primary human umbilical vein endothelial cells results in aberrant and chronic activation of STAT3, a proto-oncogene demonstrated to be pivotal to the development and progression of many cancers. I have demonstrated that KSHV infection of primary ECs results in a unique uncoupling of STAT3 activation. In latently infected ECs, I observed two populations of active STAT3, one characterized by the canonical Y705 and S727 phosphorylated STAT3 dimer and another characterized by a unique S727 monophosphorylated STAT3. These changes were found to translate into distinct changes in CCL5 and IL-6 transcript and protein expression during KSHV infection. The time-dependent changes in P-STAT3 Y705 and S727 levels in ECs observed suggest that P-Y705 mediated-gene expression may be important for the early phase of tumorigenesis, while P-S727 may be important for both the early and late phases, first to promote inflammatory conditions and then to maintain them. Expression of the latent/lytic kaposin B protein in primary endothelial cells promoted the unique monophosphorylation of STAT3 at S727 and enhanced proinflammatory gene expression. In my model (Fig. 8), I propose that kaposin B expression promotes aberrant activation of STAT3 during KSHV infections by a two-“hit” mechanism. In endothelial cells, kaposin B induces activation of an as yet unknown kinase to phosphorylate nucleus-localized STAT3 at serine 727 and MK2 to modulate inactivation of TRIM28 via serine 473 phosphorylation. TRIM28 binds directly to the central coiled-coil and DNA-binding domains of STAT3, with higher affinity for monomeric P-STAT3 S727, facilitating the recruitment of proteins involved in gene silencing, including heterochromatin protein 1 (HP1) to inhibit STAT3-mediated transcription. Phosphorylation of TRIM28 S473 by MK2 prevents TRIM28 from coordinating the assembly of gene silencing proteins and inhibiting STAT3. Thus, MK2 activity relieves TRIM28-mediated repression of STAT3, liberating monomeric nuclear P-STAT3 S727 to drive the transcription of a subset of STAT3 responsive genes that includes CCL5. The coordinated effort to modulate host kinases by kaposin B effectively manipulates the latent transcription factor STAT3, resulting in a phospho-form of STAT3 specifically linked to tumorigenesis.
Fig 8.
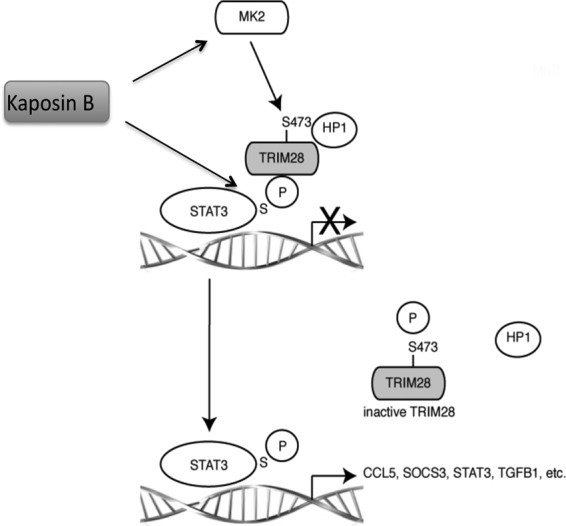
Model illustrating how kaposin B relieves TRIM28-mediated repression of STAT3. STAT3 nuclear import has been reported to occur in the absence of tyrosine phosphorylation and dimerization. Monomeric nuclear P-STAT3 S727 promotes the expression of a distinct subset of STAT3-responsive genes. STAT3 is normally held in an inactive state, unable to drive transcription, by the corepressor TRIM28. Kaposin B promotes monophosphorylation of STAT3 at serine 727 and induces activation of the host kinase MK2, which in turn mediates the phosphorylation of TRIM28 at serine 473. This phosphorylation event relieves the inhibitory activity of TRIM28, allowing P-STAT3 727 to drive transcription of a subset of STAT3-responsive genes, including CCL5.
The importance of the monophosphorylated serine 727 form of STAT3 in driving tumorigenesis is only beginning to be appreciated. While many cancers display hyperphosphorylated Y705 STAT3, recent work demonstrated that peripheral blood cells of patients with chronic lymphocytic leukemia harbor constitutively phosphorylated serine 727 STAT3 that was transcriptionally active (37, 57). Qin and colleagues elegantly demonstrated in nude mice that overexpression of a Y705F S727E form of STAT3, which results in inactivation of Y705 and constitutive activation of S727 by the phosphomimetic residue glutamic acid, results in dramatically increased tumorigenicity in NOD/SCID mice, likely by activation of STAT3-dependent oncogenes. Noncancerous prostate epithelial cells expressing the Y705F S727E form of STAT3 displayed enhanced anchorage-independent growth in soft agar and were more invasive, strongly suggesting that S727 phosphorylation of STAT3 was sufficient to activate STAT3 and drive prostate tumorigenesis (58). Furthermore, overexpression of S727A STAT3 or inhibition of STAT3 serine phosphorylation exerted a dominant-negative effect on v-Src-mediated transformation of fibroblast cells (59, 60). Others have demonstrated a non-tyrosine-phosphorylated STAT3 (U-STAT3) form that drives expression of a subset of STAT3 responsive genes in epithelial cells, including those coding for MRAS, MET, IL-8, IL-6, and CCL5 (36). Gene expression was shown to be independent of tyrosine 705 phosphorylation using a Y705F STAT3 construct. The data indicated some genes were increased in response to overexpression of wild-type STAT3 (likely mediated by STAT3 dimers), while a distinct subset of STAT3-regulated genes were increased in cells overexpressing the Y705F mutant (36, 61, 62) and not mediated by STAT3 dimers. The relative contribution of a monophosphorylated S727 form of STAT3 was not addressed in these studies. Given that STAT3 expression itself is governed by the canonical STAT3 phospho-dimer it is possible that the resultant gene expression observed in these studies was, at least in part, due to nucleus-localized S727 monophospho-STAT3.
My data support an important role for the latent/lytic protein kaposin B in driving tumorigenesis via aberrant activation of STAT3. Kaposin B expression in ECs, independent of other latent genes, induced the phosphorylation of STAT3 on S727, in the absence of Y705 phosphorylation. The levels of kaposin B in the transduced cells were ∼16-fold higher than those expressed at 2 days p.i.(data not shown), indicating that kaposin B expression during both the latent and lytic cycles, when levels are much higher, is important for activation of STAT3 during KSHV infection. Multiple kinases have been implicated in serine 727 phosphorylation, including three MAP kinases, ERK, p38, and JNK, as well as PKC-δ and mTOR, dependent on cell type and context of activation (60, 63–68). The kinase responsible for the S727 phosphorylation event induced by kaposin B in primary ECs is not known and is under investigation. Kaposin B activation of the p38 MAPK pathway (10) may mediate the S727 phosphorylation event in primary endothelial cells. Importantly, the monophosphorylated form of STAT3 appeared to be transcriptionally active. Kaposin B-expressing ECs display increased mRNA levels of a subset of STAT3-dependent genes, including a 15- to 25-fold increase in CCL5 expression that was substantially inhibited with chemical inhibitors of MK2. Kaposin B is one of three proteins expressed from the kaposin RNA transcript in both lytically and latently infected endothelial cells. Kaposin B was initially described to contribute to the proinflammatory environment of KS lesions by manipulation of the p38 MAPK/MK2 signaling pathway to promote stability of proinflammatory cytokine mRNAs (10). My studies demonstrate that the constitutively active form of MK2 (MK2EE) in ECs induced 0.4- to 7-fold and 2.4- to 3.4-fold upregulations of CCL5 and IL-6 mRNA, respectively, indicating the relative contribution of MK2-mediated stabilization of CCL5 mRNA in ECs to the upregulation observed when kaposin B was expressed, was small.
KS lesions are characterized by chronic inflammation and neoangiogenesis; CCL5 is a potent mediator of both processes (69–72). The importance of CCL5 in KS pathogenesis is underscored by the fact that both CCL5 and the cognate receptor CCR5 are upregulated in KS but not normal skin (73). CCL5 is a potent chemoattractant for many cell types, such as monocytes, NK cells (74, 75), memory T cells (75), eosinophils (76), and dendritic cells (77), many of which are present in KS lesions. CCL5 levels correlated with neovascularization in a mouse model of angiogenesis (78), and CCL5 is thought to play a pivotal role in the recruitment of endothelial progenitors that are essential for neovascularization through interaction with CCR5 (79). KS is characterized by chronic inflammation and neoangiogenesis, and the observed kaposin B-induced increase in CCL5 has the potential to mediate these effects and contribute significantly to KS tumorigenesis.
Given the important role STAT3 plays in multiple cellular functions and the implications of deregulated activation, STAT3 activity must be tightly controlled, and TRIM28 is one recently identified transcriptional repressor. TRIM28 has a higher affinity for nucleus-localized, non-tyrosine, serine-phosphorylated STAT3, the form of P-STAT3 induced by kaposin B expression. Derepression of TRIM28 activity is achieved by phosphorylation of a serine residue at position 473 (42). My data demonstrate that KSHV infection and stable expression of the latent/lytic KSHV protein kaposin B induced robust TRIM28 S473 phosphorylation in primary ECs. Little is known about the kinases responsible for S473 phosphorylation. Initial reports described inhibition of TRIM28 repressor activity by PKC-δ using the chemical inhibitor rottlerin (42). Subsequent studies demonstrated rottlerin was not an inhibitor of PKC-δ but had activity against p38-MK2 MAPKs (80) and as a result is no longer commercially available (LC laboratories). Serendipitously, I recognized that TRIM28 S473 resides in a previously unrecognized MK2 phosphorylation site (Hyd-x-R-x-x-S*). In vitro MK2 kinase assays and the use of MK2-specific chemical inhibitors, coupled with the use of MK2/MK3−/− MEFs provided clear evidence that TRIM28 was a bona fide target of the host cell kinase MK2. The partial reduction in P-TRIM28 S473 observed in MK2/MK3−/− MEFS suggests that, like many other targets, TRIM28 S473 phosphorylation is likely mediated by the cooperation of MK2 and other kinases not identified in this study. Recently, the effector checkpoint kinase Chk1 was shown to mediate P-TRIM28 S473 in U2OS osteosarcoma cells in response to DNA-damaging agents (81). Interestingly, the p38MAPK/MK2 complex is thought to work in parallel with Chk1 in response to DNA damage and shares many of the same substrates with Chk1 (82, 83), including TRIM28.
Not surprisingly, I found P-TRIM28 S473 colocalized with LANA puncta in KSHV-infected ECs. TRIM28 has been recently identified as a transcriptional repressor of KSHV lytic gene expression, associating with lytic gene promoters, playing a role in maintaining latency (84). Taken together, the observed nuclear colocalization of active MK2 with LANA puncta in latently infected ECs suggests that all of these factors come together to modify both host and viral gene expression in infected cells.
Endothelial cells play a key role in the development and function of blood and lymph vessels and through the release of soluble mediators, including cytokines, regulate blood clotting, tissue repair, inflammation, and scarring. Excessive proliferation and transformation of endothelial cells lead to pathological angiogenesis/lymphangiogenesis or vascular malfunctions, which are hallmarks of malignant disorders. Despite their involvement in malignant disorders, ECs appear to be intrinsically resistant to tumorigenesis, with only a few known malignancies of EC origin. In addition, ECs, through their secretome, have been shown to play an essential role in inhibiting the growth and invasiveness of tumor cells (85), with emerging data suggesting a constant antagonism between cancer cells and endothelial cells, which appear to function as “cellular policemen.” KSHV successfully circumvents EC resistance to tumorigenesis, instead promoting both transformation and immune evasion. How KSHV achieves this is not well understood. The aberrant and chronic activation of STAT3 induced by KSHV and the unique uncoupling of Y705 and S727 phosphorylation in infected cells, together with MK2-mediated inactivation of the STAT3 transcriptional repressor, TRIM28, likely play an important role in circumventing EC resistance to tumorigenesis, helping to drive oncogenesis. Aberrant chronic activation of STAT3, coupled with inactivation of TRIM28, may well significantly contribute to the genesis of KS and provide valid targets for the treatment of KS.
ACKNOWLEDGMENTS
I thank Rosemary Rochford, Gary Chan, and Jennifer Corcoran for helpful discussions and review of the manuscript and Craig McCormick for assistance with the in vitro kinase assay.
Footnotes
Published ahead of print 5 June 2013
REFERENCES
- 1. Boshoff C, Schulz TF, Kennedy MM, Graham AK, Fisher C, Thomas A, McGee JO, Weiss RA, O'Leary JJ. 1995. Kaposi's sarcoma-associated herpesvirus infects endothelial and spindle cells. Nat. Med. 1:1274–1278 [DOI] [PubMed] [Google Scholar]
- 2. Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 332:1186–1191 [DOI] [PubMed] [Google Scholar]
- 3. Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d'Agay MF, Clauvel JP, Raphael M, Degos L, Sigaux F. 1995. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood 86:1276–1280 [PubMed] [Google Scholar]
- 4. Boshoff C, Weiss RA. 1998. Kaposi's sarcoma-associated herpesvirus. Adv. Cancer Res. 75:57–86 [DOI] [PubMed] [Google Scholar]
- 5. Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K, Boshoff C. 1999. Distribution of human herpesvirus-8 latently infected cells in Kaposi's sarcoma, multicentric Castleman's disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. U. S. A. 96:4546–4551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boshoff C, Weiss RA. 1997. Aetiology of Kaposi's sarcoma: current understanding and implications for therapy. Mol. Med. Today 3:488–494 [DOI] [PubMed] [Google Scholar]
- 7. Ensoli B, Nakamura S, Salahuddin SZ, Biberfeld P, Larsson L, Beaver B, Wong-Staal F, Gallo RC. 1989. AIDS-Kaposi's sarcoma-derived cells express cytokines with autocrine and paracrine growth effects. Science 243:223–226 [DOI] [PubMed] [Google Scholar]
- 8. Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. 2000. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha. Cancer Res. 60:4873–4880 [PubMed] [Google Scholar]
- 9. Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, Haase AT. 1997. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 71:715–719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McCormick C, Ganem D. 2005. The kaposin B protein of KSHV activates the p38/MK2 pathway and stabilizes cytokine mRNAs. Science 307:739–741 [DOI] [PubMed] [Google Scholar]
- 11. Catlett-Falcone R, Dalton WS, Jove R. 1999. STAT proteins as novel targets for cancer therapy. Signal transducer an activator of transcription. Curr. Opin. Oncol. 11:490–496 [DOI] [PubMed] [Google Scholar]
- 12. Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H, Eckmann L, Karin M. 2009. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15:103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kujawski M, Kortylewski M, Lee H, Herrmann A, Kay H, Yu H. 2008. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Invest. 118:3367–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Levy DE, Darnell JE., Jr 2002. Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 3:651–662 [DOI] [PubMed] [Google Scholar]
- 15. Mantovani A, Allavena P, Sica A, Balkwill F. 2008. Cancer-related inflammation. Nature 454:436–444 [DOI] [PubMed] [Google Scholar]
- 16. Mantovani A, Marchesi F, Portal C, Allavena P, Sica A. 2008. Linking inflammation reactions to cancer: novel targets for therapeutic strategies. Adv. Exp. Med. Biol. 610:112–127 [DOI] [PubMed] [Google Scholar]
- 17. Yu LF, Zhu YB, Qiao MM, Zhong J, Tu SP, Wu YL. 2004. Constitutive activation and clinical significance of Stat3 in human gastric cancer tissues and cell lines. Zhonghua Yi Xue Za Zhi 84:2064–2069 (In Chinese.) [PubMed] [Google Scholar]
- 18. Bowman T, Garcia R, Turkson J, Jove R. 2000. STATs in oncogenesis. Oncogene 19:2474–2488 [DOI] [PubMed] [Google Scholar]
- 19. Chen CL, Hsieh FC, Lieblein JC, Brown J, Chan C, Wallace JA, Cheng G, Hall BM, Lin J. 2007. Stat3 activation in human endometrial and cervical cancers. Br. J. Cancer 96:591–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garcia R, Jove R. 1998. Activation of STAT transcription factors in oncogenic tyrosine kinase signaling. J. Biomed. Sci. 5:79–85 [DOI] [PubMed] [Google Scholar]
- 21. Mora LB, Buettner R, Seigne J, Diaz J, Ahmad N, Garcia R, Bowman T, Falcone R, Fairclough R, Cantor A, Muro-Cacho C, Livingston S, Karras J, Pow-Sang J, Jove R. 2002. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 62:6659–6666 [PubMed] [Google Scholar]
- 22. Turkson J. 2004. STAT proteins as novel targets for cancer drug discovery. Expert Opin. Ther. Targets 8:409–422 [DOI] [PubMed] [Google Scholar]
- 23. Turkson J, Jove R. 2000. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19:6613–6626 [DOI] [PubMed] [Google Scholar]
- 24. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, Darnell JE., Jr 1999. Stat3 as an oncogene. Cell 98:295–303 [DOI] [PubMed] [Google Scholar]
- 25. Grandis JR, Drenning SD, Zeng Q, Watkins SC, Melhem MF, Endo S, Johnson DE, Huang L, He Y, Kim JD. 2000. Constitutive activation of Stat3 signaling abrogates apoptosis in squamous cell carcinogenesis in vivo. Proc. Natl. Acad. Sci. U. S. A. 97:4227–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H. 2002. Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000–2008 [DOI] [PubMed] [Google Scholar]
- 27. Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H. 2004. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10:48–54 [DOI] [PubMed] [Google Scholar]
- 28. Yu H, Jove R. 2004. The STATs of cancer—new molecular targets come of age. Nat. Rev. Cancer 4:97–105 [DOI] [PubMed] [Google Scholar]
- 29. Aoki Y, Feldman GM, Tosato G. 2003. Inhibition of STAT3 signaling induces apoptosis and decreases survivin expression in primary effusion lymphoma. Blood 101:1535–1542 [DOI] [PubMed] [Google Scholar]
- 30. Punjabi AS, Carroll PA, Chen L, Lagunoff M. 2007. Persistent activation of STAT3 by latent Kaposi's sarcoma-associated herpesvirus infection of endothelial cells. J. Virol. 81:2449–2458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Darnell JE, Jr, Kerr IM, Stark GR. 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421 [DOI] [PubMed] [Google Scholar]
- 32. Zhong Z, Wen Z, Darnell JE., Jr 1994. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98 [DOI] [PubMed] [Google Scholar]
- 33. Wen Z, Zhong Z, Darnell JE., Jr 1995. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82:241–250 [DOI] [PubMed] [Google Scholar]
- 34. Cimica V, Chen HC, Iyer JK, Reich NC. 2011. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PLoS One 6:e20188. 10.1371/journal.pone.0020188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu L, McBride KM, Reich NC. 2005. STAT3 nuclear import is independent of tyrosine phosphorylation and mediated by importin-alpha3. Proc. Natl. Acad. Sci. U. S. A. 102:8150–8155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang J, Liao X, Agarwal MK, Barnes L, Auron PE, Stark GR. 2007. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 21:1396–1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hazan-Halevy I, Harris D, Liu Z, Liu J, Li P, Chen X, Shanker S, Ferrajoli A, Keating MJ, Estrov Z. 2010. STAT3 is constitutively phosphorylated on serine 727 residues, binds DNA, and activates transcription in CLL cells. Blood 115:2852–2863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ., III 1996. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 10:2067–2078 [DOI] [PubMed] [Google Scholar]
- 39. Tsuruma R, Ohbayashi N, Kamitani S, Ikeda O, Sato N, Muromoto R, Sekine Y, Oritani K, Matsuda T. 2008. Physical and functional interactions between STAT3 and KAP1. Oncogene 27:3054–3059 [DOI] [PubMed] [Google Scholar]
- 40. Nielsen AL, Ortiz JA, You J, Oulad-Abdelghani M, Khechumian R, Gansmuller A, Chambon P, Losson R. 1999. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 18:6385–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schultz DC, Ayyanathan K, Negorev D, Maul GG, Rauscher FJ., III 2002. SETDB1: a novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 16:919–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang CW, Chou HY, Lin YS, Huang KH, Chang CJ, Hsu TC, Lee SC. 2008. Phosphorylation at Ser473 regulates heterochromatin protein 1 binding and corepressor function of TIF1beta/KAP1. BMC Mol. Biol. 9:61. 10.1186/1471-2199-9-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Anderson R, Wang S, Osiowy C, Issekutz AC. 1997. Activation of endothelial cells via antibody-enhanced dengue virus infection of peripheral blood monocytes. J. Virol. 71:4226–4232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Komanduri KV, Luce JA, McGrath MS, Herndier BG, Ng VL. 1996. The natural history and molecular heterogeneity of HIV-associated primary malignant lymphomatous effusions. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 13:215–226 [DOI] [PubMed] [Google Scholar]
- 45. Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. 1996. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 2:342–346 [DOI] [PubMed] [Google Scholar]
- 46. Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby WF, Blackwell TK, Anderson P. 2004. MK2-induced tristetraprolin:14-3-3 complexes prevent stress granule association and ARE-mRNA decay. EMBO J. 23:1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. 1999. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 18:4969–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kedes DH, Lagunoff M, Renne R, Ganem D. 1997. Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi's sarcoma-associated herpesvirus. J. Clin. Invest. 100:2606–2610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kamitani S, Togi S, Ikeda O, Nakasuji M, Sakauchi A, Sekine Y, Muromoto R, Oritani K, Matsuda T. 2011. Kruppel-associated box-associated protein 1 negatively regulates TNF-alpha-induced NF-kappaB transcriptional activity by influencing the interactions among STAT3, p300, and NF-kappaB/p65. J. Immunol. 187:2476–2483 [DOI] [PubMed] [Google Scholar]
- 50. Anderson DR, Hegde S, Reinhard E, Gomez L, Vernier WF, Lee L, Liu S, Sambandam A, Snider PA, Masih L. 2005. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2). Bioorg. Med. Chem. Lett. 15:1587–1590 [DOI] [PubMed] [Google Scholar]
- 51. Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C, Schulz TF. 2003. Activation of mitogen-activated protein kinase and NF-κB pathways by a Kaposi's sarcoma-associated herpesvirus K15 membrane protein. J. Virol. 77:9346–9358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS, Chandran B. 2005. ERK1/2 and MEK1/2 induced by Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 79:10308–10329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Smit MJ, Verzijl D, Casarosa P, Navis M, Timmerman H, Leurs R. 2002. Kaposi's sarcoma-associated herpesvirus-encoded G protein-coupled receptor ORF74 constitutively activates p44/p42 MAPK and Akt via Gi and phospholipase C-dependent signaling pathways. J. Virol. 76:1744–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang L, Damania B. 2008. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 68:4640–4648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Boshoff C, Whitby D, Talbot S, Weiss RA. 1997. Etiology of AIDS-related Kaposi's sarcoma and lymphoma. Oral Dis. 3(Suppl 1):S129–S132 [DOI] [PubMed] [Google Scholar]
- 56. Ensoli B, Sturzl M. 1999. HHV-8 and multistep tumourigenesis. Trends Microbiol. 7:310–312 [DOI] [PubMed] [Google Scholar]
- 57. Frank DA, Mahajan S, Ritz J. 1997. B lymphocytes from patients with chronic lymphocytic leukemia contain signal transducer and activator of transcription (STAT) 1 and STAT3 constitutively phosphorylated on serine residues. J. Clin. Invest. 100:3140–3148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Qin HR, Kim HJ, Kim JY, Hurt EM, Klarmann GJ, Kawasaki BT, Duhagon Serrat MA, Farrar WL. 2008. Activation of signal transducer and activator of transcription 3 through a phosphomimetic serine 727 promotes prostate tumourigenesis independent of tyrosine 705 phosphorylation. Cancer Res. 68:7736–7741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bromberg JF, Horvath CM, Besser D, Lathem WW, Darnell JE., Jr 1998. Stat3 activation is required for cellular transformation by v-src. Mol. Cell. Biol. 18:2553–2558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Turkson J, Bowman T, Adnane J, Zhang Y, Djeu JY, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S, Jove R. 1999. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol. Cell. Biol. 19:7519–7528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang J, Chatterjee-Kishore M, Staugaitis SM, Nguyen H, Schlessinger K, Levy DE, Stark GR. 2005. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65:939–947 [PubMed] [Google Scholar]
- 62. Yang J, Stark GR. 2008. Roles of unphosphorylated STATs in signaling. Cell Res. 18:443–451 [DOI] [PubMed] [Google Scholar]
- 63. Busch S, Renaud SJ, Schleussner E, Graham CH, Markert UR. 2009. mTOR mediates human trophoblast invasion through regulation of matrix-remodeling enzymes and is associated with serine phosphorylation of STAT3. Exp. Cell Res. 315:1724–1733 [DOI] [PubMed] [Google Scholar]
- 64. Chung J, Uchida E, Grammer TC, Blenis J. 1997. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol. Cell. Biol. 17:6508–6516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jain N, Zhang T, Kee WH, Li W, Cao X. 1999. Protein kinase C delta associates with and phosphorylates Stat3 in an interleukin-6-dependent manner. J. Biol. Chem. 274:24392–24400 [DOI] [PubMed] [Google Scholar]
- 66. Lim CP, Cao X. 1999. Serine phosphorylation and negative regulation of Stat3 by JNK. J. Biol. Chem. 274:31055–31061 [DOI] [PubMed] [Google Scholar]
- 67. Lutticken C, Coffer P, Yuan J, Schwartz C, Caldenhoven E, Schindler C, Kruijer W, Heinrich PC, Horn F. 1995. Interleukin-6-induced serine phosphorylation of transcription factor APRF: evidence for a role in interleukin-6 target gene induction. FEBS Lett. 360:137–143 [DOI] [PubMed] [Google Scholar]
- 68. Yokogami K, Wakisaka S, Avruch J, Reeves SA. 2000. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 10:47–50 [DOI] [PubMed] [Google Scholar]
- 69. Denes A, Humphreys N, Lane TE, Grencis R, Rothwell N. 2010. Chronic systemic infection exacerbates ischemic brain damage via a CCL5 (regulated on activation, normal T-cell expressed and secreted)-mediated proinflammatory response in mice. J. Neurosci. 30:10086–10095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Suffee N, Hlawaty H, Meddahi-Pelle A, Maillard L, Louedec L, Haddad O, Martin L, Laguillier C, Richard B, Oudar O, Letourneur D, Charnaux N, Sutton A. 2012. RANTES/CCL5-induced pro-angiogenic effects depend on CCR1, CCR5 and glycosaminoglycans. Angiogenesis 15:727–744 [DOI] [PubMed] [Google Scholar]
- 71. Suffee N, Richard B, Hlawaty H, Oudar O, Charnaux N, Sutton A. 2011. Angiogenic properties of the chemokine RANTES/CCL5. Biochem. Soc. Trans. 39:1649–1653 [DOI] [PubMed] [Google Scholar]
- 72. Zernecke A, Shagdarsuren E, Weber C. 2008. Chemokines in atherosclerosis: an update. Arterioscler. Thromb. Vasc. Biol. 28:1897–1908 [DOI] [PubMed] [Google Scholar]
- 73. Wang HW, Trotter MW, Lagos D, Bourboulia D, Henderson S, Makinen T, Elliman S, Flanagan AM, Alitalo K, Boshoff C. 2004. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 36:687–693 [DOI] [PubMed] [Google Scholar]
- 74. Loetscher P, Seitz M, Clark-Lewis I, Baggiolini M, Moser B. 1996. Activation of NK cells by CC chemokines. Chemotaxis, Ca2+ mobilization, and enzyme release. J. Immunol. 156:322–327 [PubMed] [Google Scholar]
- 75. Schall TJ, Bacon K, Toy KJ, Goeddel DV. 1990. Selective attraction of monocytes and T lymphocytes of the memory phenotype by cytokine RANTES. Nature 347:669–671 [DOI] [PubMed] [Google Scholar]
- 76. Rot A, Krieger M, Brunner T, Bischoff SC, Schall TJ, Dahinden CA. 1992. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 176:1489–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Dieu MC, Vanbervliet B, Vicari A, Bridon JM, Oldham E, Ait-Yahia S, Briere F, Zlotnik A, Lebecque S, Caux C. 1998. Selective recruitment of immature and mature dendritic cells by distinct chemokines expressed in different anatomic sites. J. Exp. Med. 188:373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Barcelos LS, Coelho AM, Russo RC, Guabiraba R, Souza AL, Bruno-Lima G, Jr, Proudfoot AE, Andrade SP, Teixeira MM. 2009. Role of the chemokines CCL3/MIP-1 alpha and CCL5/RANTES in sponge-induced inflammatory angiogenesis in mice. Microvasc. Res. 78:148–154 [DOI] [PubMed] [Google Scholar]
- 79. Ishida Y, Kimura A, Kuninaka Y, Inui M, Matsushima K, Mukaida N, Kondo T. 2012. Pivotal role of the CCL5/CCR5 interaction for recruitment of endothelial progenitor cells in mouse wound healing. J. Clin. Invest. 122:711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Soltoff SP. 2007. Rottlerin: an inappropriate and ineffective inhibitor of PKCdelta. Trends Pharmacol. Sci. 28:453–458 [DOI] [PubMed] [Google Scholar]
- 81. Blasius M, Forment JV, Thakkar N, Wagner SA, Choudhary C, Jackson SP. 2011. A phospho-proteomic screen identifies substrates of the checkpoint kinase Chk1. Genome Biol. 12:R78. 10.1186/gb-2011-12-8-r78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. 2007. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11:175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Reinhardt HC, Yaffe MB. 2009. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21:245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chang PC, Fitzgerald LD, Van Geelen A, Izumiya Y, Ellison TJ, Wang DH, Ann DK, Luciw PA, Kung HJ. 2009. Kruppel-associated box domain-associated protein-1 as a latency regulator for Kaposi's sarcoma-associated herpesvirus and its modulation by the viral protein kinase. Cancer Res. 69:5681–5689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Franses JW, Baker AB, Chitalia VC, Edelman ER. 2011. Stromal endothelial cells directly influence cancer progression. Sci. Transl. Med. 3:66ra65. 10.1126/scitranslmed.3001542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kim SS, Chen YM, O'Leary E, Witzgall R, Vidal M, Bonventre JV. 1996. A novel member of the RING finger family, KRIP-1, associates with the KRAB-A transcriptional repressor domain of zinc finger proteins. Proc. Natl. Acad. Sci. U. S. A. 93:15299–152304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Agata Y, Matsuda E, Shimizu A. 1999. Two novel Krüppel-associated box-containing zinc-finger proteins, KRAZ1 and KRAZ2, repress transcription through functional interaction with the corepressor KAP-1 (TIF1beta/KRIP-1). J. Biol. Chem. 274:16412–16422 [DOI] [PubMed] [Google Scholar]