

Abstract
Parkinson’s disease (PD) is a neurodegenerative disorder primarily affecting the dopaminergic neurons in the nigrastriatal pathway resulting in debilitating motor impairment in both familial and sporadic cases. Histone deacetylase (HDAC) inhibitors have been recently implicated as a therapeutic candidate because of their ability to correct the disrupted HDAC activity in PD and other neurodegenerative diseases. Sodium butyrate (SB), an HDAC inhibitor, reduces degeneration of dopaminergic neurons in a mutant alpha-synuclein Drosophila transgenic model of familial PD. Chronic exposure to the pesticide rotenone also causes selective degeneration of dopaminergic neurons and causes locomotor impairment and early mortality in a Drosophila model of chemically-induced PD. This study investigated the effects of sodium butyrate on locomotor impairment and early mortality in a rotenone-induced PD model. We show that treatment with 10 mM SB-supplemented food rescued the rotenone-induced locomotor impairment and early mortality in flies. Additionally, flies with the genetic knockdown of HDAC activity through Sin3A loss-of-function mutation (Sin3Alof) were resistant to rotenone-induced locomotor impairment and early mortality. Furthermore, SB-supplemented Sin3Alof flies had a modest additive effect for improving locomotor impairment. We also show SB-mediated improvement of rotenone-induced locomotor impairment was associated with elevated dopamine levels in the brain. However, the possibility of SB-mediated protective role through mechanisms independent from dopamine system is also discussed. These findings demonstrate that HDAC inhibitors like SB can ameliorate locomotor impairment in a rotenone-induced PD model.
Keywords: Parkinson’s disease, Rotenone, HDAC inhibitors, Sodium Butyrate, Neurodegenerative disease, Drosophila disease models
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by selective loss of dopaminergic neurons in the substantia nigra pars compacta and the presence of α-synuclein inclusions, called Lewy bodies (Hisahara and Shimohama, 2010). The hallmark of PD is motor impairment due to a reduction of striatal dopamine levels. Motor impairment symptoms include tremor, rigidity, slowness of movement, and postural instability (Olanow and Tatton, 1999). The majority of cases are idiopathic, with fewer than 10% cases attributed to mutations in genes such as α-synuclein, parkin, LRRK1, PINK1 and DJ (Le Couteur et al., 2002). Currently, there is no cure for PD and available therapies only treat symptoms of motor impairment (Wu et al., 2008). Levodopa (L-dopa), a dopamine precursor, the leading treatment of PD for over 40 years (Poewe et al., 2010), improves motor impairment by increasing dopamine levels (Poewe et al., 2010). However, continued use of L-dopa leads to other motor dyskinesias that undermine the benefits of treatment (Thanvi et al., 2007). The development of effective treatment for PD is difficult because pathology is affected by several pathways that may act serially or in parallel (Mai et al., 2009).
Recent evidence suggests that the balance of histone deacetylase (HDAC) and histone acetyltransferase (HAT) activity is disrupted in PD and other neurodegenerative disorders, resulting in global decreases in acetylation (Kontopoulos et al., 2006, Saha and Pahan, 2006). The HDAC:HAT activities regulate gene expression by remodeling chromatin structure (Chuang et al., 2009). HDACs silence gene expression and enhance chromatin compaction by deacetylation of histone proteins. HDAC inhibitors can tip the balance of HDAC and HAT activity towards acetylation, resulting in relaxed chromatin structure, and enhanced gene expression (Chuang et al., 2009). The use of HDAC inhibitors has shown promise for rescue of neural degeneration through increased acetylation in HD and PD (Steffan et al., 2001, Kontopoulos et al., 2006).
Sodium butyrate (SB) is a fatty acid derivative that inhibits class I and II HDACs, but not HDAC6 (Chuang et al., 2009). SB is a small molecule that can easily cross the blood brain barrier, therefore direct injection into the brain is unnecessary because SB is readily absorbed when administered orally (Egorin et al., 1999). SB diminished apoptotic death and rescued dopaminergic neuron degeneration in a α-synuclein transgenic fly model through increased acetylation (Kontopoulos et al., 2006). SB protected dopamine neurons from neurotoxins in neuron-glia culture (Wu et al., 2008, Kidd and Schneider, 2010). In a HD Drosophila model, SB slowed degeneration caused by polyglutamine repeat expansion in vivo (Steffan et al., 2001).
Animal models provide an opportunity to better study the etiology of PD as well as potential treatments. PD can be modeled in animals through genetic manipulation or chemical exposure. Broad-spectrum pesticides, such as rotenone and paraquat, model the pathology of PD by causing dopamine neuron degeneration (Hisahara and Shimohama, 2010). Patients with PD are 2.5 times more likely to have dealt with either of these two broad-spectrum pesticides in their lifetime (Spivey, 2011). Rotenone is an inhibitor of mitochondrial complex I that causes dose-dependent ATP depletion, oxidative damage, and early mortality (Sherer et al., 2003). In Drosophila, rotenone causes locomotor impairment and dopaminergic neuron degeneration (Coulom and Birman, 2004). The rotenone-induced Drosophila model of PD is useful because it parallels the human response to L-dopa therapy (Coulom and Birman, 2004, Cannon et al., 2009). Additionally, a Drosophila model is advantageous because it allows for fast and simple testing of pharmacological agents and genetic manipulation, for instance of HDAC activity through mutant flies (e.g. Sin3Alof). The Sin3A gene expresses a corepressor of the HDAC machinery, mainly associated with class I HDACs (Steffan et al., 2001, Kontopoulos et al., 2006, Hayakawa and Nakayama, 2011).
Based on recent evidence regarding the role of HDAC inhibitors in neurodegenerative disease, we hypothesize that pharmacological and genetic reduction of HDAC activity by SB and Sin3Alof mutation respectively, would alleviate rotenone-induced locomotor impairment and early mortality in Drosophila.
2. Experimental Procedures
2.1 Drosophila stocks and drug exposure
Canton-S (CS; wild type) and Sin3Alof flies (Sin3A08269) were obtained from Bloomington Stock Center (Bloomington, IN). Fly stocks were maintained on a yeast-supplemented cornmeal-based diet (Nutri-Fly Bloomington Formula, Genesee Scientific, San Diego, CA) in a 12 h light/dark cycle at constant temperature (25 °C). Rotenone (in DMSO) and SB (in water) (Sigma, St. Louis, MO) supplemented food was prepared in dry instant fly food (Carolina Formula 4–24 Plain Medium, Burlington, NC). For all experiments, a biological replicate consisted of 20–25 adult flies (1–5 day old) reared on appropriately supplemented food. All treatments and conditions were independently repeated 3–8 times with each test done in duplicate or greater.
2.2 Locomotor impairment & survival assays
Locomotor impairment was assessed by quantifying climbing ability in a negative geotactic assay as described previously (Feany and Bender, 2000, Coulom and Birman, 2004). Flies were tested in a clear 19 cm long x 2.4 cm diameter plastic column closed at both ends, demarcated into top and bottom sections by a line drawn at 6.5 cm from the bottom of the apparatus. During their light cycle, 20–25 flies were transferred to the testing apparatus and allowed to habituate for at least 5 min. Flies were then forced to the bottom of the column by gently shaking the apparatus. After 1 min, the numbers of flies in the top and bottom sections of the apparatus were counted separately. Three trials were performed per vial, allowing 1 min of rest between trials.
Viability was calculated by counting the number of surviving flies after 3 and 7 days of treatment.
2.3 HPLC-Mass spectrometry
Five fly heads of the appropriate genotype and treatment for 3 days were homogenized in 50 μl ice cold sodium acetate buffer (pH 4.5) followed by centrifugation at 17000 g for 5 minutes. Supernatant was filtered through 0.2 μm syringe filter. Dopamine and serotonin content was determined by injecting 1 μl homogenate three times for each sample. Dopamine and serotonin standards (Sigma) were used to optimize HPLC-Mass spectrometry conditions.
Liquid chromatography was performed on an Ascentis Express C18 column (5 cm x 500 μm, 2.7 μm; Supelco-Sigma) on an 1100 series HPLC system (Agilent, Santa Clara, CA) coupled to a 6230 TOF LC/MS (Agilent) mass spectrometer. The column was eluted with a linear mobile phase gradient starting with 10% acetonitrile, 0.1% formic acid in water for 0.5 min to 90% acetonitrile, 0.1% formic acid in water at 5 minutes and held for additional 3 minutes with a flow rate of 12 μl/min. The column was equilibrated in starting elution buffer for 5 minutes between injections.
Mass spectrometry was performed in a positive ion electrospray mode with the following conditions; Drying gas temperature and flow=325 °C and 6 l/min, Sheath gas temperature and flow=400 °C and 6 l/min, Capillary voltage and current=3000 V and 4.99 μA, Nozzle voltage=1000 V, Fragmentor voltage=100 V, and Skimmer voltage=65 V. Peak area in the extracted ion chromatogram (m/z=154) and mean ± SEM peak area from three independent experiments was used to report dopamine and serotonin content.
2.4 Real-Time PCR
Total RNA (with DNAse treatment) from 6–10 heads of appropriate genotype and treatment for 3 days was isolated using RNeasy miniprep kit (Qiagen, Valencia, CA). RNA quality and yield was measured using nanodrop spectrometer (Thermo Scientific, Wilmington, DE). qRT-PCR was performed in duplicates on 50–100 ng total RNA using one step Quantifast SYBR Green RT-PCR kit (Qiagen) on StepONE Real-Time PCR system (Applied Biosystems, Foster City, CA). Dissociation curve was analyzed to ensure primer specificity. Relative normalized transcript level was determined by delta-delta Ct method. RP49 was used as the normalizing gene. Mean ± SEM TH (ple) transcript was calculated from four independent experiments.
ple primers: 5′-CTACCAGGATCAGGAGTACCA-3′ and 5′-GCGACGGAACTTGTCCTT-3′.
RP49 primers: 5′-CGGTTACGGATCGAACAAGC-3′ and 5′-CTTGCGCTTCTTGGAGGAGA-3′.
2.5 Superoxide Dismutase (SOD) assay
SOD activity assay was performed as previously described with minor modifications (Sun and Tower, 1999). Briefly, 5 flies of appropriate genotype and treatment for 3 days were homogenized in 500 μl ice-cold 50 mM phosphate buffer (pH 7.5) containing 0.1% Triton X-100, followed by centrifugation at 4 °C, 12500 g for 5 minutes. This assay measured SOD activity by measuring inhibition of quercetin oxidation by fly extract as compared to no extract control. For each assay 100 μl fly extract supernatant was added to 900 μl reaction mix containing 20 mM phosphate buffer (pH 10), 0.8 mM EDTA, 0.8 mM TEMED, and 100 μl of 1.5 mg quercetin /10 ml dimethylformamide solution (quercetin was added just before absorbance measurements). The change in absorbance at 406 nm was measured for 5 minutes. The samples were normalized for protein content, quantified by Pierce BCA protein assay (Thermo Scientific, Rockford, IL). SOD activity was reported as mean ± SEM percent inhibition of quercetin oxidation from 4 independent experiments.
2.6 Statistical analysis
Mortality data were analyzed via separate logistic regression models by considering survival (alive or dead) at either 3 or 7 days as the outcome and presented as a ratio of odds of survival as compared to corresponding control. For each genotype, SB status was used as predictor. Similarly for flies without SB, mortality was related to genotype status. Logistic regression models were also used to analyze climbing ability due to the binary nature of the outcomes – reaching the top or not - and presented as a ratio of odds of reaching the top section of the apparatus as compared to corresponding control. The same predictors were used in these analyses, either considering the effect of SB status within genotype, or considering the effect of genotype within SB status.
Due to the multiple tests being conducted for mortality and climbing ability, Bonferroni corrections were applied to all p-values. Since we are producing statistical testing results 12 times (for viability and mobility comparisons) using the same set of data, all p-values associated with motility and viability were multiplied by 12. For all analyses, the standard errors were adjusted for possible within-experiment clustering effects due to the data being collected from different independent experimental runs.
Differences among groups for dopamine, serotonin, TH mRNA, and SOD activity were tested via one-way analysis of variance (ANOVA). Post-hoc testing to determine specific group differences was done utilizing the Scheffe adjustment for multiple testing. Scheffe was utilized since it is the most conservative adjustment procedure available for comparisons made among all groups (Tabachnick and Fidell, 2007).
3. Results
3.1 SB treatment reduces rotenone-induced early mortality in Drosophila
Rotenone exposure causes early mortality in flies, likely due to oxidative damage in neuronal and non-neuronal tissues (Coulom and Birman, 2004, Hosamani et al., 2010). Therefore we tested the effect of SB treatment on rotenone-induced early mortality in flies. Wild type flies reared on 62.5 μM, 125 μM, and 250 μM rotenone-supplemented food displayed a robust early mortality as compared to vehicle treated flies in a dose dependent manner after 3 days and 7 days (Figure 1a and 1b).
Figure 1. SB treatment improves rotenone-mediated early mortality.

Wild type (CS) flies reared on 62.5 μM, 125 μM, and 250 μM rotenone for (a) 3 days and (b) 7 days had decreased viability in a dose dependent manner as compared to wild type flies reared on only vehicle- or 10 mM SB-supplemented food. Wild type flies reared on 10 mM SB- and rotenone-supplemented food had reduced rotenone-induced early mortality and showed a significant improvement in the odds of survival as compared to without SB control (After 3 days; all rotenone doses and after 7 days; 62.5 μM and 125 μM rotenone doses). Columns=average survival percentage of flies; error bars=standard error; OR; odds ratio of survival as compared to without SB control for the same rotenone dose; * p<0.05, *** p<0.001 for odds of survival; n=3–8 biological replicates set in at least duplicates.
To test the effect of SB on early mortality, we used a previously established non-toxic concentration of SB (10 mM) for flies (Pallos et al., 2008, McDonald et al., 2012). Wild type flies reared on 10 mM SB- and 62.5 μM, 125 μM, and 250 μM rotenone-supplemented food had a robust improvement in viability after 3 and 7 days and showed a significant improvement in the odds of survival as compared to flies reared on rotenone-supplemented food without SB at all rotenone doses after 3 days and at 62.5 μM and 125 μM rotenone doses after 7 days (Figure 1a and 1b).
3.2 SB treatment rescues rotenone-induced locomotor impairment
In Drosophila, rotenone exposure causes locomotor impairment at concentrations ranging from 25 μM to 500 μM (Coulom and Birman, 2004). In accordance with published findings, we observed wild type flies reared on 62.5 μM, 125 μM, and 250 μM rotenone-supplemented food displayed motor impairment. We quantified the locomotor impairment after 3 days for the 125 μM rotenone dose by a negative geotactic assay. Wild type flies reared on rotenone-supplemented food displayed a robust defect in reaching the top of the apparatus as compared to vehicle treated flies (Figure 2).
Figure 2. SB treatment improves rotenone-induced locomotor impairment.
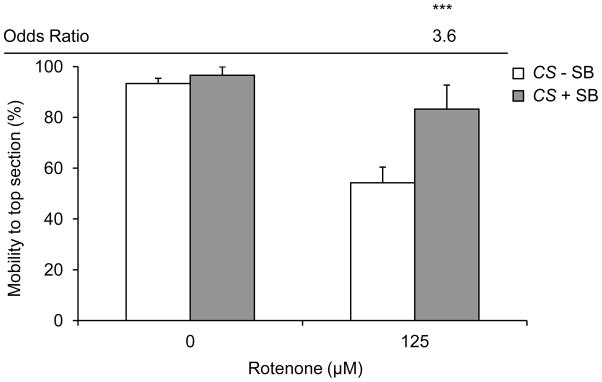
In the absence of rotenone, 10 mM SB treatment does not change negative geotactic response in wild type flies. Exposure to 125 μM rotenone for 3 days caused a robust decrease in negative geotactic response as compared to vehicle only. In the presence of rotenone, 10 mM SB treatment for 3 days improved negative geotactic response and showed a significant increase in the odds of flies reaching the top of the apparatus. Columns=average percentage of flies reaching the top section of the apparatus after 1 min; error bars=standard error; OR=odds ratio of mobility to top section as compared to without SB control for the same rotenone dose; *** p<0.001 for odds of survival; n=3–8 biological replicates set in at least duplicates.
Wild type flies reared on 10 mM SB- and 125 μM rotenone-supplemented food for 3 days had a robust improvement in climbing ability and showed a significant improvement in the odds of reaching the top section of the apparatus as compared to flies reared on rotenone-supplemented food without SB (Figure 2).
3.3 Sin3Alof mutants are resistant to rotenone-induced early mortality and locomotor impairment
To determine the effect of genetic reduction of HDAC activity on rotenone-induced defects, we measured rotenone-induced early mortality and locomotor impairment in Sin3Alof flies, which have reduced HDAC activity (Steffan et al., 2001, Kontopoulos et al., 2006). Sin3Alof flies reared on 62.5 μM, 125 μM, and 250 μM rotenone-supplemented food displayed a robust resistance to rotenone-induced early mortality after 3 and 7 days and showed a significant improvement in the odds of survival as compared to wild type flies at 62.5 μM and 125 μM rotenone doses after 3 days and at 125 μM rotenone dose after 7 days (Figure 3a and 3b).
Figure 3. Sin3Alof flies are resistant to rotenone-induced early mortality.
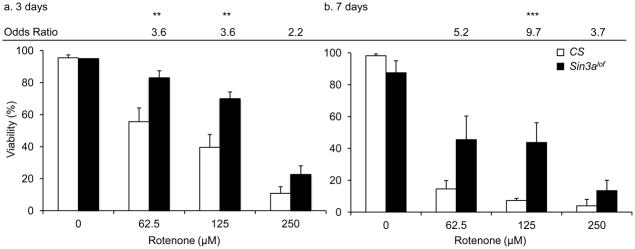
Wild type (CS) and Sin3Alof flies reared on 62.5 μM, 125 μM, and 250 μM rotenone for (a) 3 days and (b) 7 days had decreased viability in a dose dependent manner as compared to wild type flies reared on vehicle-supplemented control food. However Sin3Alof flies had reduced rotenone-induced early mortality and showed a significant increase in the odds of survival as compared to wild type flies (After 3 days; 62.5 μM and125 μM rotenone doses and after 7 days; 125 μM rotenone doses). Columns=average survival percentage of flies; error bars=standard error; Odds ratio=odds of survival ratio as compared to wild type flies for the same rotenone dose; ** p<0.01, *** p<0.001 for odds of survival; n=3–8 biological replicates set in at least duplicates.
Sin3Alof flies reared on 125 μM rotenone-supplemented food for 3 days had a robust improvement in climbing ability and showed a significant improvement in the odds of reaching the top section of the apparatus as compared to wild type flies reared on 125 μM rotenone-supplemented food (Figure 4).
Figure 4. Sin3Alof flies are resistant to rotenone-induced locomotor impairment.

Wild type (CS) and Sin3Alof flies on vehicle-supplemented control food had comparable negative geotactic response. Exposure to 125 μM rotenone for 3 days caused a significant decrease in negative geotactic response in wild type flies. However, Sin3Alof flies reared on 125 μM rotenone for 3 days had comparable negative geotactic response to Sin3Alof flies on vehicle-supplemented control food. Additionally, Sin3Alof flies reared on 125 μM rotenone for 3 days had a significant increase in the odds of reaching the top of the apparatus as compared to wild type flies on the same treatment. Columns=average percentage of flies reaching the top section of the apparatus after 1 min; error bars=standard error; Odds ratio=odds ratio for mobility to top section as compared to without SB control for the same rotenone dose; * p<0.05, ** p<0.01, *** p<0.001 for odds of survival; n=3–8 biological replicates set in at least duplicates.
Furthermore, we observed a modest additive effect of SB-supplementation and Sin3A loss-of-function on rotenone-induced early mortality. Sin3Alof flies reared on 10 mM SB- and 62.5 μM, 125 μM, and 250 μM rotenone-supplemented food showed a significant improvement in the odds of survival as compared to flies reared on rotenone-supplemented food without SB at 250 μM rotenone dose after 3 days and at 125 μM and 250 μM rotenone doses after 7 days (Figure 5a and 5b).
Figure 5. SB treatment in Sin3Alof flies shows additive improvement in rotenone-induced early mortality.

Sin3Alof flies reared on 10 mM SB- and rotenone-supplemented food had a significant increase in the odds of survival as compared to without SB control (a. After 3 days; 250 μM rotenone dose and b. after 7 days; 125 μM and 250 μM rotenone doses). Columns=average survival percentage of flies; error bars=standard error; Odds ratio=odds of survival ratio as compared to Sin3Alof flies for the same rotenone dose without SB; *p<0.05, ** p<0.01, *** p<0.001 for odds of survival; n=3–8 biological replicates set in at least duplicates.
However, we did not observe any additive effect of SB-supplementation in Sin3Alof flies on rotenone-induced locomotor impairment (not shown).
3.4 SB treatment rescues rotenone-induced dopamine deficiency
In Drosophila rotenone exposure causes brain dopamine deficiency, a hallmark of Parkinson’s disease, but does not have a significant effect on serotonin levels (Coulom and Birman, 2004). We quantified the dopamine content in head extracts after 3 days of treatment using HPLC-MS. Wild type flies reared on 125 μM rotenone-supplemented food displayed a significant dopamine deficiency as compared to vehicle treated flies (Figure 6a; ANOVA; Scheffe post hoc p<0.0001). Wild type flies reared on 10 mM SB- and 125 μM rotenone-supplemented food for 3 days had a significant improvement in dopamine content as compared to flies reared on 125 μM rotenone-supplemented food without SB (Figure 6a; ANOVA; Scheffe post hoc p<0.05).
Figure 6. SB treatment reduces dopamine deficiency in rotenone-exposed flies.
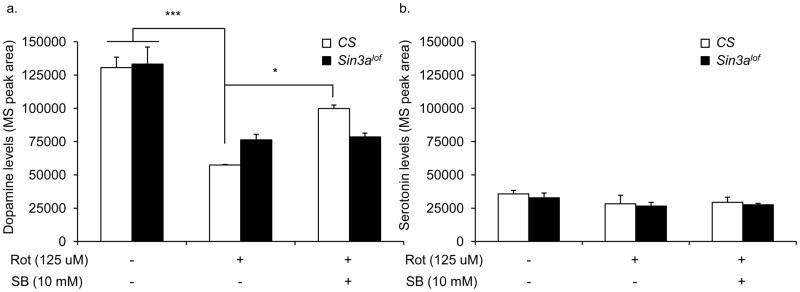
(a) Wild type (CS) and Sin3Alof flies on vehicle-supplemented control food had comparable levels of dopamine in the head extract. Exposure to 125 μM rotenone for 3 days caused a significant decrease in head dopamine levels as compared to vehicle only in both wild type and Sin3Alof flies. However, rotenone-exposed Sin3Alof flies had higher dopamine levels (not significant) as compared to rotenone-exposed wild type flies. In the presence of rotenone, 10 mM SB treatment for 3 days showed a significant improvement in head dopamine level in wild type flies only. n=3 biological replicates. (b) Serotonin levels were measured in the same groups of flies as in (a). No significant differences were observed in the serotonin content among the groups. Columns=average dopamine or serotonin content; error bars=standard error; n=3 biological replicates; * p<0.05, *** p<0.001; ANOVA; Scheffe post-hoc test; p=0.73 (serotonin).
Sin3alof flies reared on 125 μM rotenone-supplemented food showed a significant dopamine deficiency as compared to vehicle treated flies (Figure 6a; ANOVA; Scheffe post hoc p=0.002). However Sin3alof flies reared on 125 μM rotenone-supplemented food displayed a non-significant trend for moderately higher dopamine content as compared to wild type flies reared on 125 μM rotenone-supplemented food (Figure 6a; ANOVA; Scheffe post hoc p=0.3).
We also measured serotonin content in head extracts after 3 days of treatment. We did not observe any effect of rotenone, SB, or Sin3alof flies on serotonin content (Figure 6b; ANOVA; p=0.73). In accordance with previous studies, we found serotonin to be less abundant as compared to dopamine in the fly brain (Yarali et al., 2009).
3.5 SB treatment does not significantly improve tyrosine hydroxylase mRNA deficiency in rotenone-treated flies
Rotenone exposure reduces tyrosine hydroxylase (TH) transcription, the rate-limiting enzyme for dopamine biosynthesis (Kim et al., 2003, Testa et al., 2005, Cannon et al., 2009). In Drosophila TH is encoded by the pale (ple) locus (Neckameyer and Quinn, 1989). We quantified TH (ple) mRNA in head RNA extracts after 3 days of treatment using real-time PCR. We did not observe statistically significant differences in TH mRNA levels among groups (ANOVA; p=0.09). Nevertheless, we observed interesting trends in TH mRNA levels. Wild type flies reared on 125 μM rotenone-supplemented food showed reduction in TH mRNA levels as compared to vehicle treated flies (Figure 7).
Figure 7. CS (SB-treated) and Sin3Alof do not significantly improve head TH mRNA deficiency in rotenone-exposed flies.
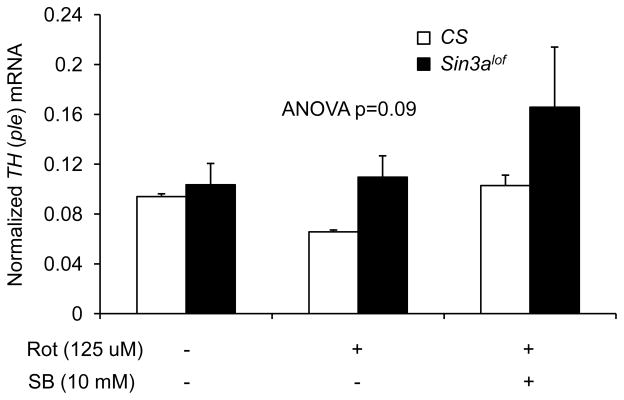
The TH mRNA levels in the head RNA extract were not statistically different between groups (n=4 biological replicates; ANOVA; p=0.09). Nevertheless there were some remarkable differences between groups. Wild type (CS) and Sin3Alof flies on vehicle-supplemented control food had comparable TH mRNA levels. Exposure to 125 μM rotenone for 3 days showed a robusr decrease in the TH mRNA levels as compared to vehicle only in wild type flies. Rotenone-exposed Sin3Alof flies had higher TH mRNA levels as compared to rotenone-exposed wild type flies. In the presence of rotenone, 10 mM SB treatment for 3 days had higher TH mRNA levels in wild type and Sin3Alof flies as compared to rotenone-exposed wild type flies. Columns=average normalized TH mRNA; error bars=standard error.
Wild type flies reared on 10 mM SB- and 125 μM rotenone-supplemented food for 3 days had a modest improvement in TH mRNA levels as compared to flies reared on 125 μM rotenone-supplemented food without SB (Figure 7). Sin3alof flies reared on 125 μM rotenone-supplemented food had a modest improvement in TH mRNA levels as compared to wild type flies reared on 125 μM rotenone-supplemented food (Figure 7).
3.6 SB treatment does not significantly improve SOD activity deficiency in rotenone-treated flies
Rotenone exposure causes mitochondrial oxidative damage and reduced SOD activity (Sherer et al., 2003, Cannon et al., 2009, Xiong et al., 2009). We quantified SOD activity in whole fly homogenates after 3 days of treatment. We did not observe statistically significant differences in SOD activity among groups (ANOVA; p=0.1). Nevertheless, we observed interesting trends in SOD activity. Wild type flies reared on 125 μM rotenone-supplemented food showed reduction in SOD activity as compared to vehicle treated flies (Figure 8).
Figure 8. CS (SB-treated) and Sin3Alof do not significantly improve SOD activity deficiency in rotenone-exposed flies.

The SOD activity in the whole fly homogenate was not statistically different between groups (n=5 biological replicates; ANOVA; p=0.1). Nevertheless there were some remarkable differences between groups. Wild type (CS) and Sin3Alof flies on vehicle-supplemented control food had comparable SOD activity. Exposure to 125 μM rotenone for 3 days showed a robust decrease in SOD activity as compared to vehicle only in wild type flies. Rotenone-exposed Sin3Alof flies had SOD activity as compared to rotenone-exposed wild type flies. In the presence of rotenone, 10 mM SB treatment for 3 days had higher SOD activity in wild type and Sin3Alof flies as compared to rotenone-exposed wild type flies. Columns=average relative percent inhibition of quercetin oxidation; error bars=standard error.
Wild type flies reared on 10 mM SB- and 125 μM rotenone-supplemented food for 3 days had a modest improvement in SOD activity levels as compared to flies reared on 125 μM rotenone-supplemented food without SB (Figure 8). Sin3alof flies reared on 125 μM rotenone-supplemented food had a modest improvement in SOD activity as compared to wild type flies reared on 125 μM rotenone-supplemented food (Figure 8).
4. Discussion
The present study is the first to examine the effect of SB, a potent HDAC inhibitor, on a rotenone-induced PD model in Drosophila. We demonstrate substantial improvement of rotenone-induced early mortality and locomotor impairment with SB supplementation. This SB-mediated improvement is associated with elevated brain dopamine levels. We also show that genetically reducing HDAC activity, by using Sin3Alof flies, results in improvement in rotenone-induced early mortality and locomotor impairment (Steffan et al., 2001, Kontopoulos et al., 2006). Furthermore, combining genetic and pharmaceutical intervention to reduce HDAC activity was additive and showed modest rescue of early mortality. The findings of this study broaden the applicability of HDAC inhibitors as an effective treatment for neurodegenerative diseases to include rotenone-induced PD.
In neurodegenerative diseases such as PD, the loss of homeostasis of acetylation due to a disrupted HDAC:HAT balance provides support for the idea that HDAC inhibitors could be therapeutic (Saha and Pahan, 2006). HDAC inhibitors have shown promise in a number of disease models, including amyotrophic lateral sclerosis, Huntington’s disease (HD), Alzheimer’s disease, stroke, and spinal muscular atrophy (Chuang et al., 2009). For example, SB has been shown to ameliorate the behavioral phenotype of HD in mice (Ferrante et al., 2003).
To date, there is limited evidence available on the effects of HDAC inhibitors in PD. More than 90% of PD cases are sporadic, and an interaction between environmental factors and individual genetic susceptibility are thought to play a major role in etiology (Cannon et al., 2009, Cicchetti et al., 2009). Therefore, in this study we focused on a well-established rotenone model of PD. Rotenone is an environmental toxin that disrupts electron transport by inhibiting mitochondrial complex I and causes oxidative damage in mitochondria associated with reduced SOD activity (Sherer et al., 2003, Cannon et al., 2009, Xiong et al., 2009). We observed a trend for a mild antioxidant effect of SB due to alleviation of rotenone-induced decline in SOD activity. Additionally, Sin3Alof flies were somewhat less deficient in SOD activity than wild type flies upon rotenone exposure. Beta-hydroxybutyrate, an endogenous HDAC inhibitor with closely related structure to SB, has an antioxidant effect in mice (Shimazu et al., 2013). Therefore it appears that the antioxidant effect of SB could contribute to the protective effects of SB on rotenone-induced toxicity.
In Drosophila rotenone exposure causes brain dopamine deficiency (Coulom and Birman, 2004). Here, SB treatment reduced rotenone-mediated dopamine deficiency in wild type flies. We also observed a trend for SB-associated alleviation of rotenone-mediated decrease in TH mRNA levels. The down regulation of TH by rotenone is relevant to a PD model because TH is the rate-limiting enzyme in synthesis of dopamine (Kim et al., 2003). Sin3alof flies were not resistant to rotenone-mediated dopamine deficit, thereby indicating involvement of an additional mechanism(s) independent of dopamine content. Nevertheless, rotenone decreases dopamine content and TH levels in cell culture, flies, and a rat model of PD (Kim et al., 2003, Coulom and Birman, 2004, Testa et al., 2005, Cannon et al., 2009, Bayersdorfer et al., 2010). To this end, many HDAC inhibitors affect TH expression. Valproic acid, an HDAC inhibitor similar in structure to SB, increased TH expression (D’Souza et al., 2009). In a mouse model of PD, valproic acid increased TH-positive cells in the substantia nigra (Kidd and Schneider, 2011). Trichostatin A, another HDAC inhibitor, also increased TH mRNA in cell lines (Kim et al., 2003). SB increases TH mRNA in neuronal and non-neuronal cells, and mice (Lewis et al., 1983, Nankova et al., 2003, Akiba et al., 2010). Therefore, it is possible that SB-mediated improvement in rotenone-induced locomotor impairment is partly a result of restoration of brain dopamine levels partially via elevation of TH mRNA levels. It should, however, be noted that dopamine itself, through disrupted homeostasis, can be cytotoxic via generation of reactive quinones and therefore contribute to rotenone-mediated oxidative damage (Emdadul Haque et al., 2003, Bayersdorfer et al., 2010). Knock down of dopamine content has been shown to reduce rotenone-mediated toxicity in flies and neuroblastoma cells (Emdadul Haque et al., 2003, Haque et al., 2003, Bayersdorfer et al., 2010).
Similarly, SB itself could have deleterious effects that are independent of rotenone-mediated insult. A recent study has shown that lifetime exposure to SB causes decreased longevity but flies reared on 10 mM SB during their senescence phase have increased longevity (McDonald et al., 2012). McDonald et al. also show that short-term exposure regimen of SB to young adults, similar to our study, has no immediate effect on viability. Therefore SB-mediated improvement of rotenone-induced early mortality may not be due to inherently increased viability due to SB exposure but may be due to other neutralizing effect of SB on rotenone toxicity. It also appears that SB-mediated improvement on rotenone-induced defects seen here is through pathway(s) in addition to TH expression, dopamine content, and oxidative damage. This notion is supported by the observed trends in SB-mediated gains in TH mRNA levels, dopamine content, and antioxidant pathway in wild type flies and partial agreement between these factors and resistance to rotenone-mediated defects in Sin3Alof flies. Accordingly, although rotenone exposure did not appear to impact brain serotonin content, rotenone-mediated locomotor defects could manifest through pathways independent of dopaminergic system and non-neuronal tissues. Therefore we cannot rule out the involvement of other pathways and cell types in improvement of rotenone-induced defects by SB and Sin3Alof. The present results also suggest that in SB-supplemented Sin3Alof flies, more robust HDAC inhibition and/or activation of additional pathway(s) could further reduce rotenone-induced mortality. It would be interesting to identify the genes and pathways affected by SB treatment that contribute towards improvement on rotenone-induced deficits.
5. Conclusion
This study provides the first evidence for SB as a therapeutic candidate for an environmental toxin-induced motor impairment observed in PD. Further studies will identify the target pathway(s) activated by SB-mediated HDAC inhibition which improve motor impairment and whether that improvement corresponds with changes in the pathology of PD. In conjunction with previous findings outlining the therapeutic attributes of HDAC inhibitors for neurodegenerative diseases, the present findings further establishes SB as a promising candidate for addressing the progressive decline of dopaminergic neurons and the debilitating motor impairment seen in PD.
Highlights.
Sodium butyrate rescues rotenone-induced locomotor defect and early death in flies.
Sodium butyrate improves rotenone-induced dopamine deficit.
Sin3A mutants (HDAC negative) are resistant to rotenone-induced locomotor defect and early death.
Acknowledgments
The authors would like to thank T. Shattuck for technical expertise in HPLC-MS and K. Roth for technical assistance. This project was supported by grants from the National Center for Research Resources, INBRE (5P20RR016463-12) and the National Institute of General Medical Sciences (8 P20 GM103423-12) from the National Institutes of Health to Colby College and Science Division Grant, Colby College to STA.
Abbreviations
- PD
Parkinson’s Disease
- HDACs
histone deacetylases
- SB
sodium butyrate
- ROT
rotenone
- L-dopa
levodopa
- TH
tyrosine hydroxylase
- SOD
superoxide dismutase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Robyn St. Laurent, Email: robyn.stlaurent@nih.gov.
Liam M. O’Brien, Email: lobrien@colby.edu.
S. Tariq Ahmad, Email: stahmad@colby.edu.
References
- Akiba Y, Cave JW, Akiba N, Langley B, Ratan RR, Baker H. Histone deacetylase inhibitors de-repress tyrosine hydroxylase expression in the olfactory bulb and rostral migratory stream. Biochem Biophys Res Commun. 2010;393:673–677. doi: 10.1016/j.bbrc.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayersdorfer F, Voigt A, Schneuwly S, Botella JA. Dopamine-dependent neurodegeneration in Drosophila models of familial and sporadic Parkinson’s disease. Neurobiol Dis. 2010;40:113–119. doi: 10.1016/j.nbd.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT. A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis. 2009;34:279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicchetti F, Drouin-Ouellet J, Gross RE. Environmental toxins and Parkinson’s disease: what have we learned from pesticide-induced animal models? Trends Pharmacol Sci. 2009;30:475–483. doi: 10.1016/j.tips.2009.06.005. [DOI] [PubMed] [Google Scholar]
- Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson’s disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Souza A, Onem E, Patel P, La Gamma EF, Nankova BB. Valproic acid regulates catecholaminergic pathways by concentration-dependent threshold effects on TH mRNA synthesis and degradation. Brain Res. 2009;1247:1–10. doi: 10.1016/j.brainres.2008.09.088. [DOI] [PubMed] [Google Scholar]
- Egorin MJ, Yuan ZM, Sentz DL, Plaisance K, Eiseman JL. Plasma pharmacokinetics of butyrate after intravenous administration of sodium butyrate or oral administration of tributyrin or sodium butyrate to mice and rats. Cancer Chemother Pharmacol. 1999;43:445–453. doi: 10.1007/s002800050922. [DOI] [PubMed] [Google Scholar]
- Emdadul Haque M, Asanuma M, Higashi Y, Miyazaki I, Tanaka K, Ogawa N. Apoptosis- inducing neurotoxicity of dopamine and its metabolites via reactive quinone generation in neuroblastoma cells. Biochimica et biophysica acta. 2003;1619:39–52. doi: 10.1016/s0304-4165(02)00440-3. [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. A Drosophila model of Parkinson’s disease. Nature. 2000;404:394–398. doi: 10.1038/35006074. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque ME, Asanuma M, Higashi Y, Miyazaki I, Tanaka K, Ogawa N. Overexpression of Cu-Zn superoxide dismutase protects neuroblastoma cells against dopamine cytotoxicity accompanied by increase in their glutathione level. Neuroscience research. 2003;47:31–37. doi: 10.1016/s0168-0102(03)00166-4. [DOI] [PubMed] [Google Scholar]
- Hayakawa T, Nakayama J. Physiological roles of class I HDAC complex and histone demethylase. Journal of biomedicine & biotechnology. 2011;2011:129383. doi: 10.1155/2011/129383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisahara S, Shimohama S. Toxin-induced and genetic animal models of Parkinson’s disease. Parkinsons Dis. 2010;2011:951709. doi: 10.4061/2011/951709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosamani R, Ramesh SR, Muralidhara Attenuation of rotenone-induced mitochondrial oxidative damage and neurotoxicty in Drosophila melanogaster supplemented with creatine. Neurochem Res. 2010;35:1402–1412. doi: 10.1007/s11064-010-0198-z. [DOI] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protection of dopaminergic cells from MPP+-mediated toxicity by histone deacetylase inhibition. Brain Res. 2010;1354:172–178. doi: 10.1016/j.brainres.2010.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd SK, Schneider JS. Protective effects of valproic acid on the nigrostriatal dopamine system in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience. 2011;194:189–194. doi: 10.1016/j.neuroscience.2011.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Park JS, Hong SJ, Woo MS, Kim SY, Kim KS. Regulation of the tyrosine hydroxylase gene promoter by histone deacetylase inhibitors. Biochem Biophys Res Commun. 2003;312:950–957. doi: 10.1016/j.bbrc.2003.11.012. [DOI] [PubMed] [Google Scholar]
- Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- Le Couteur DG, Muller M, Yang MC, Mellick GD, McLean AJ. Age-environment and gene-environment interactions in the pathogenesis of Parkinson’s disease. Rev Environ Health. 2002;17:51–64. doi: 10.1515/reveh.2002.17.1.51. [DOI] [PubMed] [Google Scholar]
- Lewis EJ, Tank AW, Weiner N, Chikaraishi DM. Regulation of tyrosine hydroxylase mRNA by glucocorticoid and cyclic AMP in a rat pheochromocytoma cell line. Isolation of a cDNA clone for tyrosine hydroxylase mRNA. J Biol Chem. 1983;258:14632–14637. [PubMed] [Google Scholar]
- Mai A, Rotili D, Valente S, Kazantsev AG. Histone deacetylase inhibitors and neurodegenerative disorders: holding the promise. Curr Pharm Des. 2009;15:3940–3957. doi: 10.2174/138161209789649349. [DOI] [PubMed] [Google Scholar]
- McDonald P, Maizi BM, Arking R. Chemical regulation of mid- and late-life longevities in Drosophila. Experimental gerontology. 2012 doi: 10.1016/j.exger.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nankova BB, Chua J, Mishra R, Kobasiuk CD, La Gamma EF. Nicotinic induction of preproenkephalin and tyrosine hydroxylase gene expression in butyrate-differentiated rat PC12 cells: a model for adaptation to gut-derived environmental signals. Pediatr Res. 2003;53:113–118. doi: 10.1203/00006450-200301000-00019. [DOI] [PubMed] [Google Scholar]
- Neckameyer WS, Quinn WG. Isolation and characterization of the gene for Drosophila tyrosine hydroxylase. Neuron. 1989;2:1167–1175. doi: 10.1016/0896-6273(89)90183-9. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annual review of neuroscience. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington’s disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poewe W, Antonini A, Zijlmans JC, Burkhard PR, Vingerhoets F. Levodopa in the treatment of Parkinson’s disease: an old drug still going strong. Clin Interv Aging. 2010;5:229–238. doi: 10.2147/cia.s6456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha RN, Pahan K. HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 2006;13:539–550. doi: 10.1038/sj.cdd.4401769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH, Miller GW, Yagi T, Matsuno-Yagi A, Greenamyre JT. Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci. 2003;23:10756–10764. doi: 10.1523/JNEUROSCI.23-34-10756.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV, Jr, de Cabo R, Ulrich S, Akassoglou K, Verdin E. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339:211–214. doi: 10.1126/science.1227166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spivey A. Rotenone and paraquat linked to Parkinson’s disease: human exposure study supports years of animal studies. Environ Health Perspect. 2011;119:A259. doi: 10.1289/ehp.119-a259a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Sun J, Tower J. FLP recombinase-mediated induction of Cu/Zn-superoxide dismutase transgene expression can extend the life span of adult Drosophila melanogaster flies. Molecular and cellular biology. 1999;19:216–228. doi: 10.1128/mcb.19.1.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabachnick BG, Fidell LS. Experimental Design Using ANOVA. Belmont: Duxbury Press; 2007. [Google Scholar]
- Testa CM, Sherer TB, Greenamyre JT. Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res. 2005;134:109–118. doi: 10.1016/j.molbrainres.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Thanvi B, Lo N, Robinson T. Levodopa-induced dyskinesia in Parkinson’s disease: clinical features, pathogenesis, prevention and treatment. Postgrad Med J. 2007;83:384–388. doi: 10.1136/pgmj.2006.054759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong N, Huang J, Zhang Z, Zhang Z, Xiong J, Liu X, Jia M, Wang F, Chen C, Cao X, Liang Z, Sun S, Lin Z, Wang T. Stereotaxical infusion of rotenone: a reliable rodent model for Parkinson’s disease. PloS one. 2009;4:e7878. doi: 10.1371/journal.pone.0007878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarali A, Krischke M, Michels B, Saumweber T, Mueller MJ, Gerber B. Genetic distortion of the balance between punishment and relief learning in Drosophila. Journal of neurogenetics. 2009;23:235–247. doi: 10.1080/01677060802441372. [DOI] [PubMed] [Google Scholar]