

Abstract
The class A macrophage scavenger receptor Msr1 (SR-A, CD204) has been reported to participate in the maintenance of immunological tolerance. We investigated the role of Msr1 in a mouse model of autoantibody-dependent arthritis. Genetic deficiency of Msr1 in K/BxN TCR transgenic mice decreased the incidence and severity of arthritis, due to decreased autoantibody production. Despite normal initial activation of autoreactive CD4+ T cells, potentially autoreactive B cells in Msr1−/− K/BxN mice retained a naïve phenotype and did not expand. This was not due to an intrinsic B cell defect. Rather, we found that macrophages lacking Msr1 were inefficient at taking up the key autoantigen glucose-6-phosphate isomerase (GPI) and that Msr1-deficient mice had elevated serum concentrations of GPI. Arthritis developed normally when bone marrow from Msr1−/− K/BxN mice was transplanted into hosts whose macrophages did express Msr1. Thus, Msr1 can regulate the concentration of a soluble autoantigen. In this model, the absence of Msr1 led to higher levels of soluble autoantigen and protected mice from developing pathogenic autoantibodies, likely due to altered cognate interactions of autoreactive T and B cells with impaired differentiation of follicular helper T cells.
Introduction
The generation of autoreactive lymphocytes is a consequence of having a diverse cellular immune repertoire capable of responding to threats of a wide range of specificities. Both cell-intrinsic and –extrinsic modes of immunological tolerance exist to constrain lymphocyte clones that recognize self-antigens (1, 2). For B cells, central tolerance occurs in the bone marrow, where most self-reactive clones are either deleted or undergo receptor editing (3-5). In the periphery, autoreactive B cells can be rendered tolerant (anergic) or induced to undergo apoptosis (6). Weakly autoreactive B cells can be maintained in a seemingly naïve state of clonal ignorance if protected from cognate interactions with activated T cells (7). Autoimmunity can arise when these B cell tolerance mechanisms fail.
The class A scavenger receptor, macrophage scavenger receptor 1 (Msr1, SR-A, CD204, encoded by the murine gene, Msr1), is a multifunctional receptor that is expressed primarily on cells of the myeloid lineage and that binds modified self- and pathogen-associated antigens (8). Several lines of evidence support a role for Msr1 in peripheral tolerance. For example, one group reported a role for Msr1 in the maintenance of peripheral tolerance via tonic cross-presentation of self-antigens “nibbled” from the membranes of adjacent cells to CD8+ T cells (9). Additionally, Msr1 may sequester sources of danger signals present among autoantigens by taking up apoptotic cellular debris. Of note, impaired clearance of apoptotic antigens has been implicated in the breakdown of tolerance in systemic lupus erythematosus (SLE) (10). Msr1-deficient mice do not develop spontaneous autoimmune diseases, however, suggesting that these reported “housekeeping” functions of Msr1 are not essential to maintain immunological tolerance and highlighting the fact that multiple layers of peripheral tolerance exist (11).
The K/BxN T cell receptor (TCR) transgenic mouse model of spontaneous autoimmune arthritis is well-suited to address questions regarding mechanisms of immunological tolerance. In this model, CD4+ T cells bearing the KRN transgene-encoded TCR recognize peptides derived from the ubiquitously expressed glycolytic enzyme, glucose 6-phosphate isomerase (GPI), presented on the MHC class II molecule I-Ag7. These activated T cells provide help to GPI-specific B cells, leading to the sustained production of high-titer arthritogenic anti-GPI autoantibodies (12, 13). Joint pathology arises via antibody-mediated activation of the innate immune system, and transfer of K/BxN serum to naïve recipient mice is sufficient to provoke transient arthritis (termed K/BxN serum-transferred arthritis) (14). Arthritis in K/BxN TCR transgenic mice develops reliably between 3-4 weeks of age, when autoreactive KRN CD4+ T cells begin to emerge from the thymus and when anti-GPI autoantibody production can be detected (12, 13), demonstrating that both T cell and B cell tolerance are breached in K/BxN mice.
Here we investigated how Msr1 impacts immunological tolerance in the K/BxN mouse model of spontaneous autoimmune arthritis.
Materials and Methods
Mice
KRN TCR transgenic mice on the C57BL/6 (B6) background (12) were a gift from Drs. Diane Mathis and Christophe Benoist (Harvard Medical School, Boston, MA) and the Institut de Génétique et de Biologie Moléculaire et Cellulaire (Strasbourg, France); B6 mice congenic for H-2g7 (B6.g7) (15) were also a gift from Drs. Mathis and Benoist. Msr1-null B6 (Msr1tm1Csk, stock no. 006096)(11), NOD/Lt (stock no. 001976) (16), Rag1-deficient B6 (Rag1tm1Mom, stock no. 002216) (17), TCR-α-deficient B6 (Tcratm1Mom, stock no. 002116) (18), and μMT (Ighmtm1Cgn, stock no. 002288) (19) mice were purchased from The Jackson Laboratory and bred in our specific-pathogen-free colonies. Msr1-deficient “K/BxN” mice (Msr1−/− K/BxN) were generated by breeding mice bearing the Msr1 knockout (KO) allele on the B6 background to KRN/B6 mice and also to B6.g7. For ease of nomenclature, we refer to the KRN+ H-2b/g7 mice as “K/BxN”. Genotyping was performed by PCR. All studies were conducted in accordance with Institutional Animal Care and Use Committee-approved protocols at the University of Minnesota (protocol nos. 0611A96106 and 0909A72086).
Antibodies and flow cytometry
The following monoclonal antibodies used for flow cytometry and/or immunofluorescent microscopy were purchased from eBioscience: B220 (RA3-6B2), CD3 (clones 17A2 and 145-2C11), CD4 (RM4-5), CD8 (53-6.7), CD11b (M1/70), CD11c (N418), CD16/32 (clone93), CD19 (1D3), CD23 (B3B4), CD38 (clone90), CD44 (IM7), CD45.1 (A20), CD45.2 (104), CD73 (ebioTy/11.8), CD90.1 (HIS51), F4/80 (BM8), Foxp3 (FJK-16s), FR4 (ebio12A5), Gr-1 (RB6-8C5), GL7-ef450, H-2Kb (AF6-88.5.5.3-PE), H-2Kd (SF1-1.1.1), IgD (11-26c), IgG1 (M1-14D12), IgM (eB121-15F9), Ki67 (SolA15), MHCII (M5/114.15.2), and PD-1 (J43). The following antibodies were purchased from BD Pharmingen: CD3 (500A2), CD24 (M1/69), CD90.2 (53-2.1), CXCR5 (2G8), GL7-FITC, H-2Kb (AF6-88.5-FITC), IgM (II/41), and TCR Vβ6 (RR4-7). Anti-CD4 (RM4-5, BioLegend) was used in some experiments. Intracellular staining using anti-Foxp3 (FJK-16s), IgG1 (RMG101, Invitrogen), and IgG (H+L) F(ab’)2 (Invitrogen) was performed using intracellular permeabilization/fixation reagents (eBioscience) per the manufacturer’s protocol. GPI-PE and GPI-AF647-PE tetramers have been described previously (20). Flow cytometry was performed using an LSRII or an LSRFortessa (BD Biosciences), and cells were analyzed using FlowJo v8.8.7 software (Tree Star). The gating scheme for all experiments included first using forward and side scatter along with Fixable Viability Dye (eBioscience) to identify live, singlet lymphocytes. Subsequent gating parameters are described in the Figures and Legends.
Immunofluorescent staining
After blocking Fc receptors with 2.4G2 (BD Pharmingen) and anti-CD64 (clone N19, SantaCruz) antibodies and blocking biotin with an avidin/biotin blocking kit (Vector Laboratories), frozen sections were stained with fluorescently-conjugated antibodies recognizing CD3, TCRβ (H57-597, BD Pharmingen), and biotinylated anti-B220 plus SA-DyLight 550 (Thermo Fisher Scientific). DAPI was used to detect nuclei. Slides were viewed on an Olympus BX51 fluorescent microscope equipped with a digital camera and DP-BSW software (Olympus).
Ag-specific B cell enrichment
GPI-specific B cells were enriched as described (20). Briefly, pooled lymph node cells and splenocytes were incubated with the Ag-specific GPI-PE and Ag-nonspecific C5A-AF647-PE (decoy) tetramers, followed by incubation with anti-PE magnetic microbeads, after which the cells were passed through a magnetic column and both the Ag-specific B cell-enriched (bound) and polyclonal (unbound) fractions were collected. Both fractions were then labeled with a cocktail of fluorescent B cell and non-B cell markers for flow cytometric analysis.
Assessment of arthritis and IgG titers
Arthritis was assessed via clinical scoring and ankle measurements, and total serum IgG and anti-GPI titers were determined as described (21, 22).
Serum-transferred arthritis
Pooled serum (150 μL/dose) from K/BxN mice was injected intraperitoneally into recipient mice on days 0 and 2 (14). The mice were monitored for the development of arthritis for 2 weeks as described above.
Anti-GPI IgG ELISPOT
Splenocytes were analyzed via ELISPOT to examine autoantibody production on a per-cell basis as previously described (23). ELISPOT plates were read using an ImmunoSpot (Cellular Technology, Ltd).
Intracellular cytokine staining
Lymph node cells were isolated, stimulated with phorbol 12-myristate 13-acetate (PMA) and ionomycin, and stained intracellularly for IL-17 and IFNγ as described (24).
Ag uptake by macrophages
Peritoneal macrophages were elicited by intraperitoneal injection of 1 mL Brewer thioglycollate medium (Fluka Analytical). Macrophages were collected 5 days later via peritoneal lavage, incubated with 5 μg GPI or BSA labeled with AF647 (Life Technologies) in complete media for 20 minutes at 37°C/5% CO2, washed, and analyzed by flow cytometry.
Western blotting
Serum samples were separated by SDS-PAGE and transferred to Immobilin-FL membranes (Millipore). GPI was detected using serum from K/BxN mice, followed by peroxidase-conjugated goat anti-mouse IgG1 (Jackson Immunoresearch), developed with ECL Prime Western Blotting Detection Reagent (GE Healthcare), and imaged quantitatively on an ImageQuant LAS4000 workstation (GE Healthcare). Because antibody heavy chain co-migrates with GPI, antibody-deficient μMT−/− mice were used to allow detection of GPI.
Determination of serum albumin concentration
Serum albumin concentrations were determined in the same mice by ELISA according to the manufacturer’s instructions (Bethyl Laboratories, Inc.) The plates were read on a Biorad Model 680 Microplate reader at 415 nm.
Cell purification and adoptive transfer
Cells were purified from spleens and lymph nodes. CD4+ T cells were purified with T cell isolation kit II (Miltenyi Biotec). B cells were purified by negative enrichment using a cocktail of biotinylated antibodies (CD4, CD8, CD11b, CD11c, F4/80, GR-1) in combination with anti-biotin beads (Miltenyi Biotec). Lymphocytes were labeled with CFSE (where indicated) and injected intravenously into recipient mice. At the experimental endpoints, the lymphocytes were analyzed by flow cytometry.
Bone marrow chimeric mice
Rag1-deficient recipient mice were sublethally irradiated with 300 rad. The following day, 10×106 bone marrow cells from Msr1-sufficient or –deficient K/BxN mice (combined with 5×106 bone marrow cells from unirradiated Rag1-deficient mice where indicated) and injected intravenously into recipient mice. The recipient mice were maintained on sulfamethoxazole and trimethoprim administered in drinking water for the duration of the study. Arthritis was measured for 10 weeks post-transplantation, after which the animals were sacrificed and serum and lymphoid organs were harvested for analysis.
Statistical analysis
Statistical differences between mean values for groups were calculated using the Student’s two-tailed t-test. Arthritis severity scores were compared with a repeated-measures analysis of variance (ANOVA). Relative risk was used to calculate differences in the incidence of arthritis between the genetic model and mixed bone marrow chimeric mice. P-values <0.05 were considered significant. Anaylsis was performed with SPSS 19.0.
Results
K/BxN mice lacking Msr1 are protected from arthritis and endocarditis
To address how Msr1 might affect the development of autoantibody-associated arthritis, we generated K/BxN TCR transgenic mice lacking Msr1 (Msr1−/− K/BxN). Surprisingly, half of the Msr1−/− K/BxN mice developed subtle to no arthritis by eight weeks of age (Fig. 1A-B and Supplemental Fig. 1A). Some of the Msr1-heterozygous K/BxN mice (Msr1+/− K/BxN) were also protected from arthritis, although to a lesser extent, indicating a gene dose effect (not shown). Protection from arthritis did not segregate by gender or cohabited littermates. The Msr1−/− K/BxN mice were also protected from mitral valve inflammation (endocarditis) (Supplemental Fig. 1A), an additional site of inflammatory attack in this model (15). To exclude the possibility that arthritis was simply delayed, we aged non-arthritic Msr1−/− K/BxN for 4 months, during which time the arthritis severity either remained consistent with the 8-week timepoint, or resolved (Supplemental Fig. 1B).
Figure 1. Msr1-deficient K/BxN mice are protected from arthritis and endocarditis.
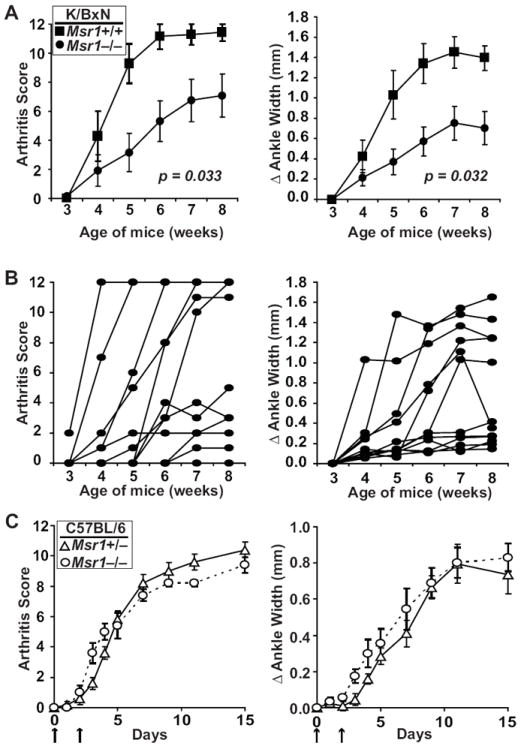
(A) Arthritis severity in Msr1+/+ (filled squares, n = 7) and Msr1−/− (filled circles, n = 12) K/BxN mice was assessed at the indicated ages and is depicted as a qualitative score (left) and quantitative measurement (right) of ankle width. Plotted values are means ± SEM; p values are shown in the panels and were determined by repeated-measures ANOVA. (B) Same parameters as A in more detail for the Msr1−/− group of K/BxN mice; each line represents one mouse. (C) Serum-transferred arthritis was induced in Msr1+/− (open triangles, n = 5) and Msr1−/− (open circles, n = 5) B6 mice by injection of arthritogenic K/BxN serum on days 0 and 2 (arrows). Plotted values are means ± SEM; p values determined by repeated-measures ANOVA were not significantly different.
The reduced incidence and severity of both arthritis and endocarditis suggested that Msr1-deficiency influenced the T and B cell-dependent initiation phase of autoimmunity in K/BxN mice. To exclude formally the possibility that Msr1 deficiency interfered with the innate immune system-mediated effector phase of arthritogenesis, we asked whether Msr1 deficiency affected the severity of arthritis induced by transfer of arthritogenic autoantibodies. Indeed, the absence of Msr1 had no observable effect on the severity arthritis in the serum-transfer system (Fig. 1C). Taken together, these findings demonstrate that Msr1 deficiency impacts the initiation phase of autoimmunity in K/BxN mice, not the effector phase. We therefore focused our attention on understanding how Msr1 deficiency affected T and B cell autoreactivity in K/BxN mice.
Decreased autoantibody production in Msr1-deficient K/BxN mice
K/BxN mice have elevated levels of total serum IgG as well as high-titer, arthritogenic anti-GPI autoantibodies (13, 14). We found that K/BxN mice lacking Msr1 had lower total IgG levels and lower anti-GPI IgG titers compared to controls. Furthermore, among the Msr1−/− K/BxN mice, those with arthritis tended to have higher total IgG levels and anti-GPI titers than those without arthritis (Figure 2), consistent with the tight link between autoantibody production and arthritis development in this model. We next sought to determine how Msr1 deficiency interfered with autoantibody production.
Figure 2. Msr1 deficiency impairs autoantibody production.
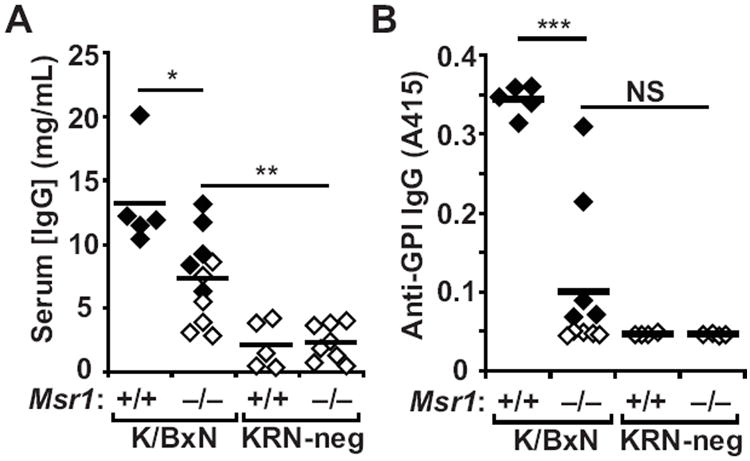
(A) Total serum IgG and (B) anti-GPI IgG were measured by ELISA in 8-week-old Msr1+/+ and Msr1−/− K/BxN mice, as well as KRN-negative Msr1+/+ and Msr1−/− mice. Each point represents one animal; filled diamonds indicate arthritic animals and open diamonds indicate non-arthritic animals; bars represent means. Data were compiled from three independent experiments. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, NS = not significant.
The T cell compartment is unaltered in Msr1-deficient K/BxN mice
Production of anti-GPI autoantibodies in K/BxN mice depends on the interaction of CD4+ T cells expressing the transgene-encoded KRN TCR with GPI-specific B cells. We therefore asked whether the impaired autoantibody production in Msr1−/− K/BxN mice was due to altered T cell development or activation, particularly in view of the restricted expression of Msr1 on macrophages, a cell type capable of presenting peptide:MHCII antigens to CD4+ T cells (8, 25). We first evaluated T cell subsets in the thymus and periphery from Msr1-deficient and control K/BxN mice, but found no difference in their numbers or relative frequencies (Figs. 3A, 3B). Peripheral CD4+ T cells from both groups of mice expressed comparable levels of the T cell activation marker, CD44, (Fig. 3C). Moreover, the number of activated CD4+ T cells producing IL-17 was equivalent (Fig. 3D), demonstrating that the activation of pathogenic helper T cells was not impaired (24, 26). Msr1 has been reported perform a variety of functions, including the capture and transfer of antigen for presentation (8, 25). To address directly whether the absence of Msr1 led to reduced antigen presentation in vivo, we adoptively transferred CFSE-labeled naïve CD4+ T cells from KRN/B6 (H-2b) mice into Msr1+/+ H-2b/g7 or Msr1−/− H-2b/g7 recipient mice and found that the transferred cells proliferated equivalently in both hosts (Fig. 3E), suggesting that the presentation of GPI peptide:I-Ag7 complexes was not impaired. We also considered the possibility that CD4+ T cells in Msr1−/− K/BxN mice were anergic and therefore unable to provide sufficient B cell help. We found, however, that activated CD4+ T cells isolated from Msr1+/+ or Msr1−/− K/BxN mice proliferated indistinguishably when transferred together into H-2b/g7 hosts (Fig. 3F). Furthermore, there was no apparent difference in the expression of the CD4+ T cell anergy markers CD73 and folate receptor 4 (FR4) in the two groups of mice (Fig. 3G) (27). Collectively, these findings suggest that the reduction in anti-GPI autoantibody production in Msr1−/− K/BxN mice was not due to impaired T cell development, activation, antigen-induced expansion, or anergy. We next focused on B cells.
Figure 3. Msr1 deficiency does not affect KRN T cell activation.
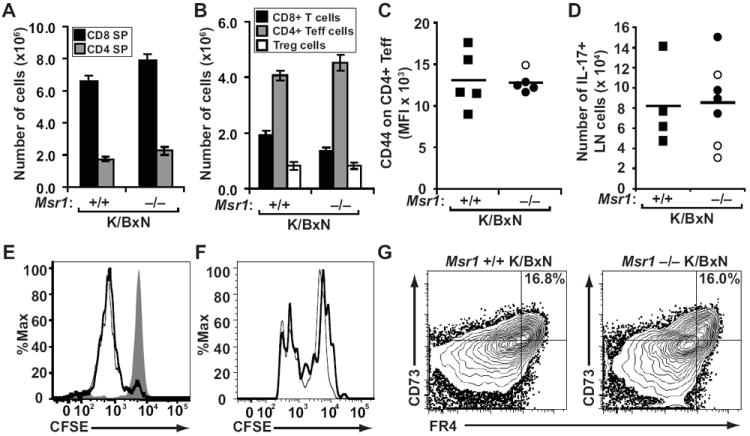
T cell subsets from (A) thymus and (B) spleen were enumerated among K/BxN mice of the indicated Msr1 genotypes. The values plotted are means ± SEM; n=5 mice per genotype. (C) Surface expression of CD44 measure by mean fluorescence intensity (MFI) on splenic CD4+ effector T cells from Msr1+/+ (squares) and Msr1−/− (circles) K/BxN mice was determined by flow cytometry. (D) The number of CD4+ lymph node T cells expressing intracellular IL-17 was determined by flow cytometry. In both C and D, filled shapes indicate individual arthritic animals, open shapes indicate non-arthritic animals; bars represent mean values. (E) CFSE-labeled naïve CD4+ T cells from KRN/B6 donor mice were adoptively-transferred into Msr1+/+ (thick line) or Msr1−/− (thin line) H-2b/g7-expressing recipient mice or control C57BL/6 (H-2b) mice (shaded histogram) and harvested 48 hours later for flow cytometric analysis. The data shown are representative of 3 separate experiments (n = 8 total mice per group). (F) CFSE-labeled, congenically-marked CD4+ T cells (105) from Msr1+/+ K/BxN mice (thick line) or Msr1−/− K/BxN mice (thin line) were adoptively transferred together into H-2b/g7-expressing hosts and harvested 3 days later for flow cytometric analysis. Data represent 2 experiments with a total of 7 recipient mice. No CFSE dilution was seen in H-2b hosts (not shown). (G) Expression of the T cell anergy markers CD73 and FR4 was evaluated among CD4+ splenocytes and lymph node cells in the indicated mice. Data represent 2 experiments with a total of 3 mice/group.
B cells remain relatively naive in Msr1−/− K/BxN mice
Genetic absence of Msr1 did not affect the total number of splenic B cells in K/BxN or C57BL/6 mice (Fig 4A). We therefore used a recently-developed tetramer enrichment strategy to enumerate antigen-specific (i.e, GPI-specific) B cells (20). We found that mice lacking the KRN transgene contained very low numbers of GPI-specific B cells. In contrast, Msr1+/+ K/BxN mice contained significantly more GPI-specific B cells relative to Msr1−/− K/BxN mice (300,000 versus 80,000, p <0.001). Notably, there was no difference in the number of GPI-specific B cells in arthritic versus non-arthritic Msr1−/− K/BxN animals (Fig. 4B). In addition, we were unable to detect anti-GPI IgG-secreting cells by ELISPOT in either arthritic or non-arthritic Msr1−/− K/BxN mice, whereas they were easily detected in Msr1+/+ K/BxN mice (Fig. 4C). We therefore investigated the expression of B cell surface markers. We found that the expression of MHCII was essentially equivalent among GPI-specific B cells from Msr1+/+ and Msr1−/− mice. In contrast, GPI-specific B cell surface expression of IgM and IgD was substantially lower in Msr1+/+ K/BxN mice relative to Msr1−/− K/BxN mice, consistent with an activated B cell phenotype in the Msr1+/+ group and a more naïve phenotype in the Msr1−/− group (Fig. 4D). Further analysis confirmed this impression. Specifically, more GPI-specific B cells in Msr1+/+ K/BxN mice demonstrated isotype switching as demonstrated by increased intracellular IgG1 expresssion and also contained a significantly larger population of GPI-specific IgMlo CD38− GL7+ germinal center cells relative to Msr1−/− K/BxN mice (Fig. 4E and 4F) (28).
Figure 4. GPI-specific B cells remain naïve in Msr1−/− K/BxN mice.
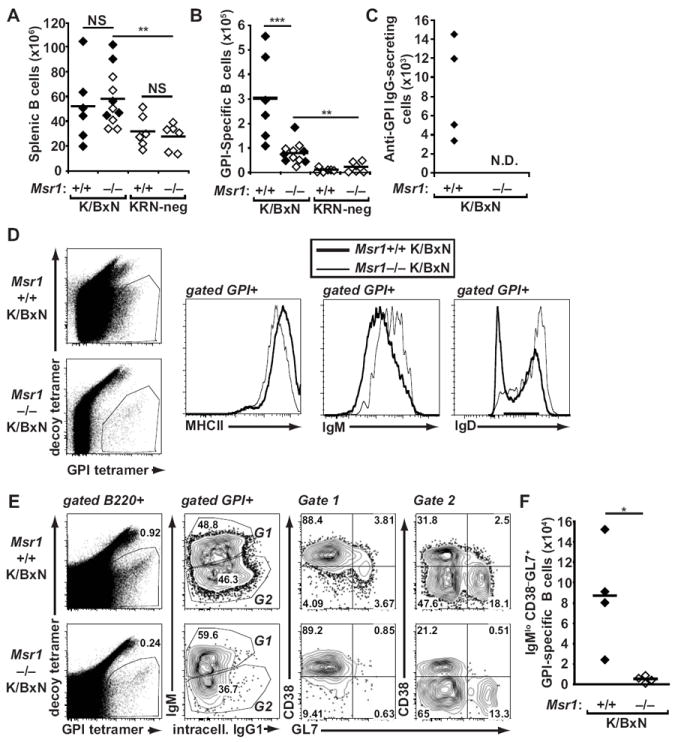
(A) Splenic B220+ B cells and (B) GPI-specific B cells from spleen and lymph nodes of 8-week-old Msr1+/+ and Msr1−/− K/BxN mice, as well as KRN-negative Msr1+/+ and Msr1−/− mice were analyzed by flow cytometry using antigen-specific and antigen-nonspecific B cell tetramers. Each point represents one animal; filled diamonds indicate arthritic animals, open diamonds indicate non-arthritic animals, and bars represent means; data were compiled from three independent experiments; **p ≤ 0.01, ***p ≤ 0.001. (C) The number of anti-GPI IgG-secreting cells was determined by ELISPOT in mice of the indicated genotypes. Data are representative of two independent experiments. In Msr1−/− K/BxN mice, these numbers were below the limit of detection for the assay (N.D. = not detected). (D) GPI-specific B cells from spleens and lymph nodes of mice of the indicated genotypes were assessed for cell surface expression of MHCII, IgM, and IgD. (E) Expression of surface IgM and intracellular IgG1 and CD38 and GL7 among GPI-specific lymph node and splenic B cells was determined in mice of the indicated genotypes. Naïve B cells are IgMhiCD38+GL7−, whereas germinal center B cells are IgMloCD38−GL7+ For D and E, the numbers indicate the percentage of cells in each quadrant or gate; results are representative of experiments performed with 3-4 mice/genotype. (F) The absolute number of GPI-specific IgMlo CD38−GL7+ germinal center B cells is depicted for the two groups of mice. Data represent two experiments with 4 mice/genotype; * p<0.05.
No intrinsic defect in Msr1-deficient B cells
We next asked why the GPI-specific B cells in Msr1−/− K/BxN mice remained relatively naïve in the presence of apparently normally-activated autoreactive CD4+ T cells. Two scenarios seemed likely – defects that were either B cell-intrinsic or B cell-extrinsic. First, it was possible that Msr1−/− B cells have an intrinsic activation defect. Arguing against this possibility is the fact that Msr1 is not expressed by B cells, in addition to a prior study demonstrating normal T cell-dependent humoral immune responses in the presence of pharmacologic blockade of Msr1 (29). Nonetheless, we formally considered the hypothesis that the B cells from Msr1−/− mice were less efficient responders than wildtype B cells. To test this, we isolated congenically-marked B cells from Msr1+/+ and Msr1−/− mice (H-2g7) and transferred them together with naïve KRN+ CD4+ T cells (H-2b) into T cell-deficient H-2b/g7 hosts, a situation known to promote the expansion of the KRN T cells (27). In this setting, GPI-specific B cells were readily detected among both the Msr1+/+ and Msr1−/− B cell populations, had gained the CD38− GL7+ germinal center phenotype, and expressed equivalent levels of the proliferation marker Ki67 (Fig. 5). We therefore concluded that Msr1-deficient B cells were not intrinsically defective in their capacity to respond to KRN+ T cells. We then focused on the second possibility: that Msr1 deficiency somehow alters the environment in which the KRN+ CD4+ T cells and GPI-specific B cells interact, leading to a B cell-extrinsic expansion defect.
Figure 5. No intrinsic defect in Msr1-deficient B cells.
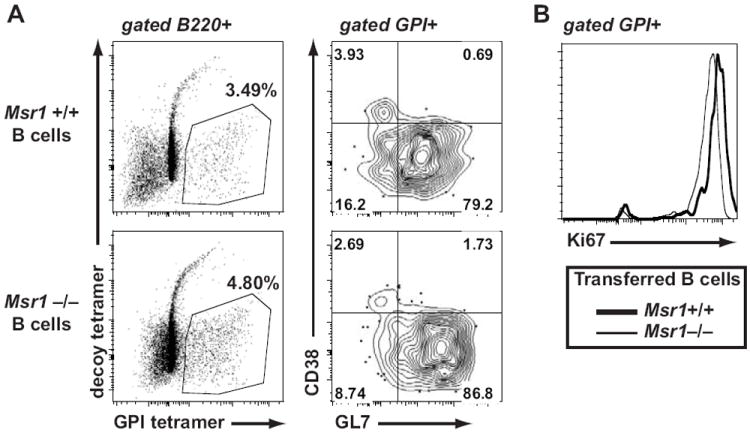
B cells (7 × 106) isolated from Msr1+/+ and Msr1−/− mice were transferred into separate H-2b/g7 T cell-deficient hosts that had received 105 KRN+ T cells one day prior. Eight days later, the congenically-marked donor B cells were analyzed by flow cytometry. GPI-specific B cells of both genotypes show similar (A) activated CD38−GL7+ phenotype and (B) Ki67 expression. In control hosts that did not receive KRN+ CD4+ T cells, too few GPI-specific B cells were recovered for analysis (not shown). The numbers indicate the percentage of cells in each quadrant; results are representative of experiments performed with 3 mice/group.
Msr1 regulates serum GPI concentration
Although normally found in the cytoplasm, low levels of soluble GPI can also be detected in the serum of mice and humans (30-32). Because Msr1 functions as a scavenger receptor, we hypothesized that Msr1 might bind and clear excess antigen (GPI) from the circulation. To address this, we incubated peritoneal macrophages from Msr1+/+ and Msr1−/− with fluorescently-labeled GPI or an irrelevant protein (bovine albumin), using fluorescence as a measure of uptake. Macrophages lacking Msr1 took up both GPI and albumin less efficiently than did wildtype macrophages, and this defect appeared more pronounced for GPI (Fig. 6A). Correspondingly, we found elevated levels of GPI in the serum of Msr1-deficient mice, which ranged between 2-20 μg/mL by Western blot analysis (Fig. 6B). Although the GPI standards detected as little as 23 ng/mL, the serum concentration of GPI in Msr1+/+ mice was below the level of detection for the assay, indicating that the serum concentration of GPI in Msr1−/− mice was a log or more higher than in Msr1+/+ mice. Deficiency of Msr1 did not impact the serum concentration of mouse albumin (42.9 ± 16 mg/mL in Msr1+/+ vs. 45.9 ± 14 mg/mL in Msr1−/−, [mean ± S.D, p = 0.76, n=5 mice/group]). We speculate that although Msr1 may be able to mediate uptake of albumin (as in Fig 6A), this effect does not measurably affect the concentration of a protein in high abundance in the serum. In contrast, the absence of Msr1-mediated uptake of a low abundance protein such as GPI can cause a detectable and immunologically significant increase in its serum concentration. These results demonstrate that Msr1 normally acts to maintain low levels of soluble GPI in the circulation.
Figure 6. Msr1-deficient mice have increased circulating GPI, leading to impaired B cell activation.
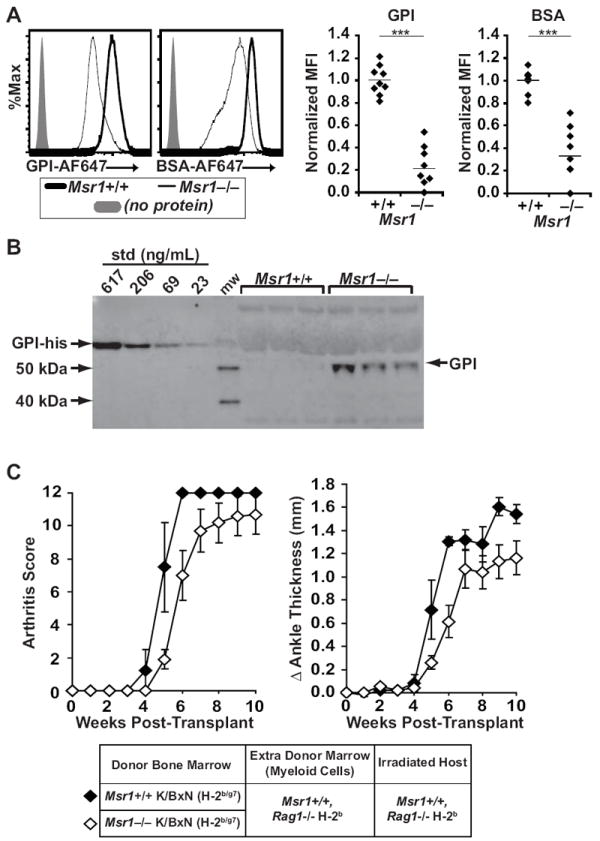
(A) Peritoneal macrophages from Msr1+/+ (thick line) and Msr1−/− (thin line) B6 mice were incubated with fluorescently-labeled GPI (left histogram), fluorescently-labeled irrelevant protein (BSA, right histogram), or no protein (grey shade) and fluorescent intensity was analyzed by flow cytometry as a measure of protein uptake. The histograms show representative plots. The right panels show MFI values for individual mice, with bars indicating mean values. (B) Serum GPI concentrations were determined by Western blot in 8-week-old Msr1+/+ and Msr1−/− μMT−/− mice using a standard curve of GPI-His. Three mice from each genotype representing the range of GPI concentration are shown. The rationale for using μMT−/− mice is explained in the text. (C)Rag1−/− recipient mice were sublethally irradiated and transplanted with a mixture of 10×106 bone marrow cells from Msr1+/+ K/BxN mice (filled diamonds, n = 4) or Msr1−/− K/BxN mice (open diamonds, n = 10) plus 5×106 bone marrow cells from Rag1−/− mice as a source of additional Msr1-sufficient myeloid cells. Arthritis was assessed via clinical arthritis scores (left) and changes in ankle thickness (right). Data are compiled from three separate experiments; plotted values are means ± SEM, and there is no statistical difference between the two groups via repeated measures ANOVA.
We hypothesized that adding Msr1-expressing antigen presenting cells (APCs) to capture and “normalize” the concentration of GPI should reverse the defect in B cell activation seen in Msr1−/− K/BxN mice. To test this, we created bone marrow chimeric mice in which bone marrow from non-arthritic Msr1−/− K/BxN or arthritic Msr1+/+ K/BxN mice was mixed with bone marrow from Rag1-deficient mice (as an additional source of Msr1-expressing APCs) and injected into sublethally-irradiated Rag1-deficient (Msr1-expressing) recipient mice. Indeed, mice transplanted with both Msr1−/− K/BxN marrow and Msr1-sufficient APCs developed arthritis equivalently to control mice (Fig. 6C). Furthermore, the incidence of arthritis among the mice transplanted with Msr1−/− K/BxN bone marrow was significantly higher than the incidence among the Msr1−/− K/BxN TCR transgenic animals (90% vs. 52.5%, p = 0.0033). In subsequent experiments in which the additional donor Rag1-deficient bone marrow was omitted, all Rag1-deficient recipients of either Msr1−/− K/BxN or Msr1+/+ K/BxN bone marrow developed arthritis, although those receiving Msr1−/− K/BxN marrow had a one-week delay in arthritis onset (Supplemental Figure 2A). The GPI-specific B cells in both groups of reconstituted mice assumed similar activated phenotypes (Supplemental Figure 2B). We found that 38-63% of the macrophages in the reconstituted mice were derived from the Msr1+/+ host (Supplemental Figure 2C), consistent with prior studies demonstrating that host macrophages are able to survive the sublethal radiation dose of 300 Rad (33, 34). As expected, the T and B cells in the reconstituted mice were entirely donor-derived (Supplemental Figure 2D). Thus, the radioresistant Msr1+/+ host macrophages were sufficient to restore B cell autoreactivity to GPI. These bone marrow transplantation experiments confirm our earlier impression that CD4+ KRN T cells from Msr1−/− K/BxN mice can provide productive B cell help and that intrinsic B cell Msr1 deficiency per se is not responsible for the reduced arthritis severity in Msr1−/− K/BxN mice. Rather, these experiments suggest that Msr1 regulates the serum concentration of the soluble self-antigen GPI.
Thus, in the presence of Msr1 on macrophages, GPI levels remain low, and KRN+ CD4+ autoreactive T cells are able to locate and activate GPI-specific B cells. In the absence of Msr1, GPI levels are higher; T cells are still activated, but are inefficient at activating GPI-specific B cells. Indeed, we found that Msr1−/− K/BxN mice had significantly fewer follicular helper T cells than did their wildtype counterparts (Figures 7A and 7B). Furthermore, immunohistologic examination of lymph nodes revealed T located in close proximity to B cells within germinal centers in Msr1+/+ K/BxN mice. In contrast, in Msr1−/− K/BxN mice, the T cells were situated in the paracortical area of the lymph node and B cells remained in primary lymphoid follicles, similar to naive C57BL/6 mice and Msr1−/− mice lacking the KRN TCR transgene (Figure 7C). Below we discuss potential mechanisms by which a relatively autoantigen-rich environment might paradoxically lead to less efficient T-B cell collaboration and decreased differentiation to follicular helper T cells.
Figure 7. Msr1-deficient K/BxN mice have few follicular helper T cells and fail to form robust germinal centers.
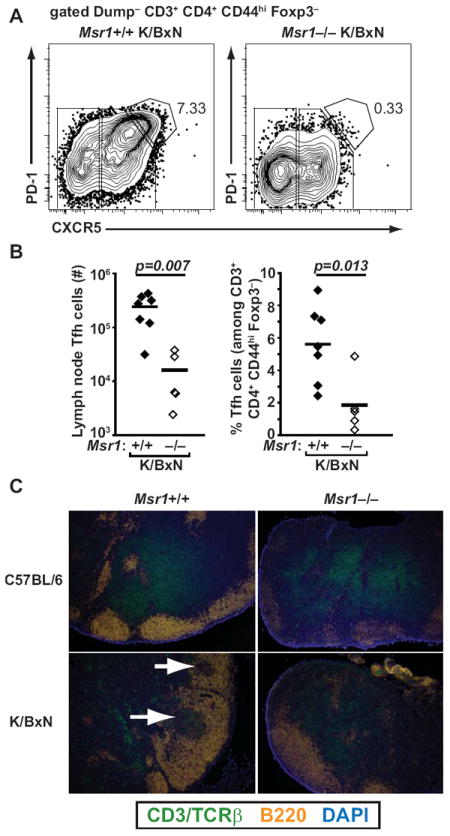
(A) Representative flow cytometric plots of lymph node cells from Msr1+/+ K/BxN and Msr1−/− K/BxN mice analyzed for the presence of CXCR5hi, PD-1hi follicular helper T (Tfh) cells. Upstream gating included dump (B220, CD11b, CD11c, F4/80) negative, CD3+, CD4+, CD44hi, Foxp3–. The number indicates the percentage of cells within the gate. (B) Numbers and percentages of CXCR5hi, PD-1hi follicular helper T cells in lymph nodes from the indicated mice, identified as in panel (A). Each point represents one mouse, line represents mean. Data compiled from two independent experiments. (C) Sections of inguinal lymph nodes from the indicated mice were analyzed for the distribution of T cells (detected by TCRβ and CD3, green) and B cells (B220, orange). Nucleated cells were detected with DAPI (blue). In Msr1+/+ K/BxN mice, T cells are in close proximity to B cells in germinal centers (lower left panel, white arrows), in contrast to Msr1−/− K/BxN mice in which the T cells remain in the paracortical area (lower right panel) as in naïve, non-arthritic animals (top two panels). Original magnification 10X. Images are representative of 3 independent experiments with lymph nodes from 2 mice/genotype.
Discussion
Here we report that Msr1 deficiency ameliorates systemic autoimmune disease in the K/BxN TCR transgenic mouse model of autoantibody-dependent arthritis. Specifically, in the absence of Msr1, circulating levels of the autoantigen GPI were increased. Despite normal activation of autoreactive CD4+ T cells, GPI-specific B cells remained relatively naïve, resulting in decreased autoantibody production and protection against arthritis development.
The increased circulating concentration of GPI in Msr1-decificent mice could theoretically impact the fate of GPI-specific B cells in a number of ways. For example, high concentrations of GPI could lead to more efficient clonal deletion of high-affinity, GPI-specific B cells or induce them to become anergic. Notably, naïve B cells may be rendered anergic, or hyporesponsive to antigenic stimuli, if they encounter their cognate antigen (signal 1) in the absence of TLR ligation or appropriate costimulation (signal 2) (6, 35). However, in Msr1−/− mice lacking autoreactive T cells, the number of GPI-specific B cells was not reduced (see Figure 4B), suggesting that clonal deletion was not more efficient. It is possible that the GPI-specific B cells in the Msr1−/− mice bind GPI with lower affinity or are relatively anergic compared to than their counterparts in Msr1+/+ mice. However, our finding that GPI-reactive B cells from Msr1−/− proliferated equivalently to those derived from Msr1+/+ mice following adoptive transfer suggests that they were neither anergic nor of substantially lower affinity.
We favor a model in which the higher concentration of circulating GPI in Msr1-deficient mice impairs B cell activation indirectly by altering the cognate interactions between autoreactive CD4+ T cells and B cells. In the Msr1−/− environment of relative GPI excess, T cells are more likely to encounter professional APCs presenting GPI peptide:MHCII complexes, resulting in T cell “stop” signals (36). Widespread, sustained arrest of the autoreactive T cells would in turn reduce the likelihood that an activated autoreactive T cell would encounter a rare GPI-reactive B cell, leading to the reduced number of follicular helper T cells that we observed. The net effect of this disruption of cognate T-B cell collaborations would be that GPI-reactive B cells would paradoxically appear to be in a clonally ignorant, naïve state despite the high concentration of soluble self antigen. As a corollary, because the precursor frequency of autoreactive (KRN+) CD4+ T cells is high in this system, our findings suggest that GPI peptide:MHCII-expressing APCs are scarce in Msr1+/+ K/BxN mice, permitting activated CD4+ T cells to move unhindered by APCs to encounter and activate GPI-specific B cells. Importantly, the proposed reduction in autoreactive T-B cell collaborations in Msr1−/− versus Msr1+/+ K/BxN mice is relative rather than absolute, meaning that autoreactive T-B cell collaborations can still occur, but less frequently and may appear to occur stochastically. Our data, including our finding that autoimmune arthritis develops in some but not all Msr1−/− K/BxN mice, are consistent with this scenario.
A similar dichotomy in arthritis susceptibility has been reported in K/BxN mice lacking interleukin-4 and in K/BxN mice lacking the neonatal Fc receptor (FcRn) (21, 37). In these studies and the present one, the development of arthritis correlated closely with the production of anti-GPI IgG autoantibodies, and even very low titers of anti-GPI IgG were sufficient to provoke arthritis (see Fig. 2B). These findings are consistent with the notion that the threshold for developing arthritis can be crossed relatively easily, and that stochastic events, such as those discussed above, likely impact if and when that crossing occurs.
Consistent with our findings, Mamula and colleagues recently noted decreased pathology and autoantibody titers in autoimmune-prone MRL mice genetically lacking Msr1 (38), perhaps attributable to defects in Msr1-mediated trafficking of antigen from B cells to macrophages, resulting in decreased T cell activation (39). In our model system, however, the loss of Msr1 did not appreciably impair T cell activation.
A main reported function of Msr1 is the clearance of apoptotic debris. Impaired clearance of apoptotic debris is implicated in the breakdown of tolerance in SLE, the prototypic autoantibody-dependent disease (10, 40). The recent description of anti-Msr1 (anti-SR-A) antibodies among patients with SLE is notable. It has been suggested that anti-Msr1 antibodies might interfere with the uptake of apoptotic debris, thereby promoting autoimmunity (41, 42). Based on these studies, one might reasonably expect autoimmunity to arise more easily in the absence of Msr1. Yet we found just the opposite – Msr1 deficiency protected against autoimmunity in K/BxN mice. This discrepancy might be related more to timing, rather than to peculiarities of the model system. That is, Msr1 deficiency early in lymphocyte development may result in impaired initial activation of autoreactive B cells, even in the presence of activated, autoreactive T cells. In contrast, in an organism with pre-existing activated, self-reactive B cells, inhibiting Msr1 and increasing the load of antigen might fuel sustained autoreactivity and inflammation (41, 42). These scenarios are consistent with Mitchison’s framework of “high zone tolerance” (43). Understanding how anti-Msr1 antibodies might influence SLE disease pathogenesis will likely require detailed knowledge of the timing of when the anti-Msr1 antibodies appear and whether or how they affect the interaction of Msr1 with specific autoantigens.
In sum, we have shown that K/BxN mice lacking Msr1 are protected from autoantibody-mediated arthritis due to decreased autoreactive B cell activation in the presence of excess soluble autoantigen. The finding that factors that increase autoantigen load might alter T-B cell collaborations and reduce rather than enhance autoimmunity furthers our understanding of the complex temporal and mechanistic balance of immunological tolerance.
Supplementary Material
Acknowledgments
We thank Christophe Benoist and Diane Mathis for mice, Sindhuja Rao, Tyler Potts, Jonathan Dexter, Gregory Schuneman, and Brandon Burbach for technical assistance, and Antonio Pagán and Marc Jenkins for helpful discussions.
This work was supported by the Lupus Foundation of Minnesota and by the University of Minnesota Department of Pediatrics. BAB was supported by NIH K08-AR054317 and an Arthritis Foundation Arthritis Investigator Award. SH was supported by NIH T32-AI007313.
Footnotes
The authors declare no competing financial interests.
References
- 1.Mathis D, Benoist C. Levees of immunological tolerance. Nat Immunol. 2010;11:3–6. doi: 10.1038/ni.1833. [DOI] [PubMed] [Google Scholar]
- 2.von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol. 2010;11:14–20. doi: 10.1038/ni.1794. [DOI] [PubMed] [Google Scholar]
- 3.Gay D, Saunders T, Camper S, Weigert M. Receptor editing: an approach by autoreactive B cells to escape tolerance. J Exp Med. 1993;177:999–1008. doi: 10.1084/jem.177.4.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemazee DA, Burki K. Clonal deletion of B lymphocytes in a transgenic mouse bearing anti-MHC class I antibody genes. Nature. 1989;337:562–566. doi: 10.1038/337562a0. [DOI] [PubMed] [Google Scholar]
- 6.Yarkoni Y, Getahun A, Cambier JC. Molecular underpinning of B-cell anergy. Immunol Rev. 2010;237:249–263. doi: 10.1111/j.1600-065X.2010.00936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pillai S, Mattoo H, Cariappa A. B cells and autoimmunity. Curr Opin Immunol. 2011;23:721–731. doi: 10.1016/j.coi.2011.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greaves DR, Gordon S. The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. Journal of lipid research. 2009;50(Suppl):S282–286. doi: 10.1194/jlr.R800066-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harshyne LA, Zimmer MI, Watkins SC, Barratt-Boyes SM. A role for class A scavenger receptor in dendritic cell nibbling from live cells. J Immunol. 2003;170:2302–2309. doi: 10.4049/jimmunol.170.5.2302. [DOI] [PubMed] [Google Scholar]
- 10.Munoz LE, van Bavel C, Franz S, Berden J, Herrmann M, van der Vlag J. Apoptosis in the pathogenesis of systemic lupus erythematosus. Lupus. 2008;17:371–375. doi: 10.1177/0961203308089990. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki H, Kurihara Y, Takeya M, Kamada N, Kataoka M, Jishage K, Ueda O, Sakaguchi H, Higashi T, Suzuki T, Takashima Y, Kawabe Y, Cynshi O, Wada Y, Honda M, Kurihara H, Aburatani H, Doi T, Matsumoto A, Azuma S, Noda T, Toyoda Y, Itakura H, Yazaki Y, Kodama T, et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature. 1997;386:292–296. doi: 10.1038/386292a0. [DOI] [PubMed] [Google Scholar]
- 12.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87:811–822. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286:1732–1735. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- 14.Korganow AS, Ji H, Mangialaio S, Duchatelle V, Pelanda R, Martin T, Degott C, Kikutani H, Rajewsky K, Pasquali JL, Benoist C, Mathis D. From systemic T cell self-reactivity to organ-specific autoimmune disease via immunoglobulins. Immunity. 1999;10:451–461. doi: 10.1016/s1074-7613(00)80045-x. [DOI] [PubMed] [Google Scholar]
- 15.Binstadt BA, Hebert JL, Ortiz-Lopez A, Bronson R, Benoist C, Mathis D. The same systemic autoimmune disease provokes arthritis and endocarditis via distinct mechanisms. Proc Natl Acad Sci U S A. 2009;106:16758–16763. doi: 10.1073/pnas.0909132106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice. Jikken dobutsu. Experimental animals. 1980;29:1–13. doi: 10.1538/expanim1978.29.1_1. [DOI] [PubMed] [Google Scholar]
- 17.Mombaerts P, Iacomini J, Johnson RS, Herrup K, Tonegawa S, Papaioannou VE. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 18.Lie BA, Ronningen KS, Akselsen HE, Thorsby E, Undlien DE. Application and interpretation of transmission/disequilibrium tests: transmission of HLA-DQ haplotypes to unaffected siblings in 526 families with type 1 diabetes. American journal of human genetics. 2000;66:740–743. doi: 10.1086/302780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- 20.Taylor JJ, Martinez RJ, Titcombe PJ, Barsness LO, Thomas SR, Zhang N, Katzman SD, Jenkins MK, Mueller DL. Deletion and anergy of polyclonal B cells specific for ubiquitous membrane-bound self-antigen. J Exp Med. 2012;209:2065–2077. doi: 10.1084/jem.20112272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akilesh S, Petkova S, Sproule TJ, Shaffer DJ, Christianson GJ, Roopenian D. The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J Clin Invest. 2004;113:1328–1333. doi: 10.1172/JCI18838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen LT, Jacobs J, Mathis D, Benoist C. Where FoxP3-dependent regulatory T cells impinge on the development of inflammatory arthritis. Arthritis Rheum. 2007;56:509–520. doi: 10.1002/art.22272. [DOI] [PubMed] [Google Scholar]
- 23.Mandik-Nayak L, Wipke BT, Shih FF, Unanue ER, Allen PM. Despite ubiquitous autoantigen expression, arthritogenic autoantibody response initiates in the local lymph node. Proc Natl Acad Sci U S A. 2002;99:14368–14373. doi: 10.1073/pnas.182549099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jacobs JP, Wu HJ, Benoist C, Mathis D. IL-17-producing T cells can augment autoantibody-induced arthritis. Proc Natl Acad Sci U S A. 2009;106:21789–21794. doi: 10.1073/pnas.0912152106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowdish DM, Gordon S. Conserved domains of the class A scavenger receptors: evolution and function. Immunol Rev. 2009;227:19–31. doi: 10.1111/j.1600-065X.2008.00728.x. [DOI] [PubMed] [Google Scholar]
- 26.Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez RJ, Zhang N, Thomas SR, Nandiwada SL, Jenkins MK, Binstadt BA, Mueller DL. Arthritogenic Self-Reactive CD4+ T Cells Acquire an FR4hiCD73hi Anergic State in the Presence of Foxp3+ Regulatory T Cells. Journal of immunology. 2012;188:170–181. doi: 10.4049/jimmunol.1101311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pape KA, Taylor JJ, Maul RW, Gearhart PJ, Jenkins MK. Different B cell populations mediate early and late memory during an endogenous immune response. Science. 2011;331:1203–1207. doi: 10.1126/science.1201730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abraham R, Choudhury A, Basu SK, Bal V, Rath S. Disruption of T cell tolerance by directing a self antigen to macrophage-specific scavenger receptors. J Immunol. 1997;158:4029–4035. [PubMed] [Google Scholar]
- 30.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3:360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 31.Neri B, Becucci A, Ciapini A, Comparini T, Guidi G, Guidi S. Chronobiological aspects of phosphohexoseisomerase in monitoring multiple myeloma. Oncology. 1983;40:332–335. doi: 10.1159/000225756. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe M, Ando Y, Todoroki H, Minami H, Hidaka H. Molecular cloning and sequencing of a cDNA clone encoding a new calcium binding protein, named calgizzarin, from rabbit lung. Biochem Biophys Res Commun. 1991;181:644–649. doi: 10.1016/0006-291x(91)91239-9. [DOI] [PubMed] [Google Scholar]
- 33.Oghiso Y, Yamada Y. Heterogeneity of the radiosensitivity and origins of tissue macrophage colony-forming cells. J Radiat Res. 1992;33:334–341. doi: 10.1269/jrr.33.334. [DOI] [PubMed] [Google Scholar]
- 34.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336:86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 35.Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature. 2012;489:160–164. doi: 10.1038/nature11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dustin ML, Bromley SK, Kan Z, Peterson DA, Unanue ER. Antigen receptor engagement delivers a stop signal to migrating T lymphocytes. Proc Natl Acad Sci U S A. 1997;94:3909–3913. doi: 10.1073/pnas.94.8.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ohmura K, Nguyen LT, Locksley RM, Mathis D, Benoist C. Interleukin-4 can be a key positive regulator of inflammatory arthritis. Arthritis Rheum. 2005;52:1866–1875. doi: 10.1002/art.21104. [DOI] [PubMed] [Google Scholar]
- 38.Raycroft MT, Harvey BP, Bruck MJ, Mamula MJ. Inhibition of Antigen Trafficking through Scavenger Receptor A. J Biol Chem. 2012;287:5310–5316. doi: 10.1074/jbc.M111.316356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Harvey BP, Quan TE, Rudenga BJ, Roman RM, Craft J, Mamula MJ. Editing antigen presentation: antigen transfer between human B lymphocytes and macrophages mediated by class A scavenger receptors. J Immunol. 2008;181:4043–4051. doi: 10.4049/jimmunol.181.6.4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202. doi: 10.1186/ar3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wermeling F, Chen Y, Pikkarainen T, Scheynius A, Winqvist O, Izui S, Ravetch JV, Tryggvason K, Karlsson MC. Class A scavenger receptors regulate tolerance against apoptotic cells, and autoantibodies against these receptors are predictive of systemic lupus. J Exp Med. 2007;204:2259–2265. doi: 10.1084/jem.20070600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen XW, Shen Y, Sun CY, Wu FX, Chen Y, Yang CD. Anti-class a scavenger receptor autoantibodies from systemic lupus erythematosus patients impair phagocytic clearance of apoptotic cells by macrophages in vitro. Arthritis Res Ther. 2011;13:R9. doi: 10.1186/ar3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mitchison NA. Induction of Immunological Paralysis in Two Zones of Dosage. Proc R Soc Lond B Biol Sci. 1964;161:275–292. doi: 10.1098/rspb.1964.0093. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.