

Abstract
A rare neurodevelopmental disorder in the Old Order Mennonite population called PMSE (polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome; also called Pretzel syndrome) is characterized by infantile-onset epilepsy, neurocognitive delay, craniofacial dysmorphism, and histopathological evidence of heterotopic neurons in subcortical white matter and subependymal regions. PMSE is caused by a homozygous deletion of exons 9 to 13 of the LYK5/STRADA gene, which encodes the pseudokinase STRADA, an upstream inhibitor of mammalian target of rapamycin complex 1 (mTORC1). We show that disrupted pathfinding in migrating mouse neural progenitor cells in vitro caused by STRADA depletion is prevented by mTORC1 inhibition with rapamycin or inhibition of its downstream effector p70 S6 kinase (p70S6K) with the drug PF-4708671 (p70S6Ki). We demonstrate that rapamycin can rescue aberrant cortical lamination and heterotopia associated with STRADA depletion in the mouse cerebral cortex. Constitutive mTORC1 signaling and a migration defect observed in fibroblasts from patients with PMSE were also prevented by mTORC1 inhibition. On the basis of these preclinical findings, we treated five PMSE patients with sirolimus (rapamycin) without complication and observed a reduction in seizure frequency and an improvement in receptive language. Our findings demonstrate a mechanistic link between STRADA loss and mTORC1 hyperactivity in PMSE, and suggest that mTORC1 inhibition may be a potential treatment for PMSE as well as other mTOR-associated neurodevelopmental disorders.
INTRODUCTION
Loss-of-function mutation of the LYK5/STRADA gene is associated with PMSE (polyhydramnios, megalencephaly, and symptomatic epilepsy syndrome) (Online Mendelian Inheritance in Man #611087), a recessive neurodevelopmental disorder characterized by intractable epilepsy and severe cognitive impairment (1). Numerous heterotopic neurons in the subcortical white matter as well as subcortical and subependymal dysplasias are observed in the brains of PMSE patients, suggesting a neural migratory defect and prompting consideration of the protein encoded by the LYK5/STRADA locus called STRADA (STE20-related kinase adaptor α) as a pivotal regulator of neuronal pathfinding and migration (2). All PMSE patients share a common homozygous 7.3-kb truncating deletion of exons 9 to 13 of the LYK5/STRADA gene, which removes the protein interacting domains that mediate complex formation with serine-threonine kinase 11 (STK11, also LKB1), a known regulator of mammalian target of rapamycin (mTOR) (3). It is estimated that about 4% of Old Order Mennonite individuals in Ohio, Pennsylvania, and New York are hemizygous for the LYK5/STRADA deletion.
The STRADA/LKB1 heteromer inhibits mTOR complex 1 (mTORC1) signaling via phospho-activation of adenosine 5′-monophosphate (AMP)–activated kinase (AMPK) and tuberous sclerosis complex 2 (TSC2) in gut, skin, and brain (2, 4–6). Previous work in our laboratory has shown that STRADA knockdown in vitro leads to enhanced mTORC1 signaling in mouse neural progenitor cells (mNPCs) and, in vivo, results in clusters of heterotopic neurons within the murine postnatal subventricular zone (SVZ) similar to heterotopia in PMSE (2). However, a direct assessment of a role for STRADA in neuronal migration and pathfinding, or of the cell signaling mechanisms responsible for linking STRADA to progenitor cell migration, has not been reported. Furthermore, the dependence of the PMSE neurological phenotype on aberrant mTORC1 signaling has not yet been established.
Loss-of-function mutations in mTOR-inhibitory genes such as TSC1 or TSC2, or gain-of-function mutations in mTOR activators such as PI3K or AKT3, result in aberrant brain development and intractable seizures (7–9). Because epilepsy in these patients is so often refractory to treatment with antiepileptic drugs, directly inhibiting the mTOR pathway might prove effective (10, 11). The mTORC1 inhibitor rapamycin (referred to as sirolimus in clinical therapy) may have the potential to reduce seizure frequency in patients with TSC, who have medically intractable epilepsy (12, 13). Recently, the sirolimus analog everolimus was associated with a reduction in subependymal giant cell astrocytoma (SEGA) volume and seizure frequency in TSC (14).
All PMSE patients develop intractable seizures and severe cognitive disability, and no effective treatment strategies have yet been reported. We propose that PMSE could serve as an effective model of mTOR-associated brain disease because all PMSE patients share the same deletion in STRADA and thus represent a homogeneous study group. Here, we use mNPCs and fibroblasts from PMSE patients to show that STRADA is a pivotal regulatory protein that governs pathfinding of migrating neural progenitor cells via an mTORC1-dependent mechanism. On the basis of these preclinical data, we then demonstrate that mTORC1 inhibition with sirolimus can reduce seizure frequency in PMSE patients.
RESULTS
STRADA knockdown in mNPCs causes an mTORC1-dependent migration defect in vitro
Stably transfected mNPC lines were established with puromycin-resistant STRADA short hairpin RNA (shRNA) (STRADA shRNA-puroR, STRADA knockdown) for STRADA knockdown and control puromycin-resistant scrambled shRNA (scrambled shRNA-puroR, scrambled). STRADA depletion (more than 90%) was confirmed by Western blot analysis (Fig. 1A). STRADA knockdown mNPCs exhibit activated mTORC1 signaling as evidenced by enhanced phosphorylation of downstream targets 4EBP1 (eukaryotic translation initiation factor 4E–binding protein 1) and ribosomal S6 protein, a substrate of the mTORC1 effector p70 S6 kinase (p70S6K) (Fig. 1A). Insulin receptor substrate 1 (IRS1) is a recognized downstream target of mTORC1 and p70S6K that signals to the actin cytoskeleton machinery via cofilin, and may play an important role in cell migration (fig. S1) (15, 16). Phosphorylation of IRS1 was up-regulated and phosphorylation of cofilin was diminished after STRADA knockdown (Fig. 1B). Aberrant IRS1 and cofilin phosphorylation was prevented by inhibition of p70S6K with a selective p70S6K kinase inhibitor, PF-4708671 (10 μM, Pfizer; p70S6Ki) (17).
Fig. 1. The migration deficit in mNPCs due to STRADA knockdown is rescued by mTORC1 or p70S6K inhibition.
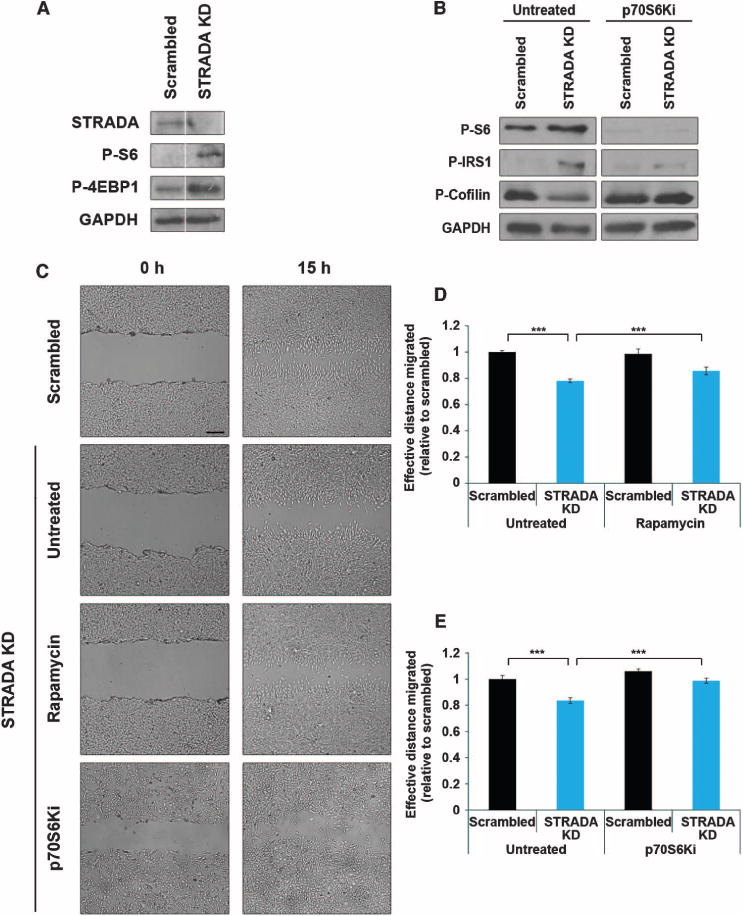
(A) STRADA depletion and enhanced mTORC1 signaling in stably transfected STRADA knockdown (STRADA KD) mNPCs used for migration assays. STRADA knockdown results in reduced expression of STRADA and enhanced phospho-S6 (P-S6) and phospho-4EBP1 (P-4EBP1), indicating mTORC1 activity relative to scrambled shRNA control. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) serves as an internal loading control. (B) p70S6K inhibition rescues aberrant signaling in STRADA knockdown mNPCs. Untreated STRADA knockdown mNPCs exhibit enhanced phospho-S6 and phospho-IRS1 (P-IRS1), with diminished inhibitory phosphorylation of cofilin, indicating aberrant signaling through mTORC1/p70S6K, which is rescued with p70S6Ki. GAPDH serves as an internal loading control. (C) Representative images depict gap closure in each transfection (scrambled shRNA or STRADA knockdown) and treatment (untreated, rapamycin, and p70S6Ki) condition from the time the scratch is made (0 hours) to the endpoint of measurement (15 hours). Scale bar, 250 μm. (D) Untreated STRADA knockdown mNPCs exhibit reduced effective distance migrated relative to scrambled control. n = 3 wells, 90 measures per condition per time point. ***P < 0.001, one-way analysis of variance (ANOVA) with Bonferroni correction. (E) Treatment with either rapamycin (D) or p70S6Ki (E) significantly increased the effective distance migrated by STRADA knockdown cells, confirming that STRADA regulates mNPC migration through mTORC1/p70S6K signaling. n = 3 wells, 90 measures per condition per time point. ***P < 0.001, one-way ANOVA with Bonferroni correction. All Western blot assays were run at the same time, single blot per protein. White line in (A) depicts empty lane between bands for each protein.
Next, we used a modified wound-healing assay, previously described in fibroblasts (18), to demonstrate that STRADA loss impairs cell migration and to test the hypothesis that failure of neural progenitor cell migration due to loss of STRADA in vitro can be rescued with inhibition of mTORC1 or p70S6K. A 200-μl micropipette tip was used to create a scratch gap in a layer of confluent mNPCs. The gap established opposing migratory leading edges so that measurements of cell migration across the gap could be determined from the time of the scratch (0 hours) to 15 hours later. Because of the inherent heterogeneity of experimental scratches created, measurements were standardized within each image region, such that effective distance migrated was recorded as half of the difference in the distance measured between cell edges at the same location between 0 and 15 hours. Knockdown of STRADA reduced the gap closure at 15 hours, compared with cells treated with scrambled shRNA, demonstrating that STRADA knockdown impairs mNPC migration in vitro (Fig. 1C).
The defect in gap closure at 15 hours caused by STRADA knockdown was prevented with rapamycin (50 nM) applied 1 hour before the initiation of the assay (−1 hour) and maintained until 15 hours (Fig. 1, C and D, and table S1). Correlatively, mNPCs treated with p70S6Ki (10 μM), applied from −1 to 15 hours throughout the course of the assay, exhibited a full rescue of the STRADA knockdown–associated migration defect, resulting in a significantly greater distance migrated by STRADA knockdown cells at 15 hours (Fig. 1, C and E, and table S1). Thus, altered migration of STRADA-depleted mNPCs in which STRADA had been knocked down is mediated through an mTORC1-and p70S6K-dependent mechanism. To rule out the possibility that STRADA knockdown or rapamycin treatment produced differences in gap closure by altering cell mitotic rate, we applied the mitotic inhibitor arabinosylcytosine (Ara-C, 20 μM) 24 hours before and during the course of migration. After Ara-C treatment, STRADA-depleted mNPCs continued to exhibit a significant migration deficit, which was rescued by rapamycin, suggesting that STRADA’s role in mNPC migration is mTORC1-dependent and not altered by changes in cell mitosis (fig. S2 and table S1).
Impaired mNPC pathfinding after STRADA knockdown is rescued by rapamycin
We next analyzed the total distance migrated and directionality of individual STRADA-depleted mNPCs in real time using video time-lapse microscopy. We hypothesized that STRADA knockdown would reduce the effective distance migrated, representing the integration of total distance and directionality, of leading-edge mNPCs. Movement of multiple individual STRADA-depleted mNPCs and mNPCs treated with scrambled shRNA was captured every 30 min for 20 hours. A blinded observer evaluated five representative leading-edge cells per video and measured for each cell the distance traveled and direction angle relative to a unit circle over each 30-min epoch. To evaluate motility and pathfinding, we calculated the total distance traveled for each cell as the sum of all distances measured over 20 hours and calculated directionality as the variance of the set of angles over that time period.
Whereas most control scrambled shRNA cells moved forward in a linear fashion, STRADA-depleted cells moved in divergent, nonlinear directions, often oriented away from the opposing cell front (Fig. 2A and movies S1 and S2). We observed that STRADA-depleted mNPCs translocated a greater total distance than cells treated with scrambled shRNA (Fig. 2B and table S1). However, compared to cells treated with scrambled shRNA, STRADA knockdown resulted in a profound impairment in directionality, with STRADA-depleted mNPCs exhibiting significantly greater directional variance than controls (Fig. 2D and table S1). STRADA-depleted mNPCs migrated, on average, a 1.5-fold greater total distance and exhibited a nearly 3-fold increase in directional variance compared to cells treated with scrambled shRNA. Thus, in the absence of functional STRADA, individual mNPCs lose the ability to follow a linear migratory pathway. Rapamycin treatment reduced total distance migrated by STRADA-depleted cells, as well as diminished directional variance compared with untreated STRADA-depleted mNPCs (Fig. 2, A, C, and E, and table S1). Overall, rapamycin enhanced linear directionality and enabled more uniform migration of STRADA-depleted mNPCs (movie S3). These results demonstrate that STRADA plays a pivotal role in establishing linear directionality and pathfinding of migrating mNPCs through an mTORC1-dependent mechanism, and suggest the important therapeutic relevance of targeting this pathway.
Fig. 2. STRADA-depleted mNPCs exhibit impaired pathfinding rescued by mTORC1 inhibition.
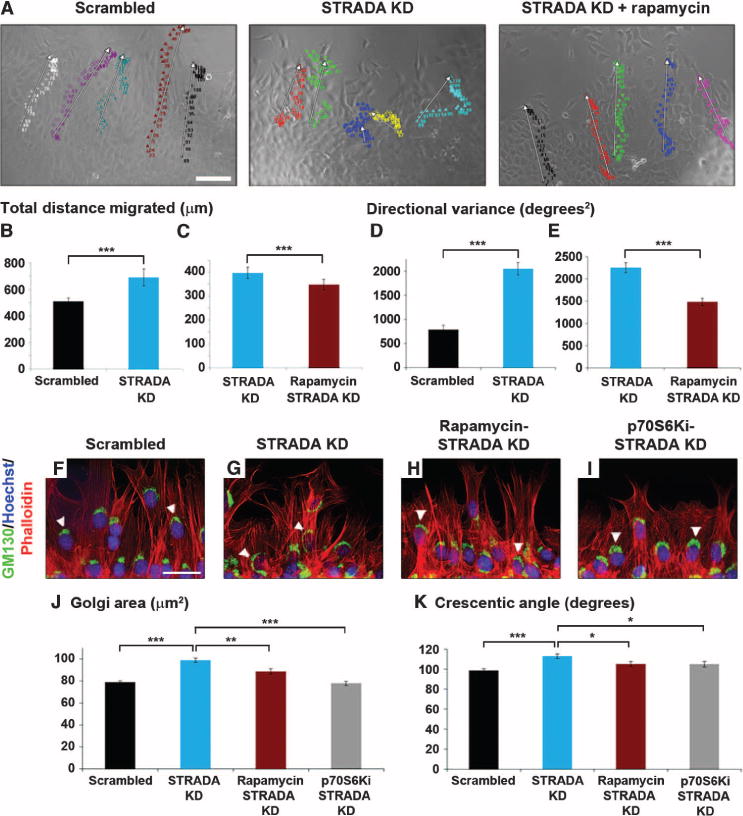
(A) Live-cell video microscopy (see also movies S1 to S3). Individual colors represent migratory paths of individual cells. White arrows indicate composite migration vectors. (Left) mNPCs treated with scrambled shRNA migrate with uniform linearity. (Middle) STRADA knockdown (STRADA KD) mNPCs migrate haphazardly without consistent linearity. (Right) Rapamycin restores linearity. Scale bar, 100 μm. (B to E) STRADA knockdown mNPCs exhibit significantly greater (B) total distance migrated and (D) directional variance; (C and E) defect was rescued with rapamycin. n = 15, untreated STRADA knockdown cells, n = 20, rapamycin or p70S6Ki treated cells. ***P < 0.001, unpaired two-tailed Student’s t test. (F to I) Migrating mNPCs were stained with GM130, Hoechst, and phalloidin. (F and G) Golgi compacts forward of the nucleus toward the leading edge, indicating cell polarization, in mNPCs treated with scrambled shRNA, and disperses around the nucleus in STRADA knockdown mNPCs. Scale bar, 50 μm. (H and I) Rapamycin (H) or p70S6Ki (I) restores Golgi compaction. White arrowheads indicate representative Golgi. (J and K) Golgi area (J) and crescentic angle (K) around the nucleus indicating Golgi compaction reveal significantly greater dispersion in STRADA knockdown mNPCs compared with scrambled shRNA–treated mNPCs; these defects are attenuated with rapamycin or p70S6Ki. n = 640 scrambled shRNA–treated cells, 600 untreated STRADA knockdown cells, 300 rapamycin-treated STRADA knockdown cells, and 360 p70S6Ki-treated STRADA knockdown cells. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA with Dunnett post hoc analysis.
STRADA regulates mNPC polarization through mTORC1/p70S6K signaling
Because pathfinding depends in large measure on appropriate establishment of cell polarity, we chose to assay mNPC polarization in the setting of STRADA depletion. Measuring the size of the Golgi apparatus provided us with a quantitative means of evaluating cell polarity because it is well established that the Golgi is compacted and localized forward of the nucleus in the direction of migration, that is, the cell’s leading edge, during active motility of fibroblasts and neurons (19), and that cell polarity depends on the intact structure, function, and localization of the Golgi apparatus (20–22). Therefore, we used Golgi compaction as a bioassay to measure polarization of mNPCs (fig. S3).
Ten hours after initiation of migration, cells were fixed and immunostained for the Golgi marker GM130, and colabeled with Hoechst nuclear stain and phalloidin for actin cytoskeleton visualization. A blinded observer measured Golgi area and the crescentic angle subtended by the Golgi around each cell’s nucleus. We hypothesized that if STRADA knockdown disrupted neural progenitor cell polarization, STRADA-depleted mNPCs would exhibit Golgi dispersion (greater area and crescentic angle). STRADA knockdown increased Golgi area and the crescentic angle of the Golgi around the nucleus compared with cells treated with scrambled shRNA (Fig. 2, F, G, J, and K, and table S1). In a separate experiment, STRADA-depleted or scrambled shRNA-treated mNPCs were treated with rapamycin (50 nM) or p70S6Ki (10 μM). Enhanced Golgi area and widened crescentic angle in STRADA-depleted mNPCs were reduced with rapamycin or p70S6Ki treatment (Fig. 2, H to K, and table S1). Treatment with p70S6Ki trended to a more robust pheno-typic rescue than rapamycin, but this was not statistically significant. Thus, reduced Golgi compaction associated with STRADA depletion suggests that STRADA may play a role in polarizing migrating mNPCs in an mTORC1- and p70S6K-dependent manner. This supports the rescue potential of mTORC1 inhibition indicated in our video migration assays.
Rapamycin prevents heterotopic neurons associated with STRADA knockdown in developing mouse cortex
Our in vitro assays provide functional confirmation that STRADA loss causes an mTORC1-dependent impairment of migration, pathfinding, and polarity. We next hypothesized that mTORC1 inhibition could rescue aberrant cortical lamination in a mouse model of PMSE (2). STRADA knockdown with green fluorescent protein (GFP)–tagged shRNA plasmids targeting STRADA by in utero electroporation (IUE) on embryonic day 14 (E14) in the mouse cortex resulted in heterotopic neurons in the ventricular zone/subventricular zone (VZ/SVZ) that fail to migrate to their appropriate destination in the cortical plate by E19. Transfected pregnant dams were treated with daily intraperitoneal injections of rapamycin (5 mg/kg; n = 6) or vehicle control solution [5% polyethylene glycol 400 (PEG-400), 5% Tween 80, 0.9% saline in sterile water; n = 5] from E15 until sacrifice at E19. Whereas vehicle-treated pups exhibited a cortical malformation with most of the STRADA-depleted neurons failing to migrate into the cortical plate by E19 (Fig. 3, A and B), rapamycin treatment rescued the cortical migratory defect and enabled STRADA-depleted neurons to reach the cortical plate (Fig. 3, C to E, and table S1). These findings support a mechanistic link between enhanced mTORC1 signaling and aberrant neuronal migration in PMSE. These results provide in vivo preclinical data suggesting the possible efficacy of rapamycin in PMSE.
Fig. 3. Inhibition of mTORC1 with rapamycin rescues the cortical lamination defect due to STRADA depletion in developing mouse brain.
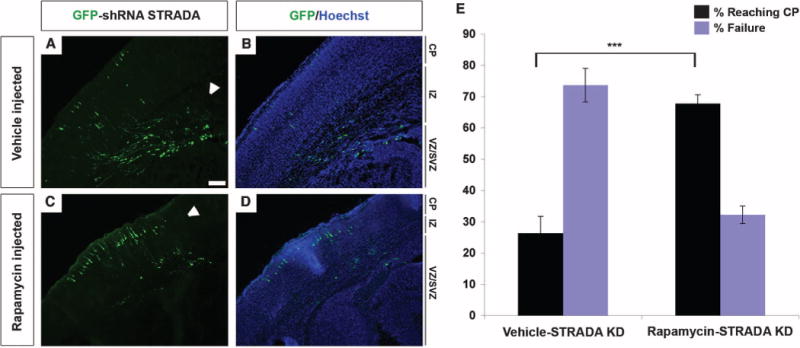
(A and B) Vehicle control injection does not alter the cortical lamination defect at E19 induced by IUE and transfection with GFP-shRNA STRADA at E14. Most of the transfected cells remain in the VZ/SVZ and fail to reach the cortical plate (CP) through translocation of the intermediate zone (IZ). Scale bar, 100 μm. (C and D) Rapamycin treatment of GFP-shRNA STRADA–transfected animals from E15 to E18 prevents the laminar defect, and most of the GFP+ cells reach the cortical plate by E19. White arrowheads indicate regions of GFP+ cells in each panel. (E) Percentage of GFP-shRNA STRADA–transfected cells reaching the cortical pate at E19 after rapamycin versus control (vehicle) treatment. ***P < 0.001, unpaired two-tailed Student’s t test.
Fibroblasts from patients with PMSE exhibit aberrant mTORC1 signaling
To confirm the effects of STRADA depletion on cell migration and mTORC1 signaling, and to test the efficacy of mTORC1 and p70S6K inhibition for rescue in a human model, we obtained fibroblasts from skin punch biopsy samples from normal controls (STRADA+/+; n = 1), heterozygous parents (STRADA+/−; n = 2), and PMSE patients (STRADA−/−; n = 3) (Fig. 4A). STRADA depletion correlated with enhanced mTORC1 activity, evaluated through P-S6 expression, in a dose-dependent manner (Fig. 4B). Compared to control cells, PMSE fibroblasts exhibit enhanced S6 and IRS1 phosphorylation and diminished cofilin phosphorylation, directly corroborating the mTORC1 signaling mechanism defined in STRADA-depleted mNPCs (fig. S4). Treatment of PMSE fibroblasts with p70S6Ki reduced S6 and IRS1 phosphorylation (Fig. 4C).
Fig. 4. PMSE fibroblasts exhibit enhanced mTORC1/p70S6K activity and impaired migration.
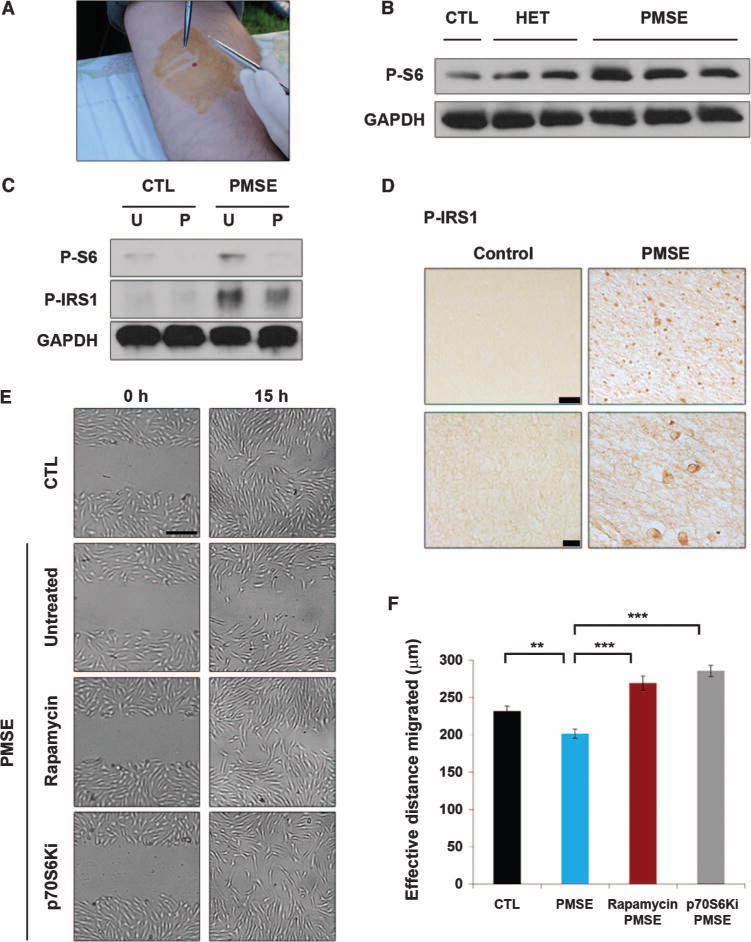
The impaired migration and enhanced mTORC1/p70S6K activity exhibited by PMSE fibroblasts were rescued by mTORC1 or p70S6K inhibition. (A) Extraction of fibroblasts from control, heterozygous parents, and PMSE patients through skin punch biopsy. (B) Enhanced mTORC1 activity in PMSE fibroblasts. Western blot analysis performed on control (CTL; STRADA+/+), heterozygous STRADA (HET; STRADA+/−), and PMSE patient (PMSE; STRADA−/−) fibroblasts reveals enhanced phospho-S6 associated with loss of STRADA. Each band represents a fibroblast line derived from a separate donor. GAPDH serves as an internal loading control. (C) Untreated PMSE fibroblasts (U) exhibit greater phospho-S6 and phospho-IRS1 than control cells, and these effects are attenuated with p70S6K inhibition (P). GAPDH serves as an internal loading control. (D) PMSE patient cortex exhibits enhanced expression of phospho-IRS1 relative to control cortex. Scale bars, 50 μm (top) and 10 μm (bottom). (E) In a wound-healing migration assay, untreated PMSE fibroblasts demonstrate impaired effective migration at 15 hours, compared with controls, corroborating the deficit associated with STRADA knockdown in mNPCs. This deficit is rescued by rapamycin or p70S6Ki treatment. Scale bar, 250 μm. (F) Quantification of PMSE fibroblast migration rescue with rapamycin or p70S6Ki. n = 3 wells, 90 measures per condition at each time point. **P < 0.01, ***P < 0.001, one-way ANOVA with Bonferroni correction.
To confirm our findings in human brain, we investigated the level of IRS1 phosphorylation in the cortex of PMSE patients and found that P-IRS1 was enhanced in PMSE cortex compared with control cortex (Fig. 4D). This suggested that enhanced mTORC1 signaling is a systemic effect seen in lymphoblasts, fibroblasts, and brain of PMSE patients (2).
PMSE patient fibroblasts exhibit a migration defect rescued by mTORC1/p70S6K inhibition
As a strategy to further corroborate the functional effects of STRADA depletion in mNPCs with different cell types from PMSE patients, we next performed a wound-healing migration assay with human PMSE and control fibroblasts. Subsets of PMSE and control fibroblasts were treated with rapamycin (100 nM) or p70S6Ki (10 μM) throughout the duration of the assay (applied at −1 hour and ending at 15 hours). The effective distance migrated by untreated PMSE fibroblasts was reduced relative to control fibroblasts, and this effect was reversed with rapamycin or p70S6Ki treatment, suggesting a dependence on mTORC1/p70S6K signaling in the human cell model (Fig. 4, E and F, and table S1). Thus, both the signaling and functional effects of STRADA loss identified in mNPCs are replicated in human PMSE cells. The parallels between signaling patterns and migration deficits in STRADA-depleted mNPCs and PMSE fibroblasts demonstrate that our in vitro and in vivo murine data provide translational insights into PMSE patho-genesis and the role of STRADA in human brain development.
Sirolimus (rapamycin) reduces seizure frequency in PMSE patients
PMSE is a rare disorder (http://www.rarediseases.org/) with assured and devastating outcomes including intractable epilepsy, severely limited cognitive development, and profound impairments of language function. Our current data provide functional and translational confirmation that loss of STRADA in mNPCs, mouse brain, and PMSE patient lymphoblasts (2), fibroblasts, and brain causes constitutively enhanced mTORC1 cascade signaling linked to impaired migration, pathfinding, and polarity, and demonstrate that inhibition of mTORC1 (or its downstream substrate p70S6K) can reverse or prevent the effects of STRADA loss. These biological effects are reminiscent of the more common neurological disorder TSC, associated with seizures, altered brain structure, and enhanced mTOR activation, which has recently been shown to respond to therapy with sirolimus (14, 23). Thus, we hypothesized that because there are no known treatments for PMSE and the neurological outcome is uniformly poor, treatment of PMSE patients with sirolimus might alter seizure frequency. Although sirolimus has no clear effect on neuronal firing patterns in vitro (24), reduction of mTOR activation has been linked to reduced seizures in animal models of TSC as well as in TSC patients. This translational approach has been previously suggested for rare and devastating diseases for which large-scale clinical trials are not feasible (25).
Five Mennonite children (current mean age, 3.0 ± 1.7 years; range, 0.7 to 4.7 years; three female; Table 1) homozygous for the LYK5/STRADA 7.3-kb deletion were treated prospectively with sirolimus from infancy and followed longitudinally at the Clinic for Special Children (Strasburg, PA). The treatment protocol was approved by the Institutional Review Board of Lancaster General Hospital, and parents of PMSE patients consented in writing to their children’s participation. Once-daily sirolimus therapy was started at a mean age of 4.8 ± 2.2 months (range, 3 to 8 months) and adjusted to achieve a trough blood concentration between 5 and 15 ng/ml (23). Sirolimus treatment was stopped temporarily during infectious illnesses and then restarted.
Table 1. STRADA-deficient PMSE patients treated with sirolimus (rapamycin) exhibit reduced seizure frequency.
Each row represents the sirolimus treatment specifications for and clinical characteristics of a single PMSE patient. The youngest child in this cohort (patient 1) has remained seizure-free at 8 months of age. Only one child in the cohort has had a single seizure in the last 12 months. Patient 3, a relative outlier with 180 seizures during his worst year, has even remained seizure-free over the last 12 months on sirolimus. All patients in this study group are on only one or two antiepileptic drugs (AEDs), a decrease from the typical four to five AEDs historically required per PMSE patient not on sirolimus. There has been no mortality within this cohort, and no patients have experienced severe adverse events causing them to terminate sirolimus treatment. Developmental quotient is defined as developmental age, determined by the Denver Developmental Screening Test II, divided by chronological age. A score of 1.0 represents development commensurate with age. Gross motor development of PMSE children is hindered by congenital absence of the anterior cruciate ligaments and precludes ambulation. All PMSE patients are mute, and we therefore determined separate developmental quotients for expressive or spoken language and receptive or understood language, the latter represented by pointing to or otherwise indicating pictures, body parts, or family members. BMI, body mass index; CBZ, carbamazepine; LEV, levetiracetam; OXC, oxcarbazepine; PB, phenobarbital; TOP, topiramate; GTC/SE, generalized tonic-clonic seizure +/− status epilepticus.
| Patient | 1 | 2 | 3 | 4 | 5 | Average (SD) |
|---|---|---|---|---|---|---|
| Age | 8 months | 1 year 7 months | 3 years 6 months | 4 years 6 months | 4 years 8 months | 3 years 0 months (1 year 8 months) |
| BMI (kg/m2) | 15.0 | 16.2 | 14.4 | 13.3 | 15.0 | 14.8 (1.1) |
| Sirolimus start (month) | 3 | 8 | 6 | 3 | 4 | 4.8 (2.2) |
| Current dose (mg/m2) | 1.0 | 1.8 | 1.1 | 3.9 | 4.9 | 2.5 (1.8) |
| Average sirolimus trough (ng/ml) | 18.5 | 1.3 | 12.9 | 4.6 | 3.6 | 8.2 (7.2) |
| AEDs | OXC | OXC PB | LEV TOP | CBZ | OXC TOP | |
| GTC/SE | 0 | 0 | 22 | 1 | 2 | 6 (11) |
| Worst no. of seizures/year | 0 | 4 | 180 | 1 | 4 | 38 (80) |
| No. of seizures in the last 12 months | 0 | 0 | 0 | 0 | 1 | 0.2 (0.4) |
| Gross motor | 0.76 | 0.44 | 0.24 | 0.14 | 0.22 | 0.37 (0.22) |
| Adaptive | 0.69 | 0.56 | 0.24 | 0.27 | 0.22 | 0.40 (0.21) |
| Expressive language | 0.63 | 0.44 | 0.14 | 0.12 | 0.14 | 0.29 (0.23) |
| Receptive language | 0.63 | 0.44 | 0.41 | 0.49 | 0.40 | 0.47 (0.10) |
| Social | 0.69 | 0.60 | 0.33 | 0.33 | 0.27 | 0.44 (0.19) |
We tracked the number of complex partial and secondarily generalized seizures and reported the per annum rate during the patient’s worst year for seizures, the total frequency of seizures in the 12 months preceding this report (that is, “current” seizure frequency), the lifetime number of secondarily generalized tonic-clonic seizures (in some cases, evolving to status epilepticus), and concurrent antiepileptic medication use. Psychomotor development was determined by serial assessments with the Denver Developmental Screening Test II and reported as developmental quotients, defined as the developmental age in each domain divided by chronological age, where a value of 1.0 represents neurological development commensurate with age. There are two relevant caveats to developmental assessments in PMSE children: (i) Gross motor development is hindered by congenital absence of the anterior cruciate ligaments, which leads to recurrent knee dislocations and in most cases precludes standing and walking, and (ii) all PMSE patients are mute, and therefore, receptive and expressive language skills are considered as separate domains.
The clinical characteristics of five Mennonite children treated with sirolimus are summarized in Table 1. There were no deaths among the five sirolimus-treated patients (six of the historical PMSE controls died by age 6), and sirolimus was well tolerated without serious adverse events causing cessation of therapy. These five treated children were compared to a group of historical PMSE patients [n = 16, ages 7 months to 28 years; see table S2; adapted from (1)], in whom 100% currently experience ongoing and intractable seizures. Although exact seizure counts were not available for each control patient, all of these patients experienced at least monthly and, in many cases, daily seizures, despite multiple (at least three) antiepileptic drugs. Seizure semiology in the control patients was mixed, with both complex partial and generalized tonic-clonic seizures. Several control patients experienced seizure clusters (several seizures in a day), as well as focal and generalized tonic-clonic status epilepticus requiring hospitalization.
During sirolimus titration, patient 2 had complex partial seizures only, whereas patients 3 to 5 had complex partial and generalized tonic-clonic seizures. Only one child (patient 5) in our cohort has had a seizure in the last 12 months, and seizures (any semiology) have stopped in four sirolimus-treated patients (Fisher’s exact test, P < 0.001; Table 1). All five patients are maintained on fewer antiepileptic drugs (one or two) than historical controls. None of the sirolimus-treated children developed infantile spasms (about 16% of historical control PMSE patients develop spasms). Of particular importance, patients 1 and 4 started sirolimus the earliest, at 3 months of age, and recurrent seizures were prevented. Indeed, patient 1 has not yet had a seizure at 8 months of age. One patient (patient 3) was an outlier with regard to epilepsy severity, having suffered about 180 seizures during his worst year (before and then while titrating sirolimus) and a total of 22 generalized tonic-clonic seizures, in some cases requiring hospitalization for status epilepticus. However, he has had no seizures of any semiology over the past 12 months while maintained on sirolimus.
When compared with historical Mennonite PMSE patients who were not treated with sirolimus, provisional developmental differences were noted in receptive language and social domains. All children were mute, and sirolimus had no clear effect on expressive language, gross motor function, or adaptive learning. However, when compared with parental assessments and developmental screening of historical control PMSE patients, parents described sirolimus-treated patients as being more interactive, socially engaged, and emotionally attached than their untreated historical counterparts. Although the oldest child in the treated cohort is only 4.7 years, receptive language is progressing without regression in all sirolimus-treated children compared to historical control patients according to parental and physician assessments. For example, during office encounters, children in the treatment cohort consistently make sustained eye contact, are emotionally engaged with parents and the examiner, and (among the three oldest children) can point to objects of interest, wave appropriately, and use hand motions to indicate basic needs. Similar behaviors were not commonly observed among untreated historical control PMSE patients (1). However, the present cohort of sirolimus-treated patients (mean age, 3.0 ± 1.7 years; range, 0.7 to 4.7 years) are younger than the 16 untreated historical control patients (age range, 0.6 to 28 years) used for developmental comparisons. Therefore, definitive judgments about sustained cognitive or behavioral benefits of sirolimus will require a longer period of clinical follow-up.
DISCUSSION
We demonstrate that loss of STRADA causes mTORC1-dependent deficits in migration, pathfinding, and polarity in vitro and altered cortical lamination in vivo that are prevented with mTORC1 pathway inhibitors. On the basis of the current data and previous findings from our laboratory (1, 2), we have clear evidence that loss of STRADA causes mTORC1 activation, seen as enhanced phosphorylation of p70S6K, ribosomal S6, and IRS1 proteins, confirming a robust effect of mTORC1 cascade activation in PMSE lymphoblasts, fibroblasts, and brain, and defining new mechanistic roles for STRADA in cortical development. As a strategy to initiate therapeutic discovery for the rare disorder PMSE, we used these preclinical signaling and functional assay data as a translational rationale to treat five PMSE patients with the mTOR inhibitor sirolimus. PMSE patients receiving sirolimus exhibited reduced seizure frequency and improved receptive language.
Our results demonstrate that STRADA functions as a pivotal regulator of polarity and pathfinding in migrating neurons by signaling through mTORC1/p70S6K to IRS1 and cofilin. Loss of STRADA after shRNA knockdown in mNPCs, or in PMSE patient cerebral cortex, leads to mTORC1 activation and enhanced phospho-inhibition of IRS1 via p70S6K. Because activation of the mTORC1 cascade by loss of STRADA leads to altered cell migration in vitro in the wound-healing assay, we propose that this signaling mechanism occurs in PMSE during brain development, resulting in failed neuronal migration and consequent heterotopic neurons in the subcortical and subependymal regions. In support of this hypothesis, we demonstrate that rapamycin prevents the formation of neuronal heterotopia after STRADA knockdown in vivo, further confirming the dependence of the PMSE phenotype on mTORC1-p70S6K signaling. These signaling and functional data provided a rational platform to initiate clinical therapy with an mTOR inhibitor in PMSE patients.
PMSE is a rare disorder, and the direct therapeutic implications of our results apply to a small number of patients. However, there is a clear clinical mandate for the development of targeted therapies to treat rare and devastating neurological diseases. Like many neurodevelopmental disorders, the functional neurological outcome of PMSE is uniformly poor, and most patients are wheelchair-bound and mute, requiring fulltime care. In a previous PMSE cohort, the fatality rate from status epilepticus or hypovolemic shock was 38% between the ages of 7 months and 6 years (1). None of the five sirolimus-treated patients died in our study, and thus, survival may be an important outcome. Sirolimus was clinically well tolerated, and four of five treated patients have been seizure-free for the past year; previously, no PMSE patient in the Mennonite community had achieved freedom from seizures, despite treatment with multiple antiepileptic drugs (1). We anticipate continued sirolimus treatment for these five patients and consideration of sirolimus for new PMSE patients. Treatment was initiated in two children by 3 months of age, and in one, only a single seizure has occurred, whereas in the other, epilepsy has not yet developed. These observations are supported by preclinical data in knockout mouse models of other mTOR-associated neurodevelopmental disorders (Tsc1 and Pten knockouts) (26–28), suggesting that initiation of mTOR inhibitors before seizure onset abrogates the seizure phenotype. Sirolimus given at an early age may have similar preventative effects in human patients and serve as a true disease-modifying therapy. The mechanism of action of sirolimus in epilepsy remains to be fully defined, and sirolimus may work by interrupting epileptogenesis rather than preventing seizure propagation. Our attempt at a new therapeutic intervention predicated on preclinical data is a tractable strategy for many rare and serious disorders. Thus, although our direct results are designed to aid PMSE patients, they provide precedent for a safe trial of mTOR inhibitors in other neurodevelopmental disorders associated with aberrant brain structure, altered cognitive abilities, and epilepsy linked to mTOR activation (mTORopathies) including TSC, focal cortical dysplasia, ganglioglioma, hemimegalencephaly, and fragile X syndrome (29).
We readily acknowledge important caveats to our results. Our preclinical data in mNPCs, whole mouse brain, and human PMSE fibroblasts support a pivotal role for mTORC1 in PMSE. Of course, in human PMSE brain, other signaling cascades may also contribute to developmental abnormalities in the cerebral cortex. Second, we do not demonstrate changes in the neuroradiological features of PMSE. In the PMSE children, we were unable to evaluate effects of sirolimus on cortical malformations, and limitations of our study precluded experiments that would directly parallel those done in mice (for example, sirolimus treatment of pregnant females). Furthermore, our data do not provide a mechanistic explanation of how mTOR inhibition might alter seizure frequency in PMSE. Additionally, our clinical cohort in no way is meant to represent a comprehensive clinical trial, and a more extensive clinical data set may be helpful, including electroencephalography and neuroimaging, to stratify and classify potential patient subgroups. We acknowledge that more expansive cognitive assessment will be necessary to prove any benefit to speech or learning in PMSE patients treated with sirolimus, and that largely subjective assessments of receptive language in treated children in an open-label approach may be biased. Although sirolimus may improve cognitive function in PMSE patients, these children are also maintained on fewer antiepileptic drugs, a variable that may also contribute to improved neurobehavioral function. However, observational improvements in receptive language and social interaction were uniform in all five patients and were in fact bolstered by objective bedside testing. Clearly, further analysis is warranted to define neurocognitive outcomes after sirolimus treatment in PMSE and other mTOR-associated developmental disorders.
MATERIALS AND METHODS
All animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and adhered to the NIH Guide for the Care and Use of Laboratory Animals. All human studies, for example, brain tissue, fibroblasts, and sirolimus treatment, were approved by the Institutional Review Board of Lancaster General Hospital (Lancaster, PA). Parents provided informed consent before their own and their children’s participation.
mNPC culture and transfection
Stable transfection of puromycin-resistant shRNA plasmids in mNPCs (provided by J. Wolfe, Children’s Hospital of Philadelphia, Philadelphia, PA) (30) was established with either puro-shRNA STRADA to create a stable STRADA knockdown line or puro-shRNA scrambled to create a control line (KM41633P; SABiosciences), as previously described (2). Cells were cultured on poly-D-lysine–coated plates (10 μg/ml; Sigma-Aldrich) in complete medium consisting of Dulbecco’s modified Eagle’s medium (DMEM)/F12 (Invitrogen) supplemented with 1% N2 (Invitrogen), 1% fetal bovine serum (FBS) (Sigma-Aldrich), 1% penicillin/streptomycin (Invitrogen), basic fibroblast growth factor (20 ng/ml) (Promega), and heparin (5 μg/ml) (Sigma-Aldrich). To maintain stable transfection, we applied selective pressure using puromycin (6 μg/ml) (Invitrogen).
Human fibroblast extraction and culture
Fibroblasts from PMSE patients (three), parents (two), and normal control (one) were obtained from skin punch biopsies at the Clinic for Special Children in Strasburg, Pennsylvania, after informed consent. None of these individuals had been treated with sirolimus. Fibroblasts were extracted from tissue samples by incubation in 0.25% trypsin/EDTA (Gibco) overnight at 4°C. The next day, epidermis was removed, and dermis was digested with collagenase P (Roche) buffered in 130 mM sodium chloride (Sigma-Aldrich), 10 mM calcium acetate (Sigma-Aldrich), and 20 mM Hepes buffer (Gibco) for 30 min at 37°C. Then, 0.5% trypsin/EDTA (Gibco) was added, and the mixture was incubated at 37°C for an additional 10 min before neutralization with fibroblast culture medium composed of DMEM (Gibco) supplemented with 10% FBS (Sigma-Aldrich), 10 mM Hepes buffer (Gibco), 1% penicillin/streptomycin [penicillin (10,000 U/ml), streptomycin (10 mg/ml); Gibco], and 1% fungizone (250 μg/ml; Gibco). Fibroblasts were pelleted through centrifugation for 5 min at 1500 rpm, and the pellet was resuspended in fibroblast culture medium to obtain the desired cells. For protein quantification assays, fibroblast medium was supplemented with the CAMKK inhibitor STO-609 (5 μM; Tocris) and the AMPK activator AICAR (2 mM; Cell Signaling) at 6 and 4 hours, respectively, before cell lysis to enable isolation of STRADA-dependent inhibition of mTORC1 signaling. Rapamycin (100 nM; Cell Signaling) or p70S6Ki (PF-4708671, 10 μM; Pfizer Pharmaceuticals) was applied for 2 hours before cell lysis. Concentration ranges for rapamycin (25 to 200 nM) and p70S6Ki (1 to 20 μM) were established as the minimal effective doses producing mTOR inhibition (defined as reduction of S6 phosphorylation) in this cell line.
Western blotting
Cells lysates were electrophoresed according to a previously established protocol (2). The following primary antibodies were used: rabbit polyclonal to STRADA (1:500; Abcam), rabbit monoclonal to phospho–S6 ribosomal protein (Ser235/236, 1:1000; Cell Signaling), rabbit monoclonal to phospho-4EBP1 (Thr37/46, 1:1000; Cell Signaling), rabbit polyclonal to phospho-IRS1 (Ser636/639, 1:500; Abcam), rabbit monoclonal to phospho-cofilin (Ser3, 1:1000; Cell Signaling), and rabbit monoclonal to GAPDH (1:1000; Cell Signaling).
Migration assay
mNPCs or human fibroblasts were plated on six-well plates, chamber slides, or laminin-coated coverslips; coated with poly-D-lysine (mNPCs; Sigma-Aldrich) or poly-L-lysine (fibroblasts; Sigma-Aldrich); and cultured in complete mNPC or human fibroblast medium (specified above). Before the migration assay, cells were cultured for 24 hours in serum-deplete medium (1 ml of complete medium/4 ml of basal medium; DMEM/F12 for mNPCs or DMEM for fibroblasts; Gibco) to attenuate basal mTORC1 activity. Serum-deplete medium was maintained for the duration of the migration assay. Rapamycin (50 nM for mNPCs, 100 nM for fibroblasts) or p70S6Ki (10 μM) was applied for 1 hour before and throughout the duration of the experiment (optimal doses to reduce S6 phosphorylation determined for each cell type). A 200-μl micropipette tip was used to create a linear scratch across a confluent monolayer of cells (18). In video migration experiments, actively dividing cells were excluded from the subset measured. Additionally, cell size was not measured in the video migration assays because cell size fluctuated on the basis of cell cycle phase and division during the 20 hours of observation period.
Treatment with Ara-C
The mitotic inhibitor Ara-C (Sigma) was used to inhibit mitosis of migrating cells and demonstrate that the migration deficit seen with STRADA knockdown and rapamycin rescue were independent of any effects on cell proliferation. Wild-type mNPCs were plated in serum-deplete medium in six-well plates and pretreated with Ara-C for 24 hours, after which a fresh dose of Ara-C was applied for 0, 15, or 20 hours to replicate the time course of the migration assay. Cells were enzymatically de-adhered with 0.25% trypsin/EDTA (Gibco) at each indicated treatment time, and trypan blue (Sigma) was used to visualize and count cells under light microscopy. The optimal effective antiproliferative, non-apoptotic Ara-C dose (20 μM) was applied to scrambled and STRADA knockdown mNPCs for 24 hours before and throughout the duration of the migration assay with rapamycin treatment (fig. S2).
Video capture in migration assay
Migration of STRADA knockdown and scrambled mNPCs was determined in chamber slides within a microincubator (model CSMI; at 37°C; Harvard Apparatus) on an inverted microscope (Nikon TE300) equipped with a digital video camera (Evolution QEi; Media Cybernetics) (31). Images were taken every 5 min in the phase-contrast channel for 20 hours. For each condition, three or four videos were taken across the scratch.
Immunocytochemistry
Scrambled and STRADA knockdown mNPCs plated on coverslips and subjected to the described migration assay were fixed in 4% paraformaldehyde (PFA) at room temperature for 20 min, permeabilized with 0.3% Triton X-100 (Sigma) in phosphate-buffered saline (Gibco), and blocked for 2 hours in 5% normal goat serum (Vector Laboratories). Cells were incubated with mouse anti-GM130 monoclonal antibody (1:100; BD Transduction Laboratories) overnight at 4°C. Alexa Fluor 488 goat anti-mouse secondary antibody (Invitrogen) was applied for 1 hour at room temperature. Cells were then blocked with 1% bovine serum albumin (Sigma-Aldrich) for 30 min. Phalloidin (1:40; Invitrogen) was applied for 20 min, followed by Hoechst nuclear stain (0.0001 μg/μl; Invitrogen). Cells were visualized and images were captured with a Leica DMI6000 B fluorescence microscope with a Leica DFC360 FX camera. Golgi-specific labeling was confirmed by costaining a subset of coverslips with rabbit anti-giantin polyclonal antibody (1:500; Abcam) and Alexa Fluor 594 goat anti-rabbit secondary antibody (Invitrogen) (fig. S5).
Human brain immunohistochemistry
A single postmortem PMSE brain of a 7-month-old female who died after status epilepticus was procured with consent from the Clinic for Special Children. Cortical dysplasia–focal epilepsy syndrome cortex served as a non-PMSE epileptic control specimen (32). The specimens were fixed in 4% PFA, embedded in paraffin, and cut into 7-μm sections. Sections were probed with rabbit polyclonal antibody to phospho-IRS1 (Ser636/639, 1:100; Abcam) and processed for immunohistochemistry as reported previously (2).
In utero electroporation
IUE was used to transfect VZ/SVZ cells in E14.0 C57BL/6J mouse embryos with GFP-tagged STRADA shRNA plasmid (8 μg/μl), as described previously (33). A subset of pregnant dams was injected intraperitoneally with rapamycin (5 mg/kg, in vehicle solution composed of 5% PEG-400, 5% Tween 80, 0.9% NaCl in sterile water; LC Laboratories) daily from E15 to E18. Animals were sacrificed with CO2 overdose on E19, at which time brains were extracted from the embryos, fixed in 4% PFA, cryoprotected in a sucrose gradient, and frozen in optimal cutting temperature solution (Tissue-Tek). Coronal sections (20 μm) were cut on a cryostat (Leica CM 1950), stained with Hoechst 33342 nuclear stain (0.0001 μg/μl; Invitrogen), and coverslipped in mounting medium (Fluoromount-G; SouthernBiotech). Images were taken with a Leica DMI6000 B fluorescence microscope with a Leica DFC360 FX camera.
Data analysis
For the still-image migration assays, three independent scratches were made per treatment condition per shRNA or disease condition, and three images were taken across each scratch at each time point. A blinded observer recorded 10 measurements per image, yielding 90 measures per time point per condition (using Photoshop CS2; Adobe or Image-Pro Plus 7.0; Media Cybernetics). Migration distances were determined by subtracting the distance between edges of the scratch at 15 hours from the distance at 0 hours and halving this value. Each set of experiments was repeated.
For video migration assays, a blinded observer evaluated five representative leading-edge cells per video and measured the distance translocated and directional angle at each 30-min time point for the same cell using Image-Pro Plus 7.0 software. Total migration distance for each cell over the 20 hours recorded was measured as the sum of all 30-min measures. Directional variance for each cell was calculated as the variance in the set of directional angles. Greater directional variance was determined to indicate reduced linear directionality.
For IUE analysis, three independent observers quantified the percentage of GFP+ cells reaching the cortical plate using the Image-Pro Plus 7.0 (Media Cybernetics) automatic count function for bright objects within an outlined region of interest, and this was verified by hand count. Localization of the cortical plate was determined by relative nuclear density.
Data are presented in table S1 as means ± SEM. Excel (Microsoft) and Prism 5 (GraphPad) software programs were used for statistical analysis. In all comparisons, a P value of less than 0.05 was considered significant. Unpaired two-tailed Student’s t tests were performed to determine significance for all individual measures, and served as the sole method of analysis for experiments set up as binary comparisons, including IUE and video migration measures. For all still migration measures and measures of Golgi area and crescentic angle, significance was determined by one-way ANOVA with Bonferroni correction for multiple comparisons in the migration assays or by Dunnett post hoc analysis to determine the extent of rapamycin or p70S6Ki rescue in the Golgi measures.
Supplementary Material
Acknowledgments
We thank C. Marshall, J. Seykora, P. Bates, B. Berg, J. Chen, D. Cook, E. Goncharova, G. Heuer, K. Haines, A. Praestgaard, V. Tsai, and B. Wells, as well as PMSE patients and their families.
Funding: This work was supported by the National Institute of Neurological Disease and Stroke at the NIH (NS045022) and the Penn-Pfizer Collaborative Program (to P.B.C.); the National Heart, Lung, and Blood Institute at the NIH (RO1 HL110551 to V.P.K.); an NIH Training Grant in Genetics (T32 GM008216 to W.E.P.); and a Ruth L. Kirschstein National Research Service Award (1F31NS078845-01 to W.E.P.).
Footnotes
Author contributions: W.E.P. and P.B.C. designed all cell culture and animal experiments. P.B.C. and K.A.S. designed, supervised, and directed sirolimus use in PMSE patients. W.E.P., K.A.O., V.P.K., D.A.G., W.H.P., J.F.B., and M.B. optimized and performed the in vitro motility and migration assays. W.E.P., W.H.P., K.O., J.F.B., and J.H. analyzed and quantified the in vitro migration, video migration, still migration, and IUE assays. W.E.P., W.H.P., J.H., and M.B. performed the Western blot assays. M.B. performed the immunohistochemistry assays. W.E.P., K.A.O., V.P.K., K.A.S., and P.B.C. analyzed the final data and wrote the paper.
Competing interests: P.B.C. is on the Board of Directors for the Tuberous Sclerosis Alliance and is a paid consultant to Novartis Oncology. The other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Puffenberger EG, Strauss KA, Ramsey KE, Craig DW, Stephan DA, Robinson DL, Hendrickson CL, Gottlieb S, Ramsay DA, Siu VM, Heuer GG, Crino PB, Morton DH. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130:1929–1941. doi: 10.1093/brain/awm100. [DOI] [PubMed] [Google Scholar]
- 2.Orlova KA, Parker WE, Heuer GG, Tsai V, Yoon J, Baybis M, Fenning RS, Strauss K, Crino PB. STRADα deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–1711. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Mäkelä TP, Alessi DR, Hardie DG. Complexes between the LKB1 tumor suppressor, STRADα/β and MO25α/β are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw RJ, Bardeesy N, Manning BD, Lopez L, Kosmatka M, DePinho RA, Cantley LC. The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell. 2004;6:91–99. doi: 10.1016/j.ccr.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355:1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 8.Lee JH, Huynh M, Silhavy JL, Kim S, Dixon-Salazar T, Heiberg A, Scott E, Bafna V, Hill KJ, Collazo A, Funari V, Russ C, Gabriel SB, Mathern GW, Gleeson JG. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poduri A, Evrony GD, Cai X, Elhosary PC, Beroukhim R, Lehtinen MK, Hills LB, Heinzen EL, Hill A, Hill RS, Barry BJ, Bourgeois BFD, Riviello JJ, Barkovich AJ, Black PM, Ligon KL, Walsh CA. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron. 2012;74:41–48. doi: 10.1016/j.neuron.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong M. Mammalian target of rapamycin (mTOR) inhibition as a potential antiepileptogenic therapy: From tuberous sclerosis to common acquired epilepsies. Epilepsia. 2010;51:27–36. doi: 10.1111/j.1528-1167.2009.02341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanopoulou AS, Gorter JA, Cepeda C. Finding a better drug for epilepsy: The mTOR pathway as an antiepileptogenic target. Epilepsia. 2012;53:1119–1130. doi: 10.1111/j.1528-1167.2012.03506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G, Dinopoulos A, Thomas G, Crone KR. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol. 2006;59:490–498. doi: 10.1002/ana.20784. [DOI] [PubMed] [Google Scholar]
- 13.Muncy J, Butler IJ, Koenig MK. Rapamycin reduces seizure frequency in tuberous sclerosis complex. J Child Neurol. 2009;24:477. doi: 10.1177/0883073808324535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363:1801–1811. doi: 10.1056/NEJMoa1001671. [DOI] [PubMed] [Google Scholar]
- 15.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 16.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. Absence of S6K1 protects against age-and diet-induced obesity while enhancing insulin sensitivity. Nature. 2004;431:200–205. doi: 10.1038/nature02866. [DOI] [PubMed] [Google Scholar]
- 17.Pearce LR, Alton GR, Richter DT, Kath JC, Lingardo L, Chapman J, Hwang C, Alessi DR. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1) Biochem J. 2010;431:245–255. doi: 10.1042/BJ20101024. [DOI] [PubMed] [Google Scholar]
- 18.Liang CC, Park AY, Guan JL. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 19.Kupfer A, Louvard D, Singer SJ. Polarization of the Golgi apparatus and the microtubule-organizing center in cultured fibroblasts at the edge of an experimental wound. Proc Natl Acad Sci USA. 1982;79:2603–2607. doi: 10.1073/pnas.79.8.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yadav S, Puri S, Linstedt AD. A primary role for Golgi positioning in directed secretion, cell polarity, and wound healing. Mol Biol Cell. 2009;20:1728–1736. doi: 10.1091/mbc.E08-10-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Solecki DJ, Govek EE, Tomoda T, Hatten ME. Neuronal polarity in CNS development. Genes Dev. 2006;20:2639–2647. doi: 10.1101/gad.1462506. [DOI] [PubMed] [Google Scholar]
- 22.Govek EE, Hatten ME, Van Aelst L. The role of Rho GTPase proteins in CNS neuronal migration. Dev Neurobiol. 2011;71:528–553. doi: 10.1002/dneu.20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisbury S, Franz DN. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358:140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rüegg S, Baybis M, Juul H, Dichter M, Crino PB. Effects of rapamycin on gene expression, morphology, and electrophysiological properties of rat hippocampal neurons. Epilepsy Res. 2007;77:85–92. doi: 10.1016/j.eplepsyres.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunoyer M. Accelerating access to treatments for rare diseases. Nat Rev Drug Discov. 2011;10:475–476. doi: 10.1038/nrd3493. [DOI] [PubMed] [Google Scholar]
- 26.Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng LH, Rensing NR, Zhang B, Gutmann DH, Gambello MJ, Wong M. Tsc2 gene inactivation causes a more severe epilepsy phenotype than Tsc1 inactivation in a mouse model of tuberous sclerosis complex. Hum Mol Genet. 2011;20:445–454. doi: 10.1093/hmg/ddq491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ljungberg MC, Sunnen CN, Lugo JN, Anderson AE, D’Arcangelo G. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crino PB. mTOR: A pathogenic signaling pathway in developmental brain malformations. Trends Mol Med. 2011;17:734–742. doi: 10.1016/j.molmed.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 30.Magnitsky S, Walton RM, Wolfe JH, Poptani H. Magnetic resonance imaging detects differences in migration between primary and immortalized neural stem cells. Acad Radiol. 2008;15:1269–1281. doi: 10.1016/j.acra.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goncharova E, Goncharov D, Noonan D, Krymskaya VP. TSC2 modulates actin cytoskeleton and focal adhesion through TSC1-binding domain and the Rac1 GTPase. J Cell Biol. 2004;167:1171–1182. doi: 10.1083/jcb.200405130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Strauss KA, Puffenberger EG, Huentelman MJ, Gottlieb S, Dobrin SE, Parod JM, Stephan DA, Morton DH. Recessive symptomatic focal epilepsy and mutant contactin-associated protein-like 2. N Engl J Med. 2006;354:1370–1377. doi: 10.1056/NEJMoa052773. [DOI] [PubMed] [Google Scholar]
- 33.Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.